

Abstract

Two factors contribute to the inefficiency associated with screening pharmaceutical library collections as a means of identifying new drugs: [1] the limited success of virtual screening (VS) methods in identifying new scaffolds; [2] the limited accuracy of computational methods in predicting off-target effects. We recently introduced a 3D shape-based similarity algorithm of the SABRE program, which encodes a consensus molecular shape pattern of a set of active ligands into a 4D fingerprint descriptor. Here, we report a mathematical model for shape similarity comparisons and ligand database filtering using this 4D fingerprint method and benchmarked the scoring function HWK (Hamza–Wei–Korotkov), using the 81 targets of the DEKOIS database. Subsequently, we applied our combined 4D fingerprint and HWK scoring function VS approach in scaffold-hopping and drug repurposing using the National Cancer Institute (NCI) and Food and Drug Administration (FDA) databases, and we identified new inhibitors with different scaffolds of MycP1 protease from the mycobacterial ESX-1 secretion system. Experimental evaluation of nine compounds from the NCI database and three from the FDA database displayed IC50 values ranging from 70 to 100 μM against MycP1 and possessed high structural diversity, which provides departure points for further structure–activity relationship (SAR) optimization. In addition, this study demonstrates that the combination of our 4D fingerprint algorithm and the HWK scoring function may provide a means for identifying repurposed drugs for the treatment of infectious diseases and may be used in the drug-target profile strategy.
Introduction
Computational methodologies utilized for in silico high throughput screening (HTS) are a critical component of drug discovery approaches.1−7 Within the available in silico HTS approaches, methodologies that combine ligand- and structure-based screening procedures find the widest application.1,8 The challenge in any HTS virtual screening (VS) platform is to develop an algorithm that is sufficiently fast and robust to evaluate many compounds while maintaining sufficient accuracy to identify a subset of biological active compounds (i.e., hits) that have diverse structural scaffolds (i.e., scaffold-hopping). We sought to employ in silico screening to evaluate the repurposing of current drugs for a new therapeutic target.9−11 Drug-repurposing maximizes the potential value of each hit by screening well-known compounds that have minimal toxicity and/or few side-effects.12−14
Comparative studies of well-established ligand- and docking-based approaches concluded that shape-based ligand screening yielded markedly better outcomes than protein docking schemes.15−18 A ligand-based computational method involved two essential elements: [1] an efficient similarity measure and [2] a reliable scoring method. The similarity measure varied among different methods and focused on three factors: pharmacophores, molecular shapes, and molecular fields. The molecular-shape approaches maximized the overlap of shapes and determined a similarity value based on the degree of shape overlap. Over the years, despite the investment made in developing scoring functions for molecular-shape approaches, none possessed accuracy and general applicability. Every scoring function had its advantages as well as its limitations. Consequently, investigators turned to the consensus-scoring technique that improved the probability of finding solutions by combining the scores from multiple scoring functions or using different reference molecules.15,19−22
We recently developed an efficient 3D shape-based similarity algorithm encoding the consensus molecular shape pattern of a set of active ligands into one descriptor, called the 4D fingerprint (Figure 1). The 4D fingerprint formalism was originally proposed by Hopfinger and co-workers and developed the quantitative structure–activity relationships (4D-QSAR) model.23 The 4D-QSAR model estimates molecular similarity measures as a function of conformation, alignment, and atom type.24 The resulting descriptors values were the occupancy measures for the atoms in the investigated set of bioactive molecules. While the similarity measures achieved excellent predictions for a variety of enzyme inhibitors,25−27 the weakness of this approach lies with the occupancy measures for the atoms (or pharmacophoric groups) which may also be present in similar, “inactive” compounds.28
Figure 1.

Ligand and structure shape-based VS approach using the 4D fingerprint. The resulting 4D fingerprint encoded in the 3D shape of the candidate ligand Bi is docked and ranked using the HWK scoring function. The application of the 4D fingerprint to the ligand Bi decreases the interaction (purple arrow) with the receptor.
The 4D fingerprint approach implemented in the Shape-Approach-Based Routines Enhanced or SABRE program possessed a number of attractive advantages over other VS methods.29,30 First, it depended explicitly on 3D shape, not on the underlying chemical structure, and thus it excelled in identifying novel chemical scaffolds based on a set of known active ligands (scaffold-hopping). The iterative 4D fingerprint approach was particularly robust for several reasons: (i) the 4D fingerprint descriptors were very sensitive to the details of molecular shape of active ligands, reducing the need to use multiple conformers of multiple query structures; (ii) the method excel by the incorporation of the spatial distributions of chemical features of similar inactive ligands during the optimization and screening procedures; (iii) the algorithm was fast and had the ability to scan a library of millions of compounds in a matter of hours. The method unified ligand- and structure-based 4D fingerprint VS approaches by docking the shape filtered ligand structures into the receptor-binding cavity. Finally, running searches using this methodology was remarkably easy and required only that the end-user supply a query structure and runtime parameters to control the number of hits that were returned. Despite these advantages, the 4D fingerprint method, as previously reported, suffered from a weakness in the empirical HWZ scoring function17 for ranking and selecting the active ligands from large databases. To remedy this deficiency, we modified the shape-based VS algorithm of the SABRE program and implemented a new, robust scoring function that accommodated the diversity of ligand scaffolds with an accuracy that exceeded our prior efforts.
Tuberculosis (TB) is a chronic and complex disease resulting from infection with the bacterium Mycobacterium tuberculosis. TB remains an important public health problem worldwide, with 8.6 million estimated cases and 1.3 million deaths attributed to the disease in 2012.31 In order to combat the spread of TB—particularly resistant strains of M. tuberculosis—it is necessary to identify new molecular targets for TB drugs and develop new, more efficient, methods for screening ligands as potential drug candidates than methods used in the past. Historically, high-throughput screening (HTS) approaches coupled with in vitro testing served to identify promising hits with anti-TB activity. While successful in some cases, the HTS approach frequently failed in the antibacterial drug discovery area due to the poor ADMET properties and insufficient or improper molecular diversity of the compounds screened.32,33 The mycobacterial ESX secretion system, also referred to as the type VII secretion system, represents a promising, new target for TB drug development.34,35 The ESX secretion system is a specialized system unique to mycobacteria that secrete a large number of proteins necessary for M. tuberculosis virulence.36−38 Each ESX secretion system includes a membrane-associated subtilisin-like protease, called the mycosin: MycP1–MycP5 (numbered according to gene cluster). MycP1 from the ESX-1 system hydrolyzes the ESX associated protein B (EspB) during secretion,39−41 and this processing affects virulence in a mouse model of TB infection.42 The recent description of the molecular structure and substrate specificity of MycP143−45 prompted interest in MycP1 as a promising target for structure-based drug design.
Recently, we applied the combined ligand- and structure-based virtual screening procedure and 4D fingerprint algorithm to identify new inhibitors for MycP1 protease.30 The study reported here extends our previous work and reports a rational approach for ranking the ligand databases and demonstrates the performance of the novel HWK scoring function using 81 targets from the DEKOIS database.46 Validation of the efficiency of the VS method for scaffold-hopping and drug repurposing involved the application of this methodology for the identification of diverse inhibitor scaffolds against MycP1 protease and experimental testing of some of these scaffolds in an in vitro enzyme assay.
Methods
We have recently developed a ligand and structure shape-based VS algorithm implemented within the SABRE program.29,30 Unlike other ligand-based shape overlapping methods,47−49 our approach efficiently detected the key pharmacophore groups of the active ligands responsible for binding to the target. The main advance of our methodology resided in the consideration of “virtual” but similar inactive structures (decoys) during the consensus molecular-shape detection process (Figure 1). After similarity scoring, the selected structures were ranked according to their shape complementarity in the receptor-binding site. This report highlights the major steps of the algorithm and describes the approach used for the development of this scoring function.
Enhanced Molecular Shape-Density Model
The molecular shape density function φ(r) of a ligand is expressed in terms of the shape density functions of individual atoms and their overlap
| 1 |
in which each atom i with coordinates Ri = (Xi, Yi, Zi) is described by a spherical Gaussian:50−53
| 2 |
where σi is the van der Waals radius of the atom i. The molecular volume V of the ligand is defined as54
| 3 |
The volume vi of an atom i is
| 4 |
The intersection volume of atom pairs is defined as
| 5 |
The overlap volume of two molecules A and B is defined as
| 6 |
In the SABRE algorithm, the shape-density model is enhanced and defined as a linear combination of weighted atomic Gaussian functions.18,29,30 Thus, the molecular shape-density is the sum of all individual weighted pharmacophore densities, and the molecular volume is defined as
| 7 |
where Vkpharm–k = ∑i∫dr ρki(r) is the partial volume of the pharmacophore group k and is defined as a linear combination of atomic Gaussian functions. The optimal coefficients {Ck} are determined by iteratively adjusting the coefficients of the set of known active ligands {Ai} in the presence of virtual decoy structures {Bi} (virtual decoys are inactive similar compounds that are not necessarily synthetically feasible or identified in the previous VS rounds) until they satisfy these two criteria:
| 7a |
This algorithm quickly builds a consensus molecular-shape pattern in which the optimal coefficients {Ck} define a 4D fingerprint of the entire set of active ligands that also effectively excludes structurally similar, but inactive ligands (decoys) (Figure 1).29,30
Rational Approach for Developing a Robust and Efficient Scoring Function
Given a set of known active ligands {Ai} with volumes V{Ai} and a query structure A with the volume VA, we effect the shape-filtering and ranking the candidate molecule Bi with volume VBi.
During the VS process, we observe two trends: (i) VBi ≤ VA or (ii) VBi ≥ VA. As a result, we can rank the structure Bi according to either the condition (i), (ii), or the combination of (i) and (ii) for two different ligands Bi and Bj. Thus, we have two possible outcomes:
-
(1)The volume of the candidate structure Bi is smaller than the volume of the query A: VBi ≤ VA. The maximal overlap volume VABi for the two structures is restricted as VABi ≤ VBi and rewritten as

8 -
(2)The volume of the structure Bi is larger than the volume of the query A: VBi ≥ VA and VABi ≤ VA is rewritten as

9 - (3)
and we obtain the Tanimoto scoring function:55
| 10 |
We rewrote eqs 8 and 9 as smooth Gaussian distributions and defined the scoring function HWK (Hamza–Wei–Korotkov) that converges to one for optimal similarity:
| 11 |
| 12 |
The Tanimoto function and eqs 11 and 12 clearly reveal that the ranking of the candidate structure Bi is determined by several inhomogeneous criteria. For a fixed overlap volume, eq 11 gives the highest score for the ligand Bi with a smallest size even if it possess less similar chemical features than other ligands with larger volume sizes. Second, the VABi term takes into account the overlap volume of the full ligand size instead the volume of the key chemical features present in both the query A, and candidate ligand Bi. This result in a higher ranking for the ligand Bi with largest size since the overlap volume varies with the full size of the ligand. Third, we recently demonstrated the weakness of the Tanimoto scoring function when used for filtering the 3D shape of the ligands and found that the Tanimoto function only efficiently ranks the ligands with comparable volume size to the query.17
These drawbacks can be overcome by taking into account the specific atom-type information, such as the consensus molecular shape pattern or “4D fingerprint” of the set of known active ligands. According to the 4D fingerprint approach (eq 7), the volume of the query A and the set of active ligands {Ai} are defined as
| 13 |
The optimization of the coefficients {ck} leads to the residual volume Vε ≪ VAPHARM, and the value of the VA term in eq 7 ranges across the interval [min V{Ai}PHARM; maxV{Ai}].
In the following demonstration, we assumed that the candidate Bi is an active ligand and similar to the query A in the set of active ligands {Ai}, since if Bi is dissimilar the overlap volume converges to a small value. We have
Using the computed 4D fingerprint coefficients, the volume of Bi is written as
| 14 |
the volume of Bi* is defined by eq 7 and is equals to the sum of the weighted partial volumes of the key pharmacophore groups and its volume size is in the interval of the set of active ligand volumes V{Ai}*.
Three possible scenarios exist:
-
(1)
VBi* ≤ VA
In this case, VBi* ∈ [min(V{Ai}); VA*] and the optimal similarity value is reached for
-
(2)
VBi* ≥ VA
In this case, VBi* ∈ [VA; max(V{Ai}*)] and the optimal similarity is reached for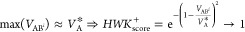
- (3)
The optimal similarity is reached for Ti = VBi*/VA ≈ Tj = VA*/VBj → 1.
As a result, the candidate ligands with a volume size slightly smaller or larger than the query volume are ranked equivalently when using the 4D fingerprint method with the Tanimoto equation. Finally, we observe that the weighting coefficients of the 4D fingerprint adjust and group the unknown “active” candidate structures with miscellaneous volume sizes and scaffolds into three classes relative to the query size. This confers the advantage of ranking more effectively these three types of shape using the HWK scoring function (HWK–, HWK+, HWKTanimoto).
Shape-Fitting Procedure
The docking approach described in our previous work combined the performance and speed of the ligand-based 4D fingerprint method with the shape characteristic of the receptor binding site.29 The current SABRE docking algorithm encodes both the 4D fingerprint and the novel HWK scoring function, and it generates alignments where patterns with similar binding character are oriented in a similar fashion in the binding site of the receptor (Figure 1). During the rigid docking process, the SABRE program takes into account only the pharmacophore groups present in the VBiPHARM(VBi*) that interact in designated ways with key receptor atoms. Five important chemical features were assigned to an atom type: hydrogen bond donor, hydrogen bond acceptor, acidic center (negatively charged at physiological pH 7), basic center (positively charged at pH 7), and metal-chelation. The main novelty of the SABRE docking approach is that the pairwise interaction between the key pharmacophoric groups (defined by the 4D fingerprint, Figure 1) of the ligand and the receptor atoms are calculated using the Gaussian function Gij. The pairwise interaction of atoms i (ligand atoms) and j (receptor atoms) is defined as
| 15 |
where DEqtype is the standard distance between the heavy atoms i and j for each “type” of interaction (i.e., hydrogen bond interaction, electrostatic interactions) and di,j is the distance between the two atom centers. The parameter ωtype is a freely adjustable parameter and controls the distribution of the Gaussian function. The parameter λtype controls the weight of the interaction type and depends on the 4D fingerprint coefficients.
The pairwise interaction is attractive for λtype > 0 and repulsive for λtype < 0. The total of n pairwise interactions between the pharmacophoric groups of the ligand and the key receptor atoms is defined by the geometric mean Gi,jTOTAL(di,j) as
| 16 |
Thus, the combination of the total pairwise interactions Gi,jTOTAL(di,j) and eqs 10–12 takes into account both the 4D fingerprint and the key interacting pharmacophoric groups of the ligand and leads to improved enrichment of the VS process. The HWKDock scoring function of the SABRE docking method is summarized by the three equations:
| 17 |
| 18 |
| 19 |
It is interesting to note that a ligand with high similarity score (eqs 10–12) is reranked with a lower score if its chemical features are close to repulsive receptor atoms. Therefore, our scoring strategy developed in the docking method combines the fast and efficient ligand-shape-based 4D fingerprint VS with an extremely quick calculation of the interactions between the ligand pharmacophoric groups and the key receptor atoms. In addition, we observe in Figure 1 that the interaction (purple arrow) involving the pharmacophore groups present in both active and decoy structures become negligible. Analysis of the HWKDock function (eqs 17–19) highlights a new strategy for improving scaffold-hopping and drug repurposing performances. During the VS campaign, the shape size of the query is fundamental and orients the choice of the scoring equations. Thus, if the shape of the query is small and does not completely fill the receptor binding cavity, the HWKDock+ is appropriate to identify structural hits with either comparable or larger volume sizes than the query volume. However, the hits with volume sizes ranging in the interval [min(V{Ai}); VA*)] of the set of active ligands (eq 14) are also ranked with a high HWKDock score. In contrast, if the shape of the query complement the receptor binding cavity, the HWKDock– is better suited to identify hits with smaller volume sizes while keeping high overlap volume with the pharmacophore groups of the query. Finally, the HWKDock is effective to identify hits with comparable query shape (i.e., rather smaller or larger structural sizes that fit into the receptor binding cavity while retaining structural diversity). Consequently, three different classes of hits emerge based on the equations of the HWK function that selected them. In each list, the compounds are first ranked according to query similarity using the 4D fingerprint approach, and the diversity is achieved by selecting compounds ranked highly using one of these scoring equations.
Evaluation of VS Efficiency and Robustness Using the Novel HWK Scoring Function
The DEKOIS (version 2.0) database of annotated active compounds and decoys was used to validate the HWK scoring function.46 The DEKOIS database is a publicly available VS test database consisting of 81 targets. For our purposes, the ratio of the number of decoys to the number of active ligands was fixed at 30. We used the DEKOIS database instead of the 40 targets of the DUD database for our VS test in order to measure the robustness of the scoring function when screening a large number of targets. This is one of the most commonly encountered measures for estimating prediction accuracy of VS algorithms.
The effectiveness of the SABRE program was evaluated using the enrichment factor (EF) metric at a given percentage of the database screened,56−58 and the area under the ROC (receiver operator characteristics). To test the efficiency of the HWK scoring function defined by eqs 10–12, we screened each target 10 times using a different set of five randomly selected active ligands (as templates) and reported both the highest performance (ROC AUC value) and enrichment factor EF at 1% for the 81 targets. Screening results using the empirical HWZ scoring function were reported for comparison.17 For each screening test, the five template structures where first removed from the list of active ligands.
Identification of Potential Inhibitors of MycP1 Protease
The detailed VS procedure was described in our previous work30 and summarized in the Supporting Information (Supplementary Figure SI-1). Briefly, the 4D fingerprint algorithm defined in the 3D-shape-based similarity method of SABRE was used as the first filter of the NCI (National Cancer Institute) from the NCI Open Database Compounds (Release 4, ∼265 000 structures) and FDA (Food and Drug Administration, 1217 compounds) database downloaded from the ZINC database.59 It is important to note that the 4D fingerprint was generated using both the previously identified leads (active ligands) and inactive compounds (considered as decoys).30 The multiple conformation states of each ligand in the database were generated using OMEGA (OpenEye Scientific Software).60−62 Thereafter, we utilized the “docking option” of the SABRE program to place the filtered conformations of each ligand into the active site of the MycP1 (PDB ID: 4HVL) and ranked them using the HWK scoring function.
Drug Repurposing Approach Using the 4D Fingerprint
According to eq 14, the candidate Bi structure is similar to the set of known active ligands {Ai} if VBi* ∈ [min(V{Ai}); max(V{Ai}*)] and VBi ≈ VAPHARM ≈ VAB, which results in HWK converging to 1. The volume VA is defined by the 4D fingerprint coefficients {Ck} that encoded the chemical features of the consensus molecular shape pattern of known active ligands (eq 7) for the specific binding target. Therefore, this suggests that the best fitted and most highly ranked ligands from the VS of the database have similar 4D fingerprint coefficients and thus should interact with the receptor of the known active ligands (concept of the drug-target profile). It is important to note that this approach considers only the 4D fingerprint and the fast-fitting method implemented in the SABRE program. To validate the effectiveness of SABRE for drug repurposing, we conducted a ligand- and structure-based VS procedure using the FDA database.
In Vitro Assay of MycP1 Inhibitors
Recombinant Mycobacterium thermoresistibile MycP1 was expressed and purified as reported previously.43 A quenched, fluorescent peptide assay was used to measure the activity of MycP1 in the presence of inhibitors. MycP1 was used to digest 20 μM of the fluorescent substrate, AbzAVKAASLGK(Dnp)OH (GenScript Inc.). Potential MycP1 inhibitors identified by SABRE were diluted to a concentration of 150 μM, and assays were measured in 96-well format. Compounds that were considered hits showed less than 50% activity compared to controls (DMSO-buffer blank). The same in vitro assay was used to measure the inhibitory concentration 50% (IC50) of the most promising hits. For IC50 measurements, inhibitors were added at 0, 5, 10, 50, 100, 200, 350, and 500 μM concentrations. Initial rates of fluorescent peptide hydrolysis were measured then incorporated into dose–response curves using GraphPad Prism.
Results and Discussion
Performance of the SABREHWK Scoring Function
We evaluated the accuracy of the HWK scoring function and compared the current values to those obtained with the empirical HWZ scoring function17 using the multi conformational states of the decoys and active ligands of the 81 targets from the DEKOIS database. The merits of scoring function became clear as it accurately ranked compounds with subtle structural changes. In the present VS trial, we selected query molecules according to the procedures presented in Kirchmair et al.63 and used to describe the performance of the algorithms.29,49
As shown in Figure 2, we evaluated the AUC for each target of the DEKOIS database and the average AUC using the SABREHWK and SABREHWZ scoring functions. The average AUC value of the best performing query for the 81 DEKOIS targets using HWK and HWZ scoring functions was 0.875 ± 0.054 and 0.851 ± 0.054, respectively. The two scoring functions had similar overall performance for some of the 81 targets based on the AUC metric; however, an analysis of the complete set of targets revealed that SABREHWK performed more consistently in terms of AUC with an average AUC ≥ 0.9 for 26 targets and 0.9 > AUC ≥ 0.8 for 51 targets than SABREHWZ. Moreover, SABREHWK did not fail for any of the 81 targets screened. In comparison, SABREHWZ ranked the screening results with an AUC > 0.9 for only 17 of the 81 DEKOIS targets (11 targets out of 81 have AUC ≤ 0.8). The detailed results for each target are displayed in Table SI-1. One of the advantages of the SABREHWK approach is that the VS performance combining the 4D fingerprint and the novel HWK scoring function depended less on the screened targets, as already observed in our previous benchmark tests using the HWZ function with the 40 DUD targets.17,18,29
Figure 2.

Comparison of the areas under the ROC curves (AUC) of the 81 DEKOIS databases using the SABREHWK and SABREHWZ scoring functions.
Analysis of the Enrichment Factor Using the SABREHWK Scoring Function
The efficiency of the SABREHWK scoring function was evaluated using the enrichment factor at 1% (EF1%), and the results were also compared to those using the SABREHWZ function (Figure 3 and Table SI-1). The average EF1% values for the 81 targets using the novel HWK and empirical HWZ score-based virtual screening were 21.8 ± 5.0 and 15.5 ± 5.9, respectively. The SABREHWK method performed more consistently resulting in an EF1% less than 10% for only two targets, whereas the results using the SABREHWZ method provided enrichment factors below 10% for 24 targets. Thus, the enrichments achieved with SABREHWK are considerably better than those obtained with the empirical HWZ scoring function, indicating that the novel scoring function was more efficient in identifying hits with notably different scaffolds compared to the query structure. Therefore, on the basis of the AUC and enrichment factor EF values, these results indicated that the novel HWK score demonstrated an improved and robust VS performance, albeit with the caveat that we used only 81 targets in this study.
Figure 3.

Comparison of the Enrichment Factor EF at 1% of the 81 DEKOIS databases using the SABREHWK and SABREHWZ scoring functions.
Identification of Novel Inhibitors of MycP1 Protease
The SABRE program was generally applicable for ranking any bioactive scaffold classes with the exception of inactive decoys. The recognition of a wide variety of structurally different ligand classes was an important goal of our virtual screening strategy. The MycP1 protease represented a challenge for both ligand- and structure-based virtual-screening approaches. Indeed, only the crystal structure of the apo form of the enzyme was available, and the protein active site is relatively large, which decreased the probability of successfully identifying and ranking the correct pose of the screened ligands. The structures of MycP1 inhibitors that we previously reported were available, and visual analysis of their putative poses in the active site revealed that the binding mode of compound 1 was reasonable.30 These compounds have chemical features that enabled SAR (structure–activity relationship) studies and generated a novel 4D fingerprint. For the purpose of SAR, an intuitive strategy for scaffold-hopping used the hit compound 1 as query and the HWKDock+ scoring function to rank hits with larger volume sizes from the NCI database. We constructed such a ranking of the best 1000 structures (top-1000) according to the docking-score function. The pharmacophore model reduced the number of these structures to a small subset of promising MycP1 lead candidates. The 135 hits derived from the NCI database were superimposed with the binding query (compound 1) and visually inspected. Forty molecules were selected from the hits and tested in vitro for inhibitory activity against MycP1. Notably, 9 compounds out of the 40 were able to inhibit MycP1 by more than 50% when added at 150 μM (Table 1 and Figure 4) and one compound showed an IC50 less than 100 μM. Compound 2 inhibited MycP1 activity in the low micromolar range with an IC50 of 76.8 μM and does not includes substructures described as Pan Assay Interference Compounds (PAINS).64
Table 1. Experimentally Determined Inhibitory Activity of the 13 Compounds Selected from the Virtual Screening.
| compound | name | % inhibition at 150 μM | IC50 (μM)a | PAINS filterc | |
|---|---|---|---|---|---|
| NCI database | 1 (query) | NSC-357905 | 73.0% | 48.0b | pass |
| 2 | NSC-67021 | 71.5% | 76.8 | pass | |
| 3 | NSC-270375 | 75.8% | pass | ||
| 4 | NSC-67931 | 63.8% | pass | ||
| 5 | NSC-356820 | 68.5% | fail | ||
| 6 | NSC-206155 | 54.7% | pass | ||
| 7 | NSC-614859 | 54.3% | pass | ||
| 8 | NSC-207092 | 55.6% | fail | ||
| 9 | NSC-111151 | 52.5% | fail | ||
| 10 | NSC-641874 | 57.3% | pass | ||
| FDA database | 11 | Hydroxystilbamidine | 79.1% | 85.6 | pass |
| 12 | Diminazene | 58.0% | fail | ||
| 13 | Thiacetazone | 80.1% | pass |
Figure 4.

Structural scaffold of MycP1 inhibitors identified during the VS of the NCI database.
The VS procedure identified these compounds based on their common chemical features (4D fingerprint) present in the subset of known active ligand structures and their fit in the binding pocket. The coefficients of the 4D fingerprint efficiently encoded the spatial distributions of pharmacophoric points providing the alignment of compounds relative to the binding site surfaces. Each point accounted for an important chemical feature such as hydrogen bond donors/acceptors and negative/positive charged groups. The basic physicochemical features of the known MycP1 compounds included the potential to establish hydrogen bonds as donors with Thr156 and Ser202, Glu203 and Thr333 residues. (Figure 5). Furthermore, analysis of the docking results revealed that the lead compound 2 fit well within the binding site cavity. The compound 2 formed hydrogen bonds with the Thr156, Thr333, Ser202, and Glu203 residues of MycP1. The results were in agreement with our previous docking studies pointing out Ser202 and Thr156 as key residues to stabilize the ligand scaffold in the MycP1 catalytic binding site.30 A detailed analysis of the docking mode of the 11 compounds (Figure 4) revealed a close match between the pattern of hydrophobic and hydrogen bond donor pharmacophoric points of these hits compared to the pharmacophore model defined in our previous study.30
Figure 5.
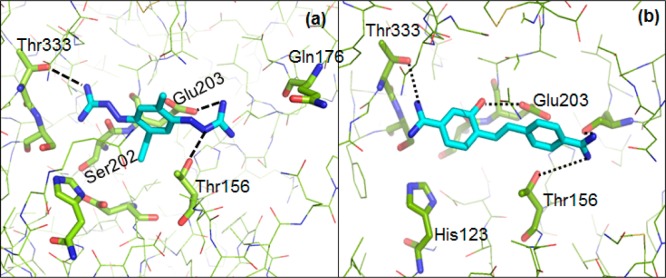
Stick view of the binding compound 2 (NSC-67021) and 11 (Hydroxystilbamidine) in the MycP1 active site.
As shown in Table 2, the VS procedure ranked 9 lead compounds at different cut-offs among the initial 1000 docked structures. Among the top-30, one lead compound was present, and this outcome corresponded to 11% coverage. Furthermore, the 9 lead compounds were among the top-300 of the filtered NCI database. These results highlight the merits of our 4D fingerprint VS approach when combined with the novel HWK scoring function. We also compared this simple approach to a complex approach including other likely query conformations. We modeled three plausible binding poses of the compound 1 (query) with different conformations in MycP1 cavity and redocked the top-1000 ligands using these three conformations, as shown in Table 2. The fusion approach markedly improved the percentage of retrieved lead compounds in the top-75 and further underscored the potential of the 4D fingerprint and HWKDock scoring VS procedure in the identification of lead compounds using the structure of unliganded receptor.
Table 2. Percent of Lead Compounds Recovered at Different Cutoffs of the Final Docked and Ranked Structures.
Total lead structures = 9.
% coverage of leads = (number of leads in the top/total lead) × 100.
Assessment of the 4D Fingerprint and HWK Scoring Function for Drug Repurposing
The integration of this newly generated computational method, which combined the 4D fingerprint and the HWK scoring function with in vitro enzyme inhibition studies, was a useful approach for evaluating current drugs, already on the market for a particular therapeutic purpose as potential agents for treating TB. To demonstrate the applicability of this integrated virtual and experimental screening for drug-repurposing, we undertook the virtual screening of the FDA-approved drug database consisting of 1,217 compounds (corresponding to 3358 structures including tautomers) using the 4D fingerprints previously generated during NCI database screening. Hits were evaluated using the aforementioned MycP1 enzyme assay. In order to increase the structural diversity of the compounds identified by this process, we conducted the VS procedure three times using compounds 1, 8, and 9 as query for each VS round. The choice of the structural query was critical to the success of this approach. As described in Methods, the scaffold diversity depended on the selected HWK– or HWK+ or HWKT (Tanimoto) scoring equations, which also depended on the query size. Thus, compound 1, discovered in our previous work, was used as query since it has the highest affinity to MycP1. The compounds 8 (larger volume than that of compound 1) and 9 (smaller volume than that of compound 1) were selected based on their differential volumes and structural diversity compared to the structure of 1. The goal of our screen was to find hits with diverse structural scaffolds and comparable volume sizes to the queries. Thus, the resulting docked ligands of the FDA database were ranked using the HWKDockTanimoto scoring function (eq 19).
Since the screening process of the NCI database and the benchmark test using the 4D fingerprint of SABRE program demonstrated high enrichment factor at 1% of the screened database, we visually inspected the binding mode of the best 30 structures (∼1%) identified within the FDA database. We focused, in particular, on four compounds based on their high HWKDockTanimoto docking score and their interactions with the key residues (Thr156 and Ser202) of the MycP1 binding cavity. These four compounds were chosen for in vitro inhibition assays, and three out of the four selected compounds exhibited more than 50% inhibition of MycP1 when used at 150 μM (Table 1), which validated the merits of our VS approach. Finally, the active compounds were filtered for Pan Assay Interference Compounds (PAINS) (Table 1) and showed that the hydroxystilbamidine scaffold may be used as starting structure for further optimization. An analysis of the three ranked FDA approved drugs showed that the compounds 11, 12, and 13 were ranked by SABRE near the top at positions 2, 24, and 3 out of 1217. The high score for compounds 11 and 13 was attributable to the HWKDock scoring function, which took into account the ligand similarity as well as the optimal ligand/receptor pharmacophore model (eq 19). Out of the three, compound 11 had the greatest effect, with an IC50 of 85.6 μM, whereas the two other leads had IC50 > 100 μM. In addition, we noted the low structural similarity between the three identified FDA compounds (Figure 6). As observed for the other leads, compound 11 formed hydrogen bonds with the Thr156, Glu203, and Thr333 residues of the MycP1 active site (Figure 5). Interestingly, compound 11 is typically used as a histochemical stain to understand the distribution and localization of biomarkers,65 and these results suggested that it or its analogs may be repurposed for inhibition of MycP1. More importantly, these preliminary findings show that the SABRE algorithm with HWK scoring provides an efficient means for the identification of new uses for current drugs and encourages us to pursue the applicability of methodology in drug repurposing strategy for other medically relevant drug targets.
Figure 6.

Structural scaffolds of MycP1 inhibitors identified during the VS of the FDA database. The percentage of inhibition and IC50 are displayed.
Assessment of Lead Scaffold Diversity
Published data suggested counting hits only when the chemotype of a molecule is not equal to a template chemotype or any other chemotype that already exists in the hit list.66 This approach resulted in a chemotype enrichment that emphasizes discovery of ligands with different chemotype properties. We assessed the novelty of the confirmed 12 hits by comparing their structural similarities with a “simple 2D descriptor”.67 We computed the pairwise similarity index using the molecular access system MACCS structural keys (MACCS, 166 bits) of our 13 compounds (query +12 leads) and represented the structural diversity using the heat map (Figure 7). The MACCS similarity indexes were calculated using Openbabel.68 The map visualizes 15 × 15 = 225 pairwise comparisons and was color-coded by similarity values ranging from red (low similarity value) to dark blue (high similarity value). We observed only two lobes in dark blue consistent with high similarity between the compounds (MACCS index > 0.8) and most of the compounds were dissimilar. This result supported the increased structural diversity (MACCS index < 0.6) of the new lead compounds using the combined 4D fingerprint and HWK scoring function. Considering the high degree of substructure encoded in each VS round, it was not surprising that the 4D fingerprint algorithm performed well at finding diverse chemotypes.
Figure 7.
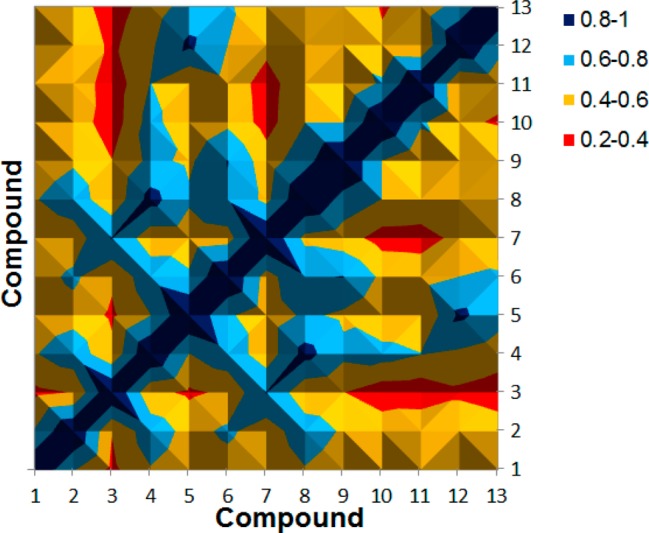
Heat map of the MACCS similarity index for the 13 compounds (12 leads + query).
Conclusion
We report a rational method for the design of novel scoring function HWK and validated its performance using a large number of targets from the DEKOIS database. The VS approach test using the 4D fingerprint and the HWK scoring function provided high enrichment factors in detecting active compounds at early stage of the 81 screened databases. We validated the efficiency of the combined 4D fingerprint and HWK scoring function in scaffold-hopping strategy through the identification of nine novel lead compounds in a short hit list from the VS of the NCI database. The result of the VS round ranked these compounds in the top-300 of the database, and one of them displayed an IC50 comparable to that of the reference structure.
In the absence of new drugs for infectious diseases like TB, it made sense to develop a VS strategy capable of exploring databases of current drugs used to treat diseases other than infectious diseases and potentially repurpose some of them for TB treatment.9−11 The merit of this approach lies in the obvious point that these commercially available drugs lack significant toxicity or side-effects.12−14 To test this notion, the screening of the FDA database using our screening approach identified three FDA-approved compounds as potential lead structures. One of these compounds displayed an IC50 of 85.6 μM against MycP1 protease. The distributions of pairwise structural similarities presented in the heat map revealed that the 13 lead compounds resulting from the VS of NCI and FDA databases were structurally diverse. In summary, this study represents the comprehensive quantification of VS approach for scaffold-hopping and drug repurposing and provides a solid strategy for the discovery of new classes of MycP1 inhibitors.
Acknowledgments
We thank Timothy J. Evans for assistance with protein purification. We acknowledge the Drug Synthesis and Chemistry Branch of National Cancer Institute for supplying the compounds used in the virtual screening and in vitro testing. We thank the University of Kentucky Information Technology department and Center for Computational Sciences for computing time on the Lipscomb High Performance Computing Cluster. We acknowledge the use of OpenEye Scientific Software via free academic licensing program. We acknowledge the University of Kentucky Organic Synthesis Core that is partially supported by grant 1P30GM110787 from the National Institute of General Medical Sciences (to Louis B. Hersh). This study was supported by NIH/NIGMS grant P20GM103486 (to K.V.K.), and its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH or the NIGMS.
Supporting Information Available
Detailed information about the enrichment factor at top 1% and the ROC (AUC) values for the 81 DEKOIS targets using SABRE program and rank of lead compounds from NCI and FDA databases. These information are available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
# Department of Molecular Physiology and Biological Physics and The Myles H. Thaler Center for AIDS and Human Retrovirus Research, University of Virginia, Charlottesville, Virginia 22908, United States.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Polgar T.; Keseru G. M. Integration of virtual and high throughput screening in lead discovery settings. Comb. Chem. High Throughput Screen. 2011, 14, 889–897. [DOI] [PubMed] [Google Scholar]
- Langer T.; Hoffmann R. D. Virtual screening: an effective tool for lead structure discovery?. Curr. Pharm. Des. 2001, 7, 509–527. [DOI] [PubMed] [Google Scholar]
- John S.; Thangapandian S.; Sakkiah S.; Lee K. W. Discovery of potential pancreatic cholesterol esterase inhibitors using pharmacophore modelling, virtual screening, and optimization studies. J. Enzyme Inhib. Med. Chem. 2011, 26, 535–545. [DOI] [PubMed] [Google Scholar]
- Bi J.; Yang H.; Yan H.; Song R.; Fan J. Knowledge-based virtual screening of HLA-A*0201-restricted CD8(+) T-cell epitope peptides from herpes simplex virus genome. J. Theor. Biol. 2011, 281, 133–139. [DOI] [PubMed] [Google Scholar]
- Akula N.; Zheng H.; Han F. Q.; Wang N. Discovery of novel SecA inhibitors of Candidatus Liberibacter asiaticus by structure based design. Bioorg. Med. Chem. Lett. 2011, 21, 4183–4188. [DOI] [PubMed] [Google Scholar]
- Englebienne P.; Moitessier N. Docking ligands into flexible and solvated macromolecules. 4. Are popular scoring functions accurate for this class of proteins?. J. Chem. Inf. Model. 2009, 49, 1568–1580. [DOI] [PubMed] [Google Scholar]
- Bleicher K. H.; Green L. G.; Martin R. E.; Rogers-Evans M. Ligand identification for G-protein-coupled receptors: a lead generation perspective. Curr. Opin. Chem. Biol. 2004, 8, 287–296. [DOI] [PubMed] [Google Scholar]
- Gohlke H.; Klebe G. Approaches to the description and prediction of the binding affinity of small-molecule ligands to macromolecular receptors. Angew. Chem., Int. Ed. Engl. 2002, 41, 2645–2676. [DOI] [PubMed] [Google Scholar]
- Boguski M. S.; Mandl K. D.; Sukhatme V. P. Repurposing with a difference. Science 2009, 324, 1394–1395. [DOI] [PubMed] [Google Scholar]
- D’Oca G. Program aims to find new use for old drugs. Future Med. Chem. 2013, 5, 1372–1372. [Google Scholar]
- Conticello C.; Martinetti D.; Adamo L.; Buccheri S.; Giuffrida R.; Parrinello N.; Lombardo L.; Anastasi G.; Amato G.; Cavalli M.; Chiarenza A.; De Maria R.; Giustolisi R.; Gulisano M.; Di Raimondo F. Disulfiram, an old drug with new potential therapeutic uses for human hematological malignancies. Int. J. Cancer 2012, 131, 2197–2203. [DOI] [PubMed] [Google Scholar]
- Gleeson M. P.; Hersey A.; Montanari D.; Overington J. Probing the links between in vitro potency, ADMET and physicochemical parameters. Nat. Rev. Drug Discovery 2011, 10, 197–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald M. L.; Lamerdin J.; Owens S.; Keon B. H.; Bilter G. K.; Shang Z.; Huang Z.; Yu H.; Dias J.; Minami T.; Michnick S. W.; Westwick J. K. Identifying off-target effects and hidden phenotypes of drugs in human cells. Nat. Chem. Biol. 2006, 2, 329–337. [DOI] [PubMed] [Google Scholar]
- Bender A.; Scheiber J.; Glick M.; Davies J. W.; Azzaoui K.; Hamon J.; Urban L.; Whitebread S.; Jenkins J. L. Analysis of pharmacology data and the prediction of adverse drug reactions and off-target effects from chemical structure. ChemMedChem. 2007, 2, 861–873. [DOI] [PubMed] [Google Scholar]
- Venkatraman V.; Perez-Nueno V. I.; Mavridis L.; Ritchie D. W. Comprehensive comparison of ligand-based virtual screening tools against the DUD data set reveals limitations of current 3D methods. J. Chem. Inf. Model. 2010, 50, 2079–2093. [DOI] [PubMed] [Google Scholar]
- Giganti D.; Guillemain H.; Spadoni J.-L.; Nilges M.; Zagury J.-F.; Montes M. Comparative evaluation of 3D virtual ligand screening methods: impact of the molecular alignment on enrichment. J. Chem. Inf. Model. 2010, 50, 992–1004. [DOI] [PubMed] [Google Scholar]
- Hamza A.; Wei N. N.; Zhan C. G. Ligand-based virtual screening approach using a new scoring function. J. Chem. Inf. Model. 2012, 52, 963–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamza A.; Wei N.-N.; Hao C.; Xiu Z.; Zhan C.-G. A novel and efficient ligand-based virtual screening approach using the HWZ scoring function and an enhanced shape-density model. J. Biomol. Struct. Dyn. 2012, 31, 1236–1250. [DOI] [PubMed] [Google Scholar]
- Hert J.; Willett P.; Wilton D. J.; Acklin P.; Azzaoui K.; Jacoby E.; Schuffenhauer A. New methods for ligand-based virtual screening: use of data fusion and machine learning to enhance the effectiveness of similarity searching. J. Chem. Inf. Model. 2006, 46, 462–470. [DOI] [PubMed] [Google Scholar]
- Wang R. X.; Wang S. M. How does consensus scoring work for virtual library screening? An idealized computer experiment. J. Chem. Inf. Comput. Sci. 2001, 41, 1422–1426. [DOI] [PubMed] [Google Scholar]
- Charifson P. S.; Corkery J. J.; Murcko M. A.; Walters W. P. Consensus scoring: a method for obtaining improved hit rates from docking databases of three-dimensional structures into proteins. J. Med. Chem. 1999, 42, 5100–5109. [DOI] [PubMed] [Google Scholar]
- Hert J.; Willett P.; Wilton D. J. Comparison of fingerprint-based methods for virtual screening using multiple bioactive reference structures. J. Chem. Inf. Comput. Sci. 2004, 44, 1177–1185. [DOI] [PubMed] [Google Scholar]
- Hopfinger A. J.; Wang S.; Tokarski J. S.; Jin B. Q.; Albuquerque M.; Madhav P. J.; Duraiswami C. Construction of 3D-QSAR models using the 4D-QSAR analysis formalism. J. Am. Chem. Soc. 1997, 119, 10509–10524. [Google Scholar]
- Pan D. H.; Iyer M.; Liu J. Z.; Li Y.; Hopfinger A. J. Constructing optimum blood brain barrier QSAR models using a combination of 4D-molecular similarity measures and cluster analysis. J. Chem. Inf. Comput. Sci. 2004, 44, 2083–2098. [DOI] [PubMed] [Google Scholar]
- Senese C. L.; Duca J.; Pan D.; Hopfinger A. J.; Tseng Y. J. 4D-fingerprints, universal QSAR and QSPR descriptors. J. Chem. Inf. Comput. Sci. 2004, 44, 1526–1539. [DOI] [PubMed] [Google Scholar]
- Iyer M.; Hopfinger A. J. Treating chemical diversity in QSAR analysis: modeling diverse HIV-1 integrase inhibitors using 4D fingerprints. J. Chem. Inf. Model. 2007, 47, 1945–1960. [DOI] [PubMed] [Google Scholar]
- Pasqualoto K. F. M.; Ferreira E. I.; Santos O. A.; Hopfinger A. J. Rational design of new antituberculosis agents: receptor-independent four-dimensional quantitative structure-activity relationship analysis of a set of isoniazid derivatives. J. Med. Chem. 2004, 47, 3755–3764. [DOI] [PubMed] [Google Scholar]
- Andrade C. H.; Pasqualoto K. F. M.; Ferreira E. I.; Hopfinger A. J. 4D-QSAR: perspectives in drug design. Molecules 2010, 15, 3281–3294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei N.-N.; Hamza A. SABRE: Ligand/structure-based virtual screening approach using consensus molecular-shape pattern recognition. J. Chem. Inf. Model. 2014, 54, 338–346. [DOI] [PubMed] [Google Scholar]
- Hamza A.; Wagner J. M.; Evans T. J.; Frasinyuk M. S.; Kwiatkowski S.; Zhan C.-G.; Watt D. S.; Korotkov K. V. Novel mycosin protease MycP1 inhibitors identified by virtual screening and 4D fingerprints. J. Chem. Inf. Model. 2014, 54, 1166–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WorldHealthOrganization. WHO global tuberculosis report. 2013, http://www.who.int/tb/publications/global_report/en/ (accessed on July 2, 2014).
- Payne D. J.; Gwynn M. N.; Holmes D. J.; Pompliano D. L. Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat. Rev. Drug Discovery 2007, 6, 29–40. [DOI] [PubMed] [Google Scholar]
- Fischbach M. A.; Walsh C. T. Antibiotics for emerging pathogens. Science 2009, 325, 1089–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J. M.; Pojer F.; Blasco B.; Cole S. T. Towards anti-virulence drugs targeting ESX-1 mediated pathogenesis of Mycobacterium tuberculosis. Drug Discovery Today Dis. Mech. 2010, 7, e25–e31. [Google Scholar]
- Bottai D.; Serafini A.; Cascioferro A.; Brosch R.; Manganelli R. Targeting type VII/ESX secretion systems for development of novel antimycobacterial drugs. Curr. Pharm. Des. 2014, 20, 4346–4356. [DOI] [PubMed] [Google Scholar]
- Stanley S. A.; Raghavan S.; Hwang W. W.; Cox J. S. Acute infection and macrophage subversion by Mycobacterium tuberculosis require a specialized secretion system. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 13001–13006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoop E. J. M.; Bitter W.; van der Sar A. M. Tubercle bacilli rely on a type VII army for pathogenicity. Trends Microbiol. 2012, 20, 477–484. [DOI] [PubMed] [Google Scholar]
- Houben E. N.; Korotkov K. V.; Bitter W. Take five — type VII secretion systems of Mycobacteria. Biochim. Biophys. Acta 2014, 1844, 1707–1716. [DOI] [PubMed] [Google Scholar]
- McLaughlin B.; Chon J. S.; MacGurn J. A.; Carlsson F.; Cheng T. L.; Cox J. S.; Brown E. J. A Mycobacterium ESX-1-secreted virulence factor with unique requirements for export. PLoS Pathog. 2007, 3, 1051–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J.; Laine O.; Masciocchi M.; Manoranjan J.; Smith J.; Du S. J.; Edwards N.; Zhu X.; Fenselau C.; Gao L.-Y. A unique Mycobacterium ESX-1 protein co-secretes with CFP-10/ESAT-6 and is necessary for inhibiting phagosome maturation. Mol. Microbiol. 2007, 66, 787–800. [DOI] [PubMed] [Google Scholar]
- Chen J. M.; Zhang M.; Rybniker J.; Boy-Röttger S.; Dhar N.; Pojer F.; Cole S. T. Mycobacterium tuberculosis EspB binds phospholipids and mediates EsxA-independent virulence. Mol. Microbiol. 2013, 89, 1154–1166. [DOI] [PubMed] [Google Scholar]
- Ohol Y. M.; Goetz D. H.; Chan K.; Shiloh M. U.; Craik C. S.; Cox J. S. Mycobacterium tuberculosis MycP1 protease plays a dual role in regulation of ESX-1 secretion and virulence. Cell Host Microbe 2010, 7, 210–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner J. M.; Evans T. J.; Chen J.; Zhu H.; Houben E. N.; Bitter W.; Korotkov K. V. Understanding specificity of the mycosin proteases in ESX/type VII secretion by structural and functional analysis. J. Struct. Biol. 2013, 184, 115–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomonson M.; Huesgen P. F.; Wasney G. A.; Watanabe N.; Gruninger R. J.; Prehna G.; Overall C. M.; Strynadka N. C. Structure of the mycosin-1 protease from the mycobacterial ESX-1 protein type VII secretion system. J. Biol. Chem. 2013, 288, 17782–17790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun D.; Liu Q.; He Y.; Wang C.; Wu F.; Tian C.; Zang J. The putative propeptide of MycP1 in mycobacterial type VII secretion system does not inhibit protease activity but improves protein stability. Protein Cell 2013, 4, 921–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer M. R.; Ibrahim T. M.; Vogel S. M.; Boeckler F. M. Evaluation and optimization of virtual screening workflows with DEKOIS 2.0 — a public library of challenging docking benchmark sets. J. Chem. Inf. Model. 2013, 53, 1447–1462. [DOI] [PubMed] [Google Scholar]
- Mavridis L.; Hudson B. D.; Ritchie D. W. Toward high throughput 3D virtual screening using spherical harmonic surface representations. J. Chem. Inf. Model. 2007, 47, 1787–1796. [DOI] [PubMed] [Google Scholar]
- Dror O.; Schneidman-Duhovny D.; Inbar Y.; Nussinov R.; Wolfson H. J. Novel approach for efficient pharmacophore-based virtual screening: method and applications. J. Chem. Inf. Model. 2009, 49, 2333–2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan X.; Li J.; Liu Z.; Zheng M.; Ge H.; Xu J. Enhancing molecular shape comparison by weighted Gaussian functions. J. Chem. Inf. Model. 2013, 53, 1967–1978. [DOI] [PubMed] [Google Scholar]
- Mezey P. G. Topological shape analysis of chain molecules: an application of the GSTE principle. J. Math. Chem. 1993, 12, 365–373. [Google Scholar]
- Walker P. D.; Arteca G. A.; Mezey P. G. A complete shape characterization for molecular charge densities represented by Gaussian-type functions. J. Comput. Chem. 1991, 12, 220–230. [Google Scholar]
- Grant J. A.; Pickup B. T. A Gaussian description of molecular shape. J. Phys. Chem. 1995, 99, 3503–3510. [Google Scholar]
- Grant J. A.; Gallardo M. A.; Pickup B. T. A fast method of molecular shape comparison: a simple application of a Gaussian description of molecular shape. J. Comput. Chem. 1996, 17, 1653–1666. [Google Scholar]
- Grant J. A.; Pickup B. T. A Gaussian description of molecular shape. J. Phys. Chem. 1996, 100, 2456–2456. [Google Scholar]
- Rogers D. J.; Tanimoto T. T. A computer program for classifying plants. Science 1960, 132, 1115–1118. [DOI] [PubMed] [Google Scholar]
- Jacobsson M.; Liden P.; Stjernschantz E.; Bostrom H.; Norinder U. Improving structure-based virtual screening by multivariate analysis of scoring data. J. Med. Chem. 2003, 46, 5781–5789. [DOI] [PubMed] [Google Scholar]
- Hecker E. A.; Duraiswami C.; Andrea T. A.; Diller D. J. Use of catalyst pharmacophore models for screening of large combinatorial libraries. J. Chem. Inf. Comput. Sci. 2002, 42, 1204–1211. [DOI] [PubMed] [Google Scholar]
- Diller D. J.; Li R. X. Kinases, homology models, and high throughput docking. J. Med. Chem. 2003, 46, 4638–4647. [DOI] [PubMed] [Google Scholar]
- Irwin J. J.; Shoichet B. K. ZINC — A free database of commercially available compounds for virtual screening. J. Chem. Inf. Model. 2005, 45, 177–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OMEGA, version 2.5.1; OpenEye Scientific Software; Santa Fe, NM; http://www.eyesopen.com. [Google Scholar]
- Hawkins P. C. D.; Skillman A. G.; Warren G. L.; Ellingson B. A.; Stahl M. T. Conformer generation with OMEGA: algorithm and validation using high quality structures from the Protein Databank and Cambridge Structural Database. J. Chem. Inf. Model. 2010, 50, 572–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins P. C. D.; Nicholls A. Conformer generation with OMEGA: learning from the data set and the analysis of failures. J. Chem. Inf. Model. 2012, 52, 2919–2936. [DOI] [PubMed] [Google Scholar]
- Kirchmair J.; Distinto S.; Markt P.; Schuster D.; Spitzer G. M.; Liedl K. R.; Wolber G. How to optimize shape-based virtual screening: choosing the right query and including chemical information. J. Chem. Inf. Model. 2009, 49, 678–692. [DOI] [PubMed] [Google Scholar]
- Baell J. B.; Holloway G. A. New substructure filters for removal of Pan Assay Interference Compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [DOI] [PubMed] [Google Scholar]
- Schmued L. C.; Fallon J. H. Fluoro-gold: a new fluorescent retrograde axonal tracer with numerous unique properties. Brain Res. 1986, 377, 147–154. [DOI] [PubMed] [Google Scholar]
- Good A. C.; Hermsmeier M. A.; Hindle S. A. Measuring CAMD technique performance: A virtual screening case study in the design of validation experiments. J. Comput. Aided Mol. Des. 2004, 18, 529–536. [DOI] [PubMed] [Google Scholar]
- Jain A. N.; Nicholls A. Recommendations for evaluation of computational methods. J. Comput. Aided Mol. Des. 2008, 22, 133–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Boyle N. M.; Banck M.; James C. A.; Morley C.; Vandermeersch T.; Hutchison G. R. Open Babel: an open chemical toolbox. J. Cheminform. 2011, 3, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.