

Abstract
A limitation of traditional molecular dynamics (MD) is that reaction rates are difficult to compute. This is due to the rarity of observing transitions between metastable states since high energy barriers trap the system in these states. Recently the weighted ensemble (WE) family of methods have emerged which can flexibly and efficiently sample conformational space without being trapped and allow calculation of unbiased rates. However, while WE can sample correctly and efficiently, a scalable implementation applicable to interesting biomolecular systems is not available. We provide here a GPLv2 implementation called AWE-WQ of a WE algorithm using the master/worker distributed computing WorkQueue (WQ) framework. AWE-WQ is scalable to thousands of nodes and supports dynamic allocation of computer resources, heterogeneous resource usage (such as central processing units (CPU) and graphical processing units (GPUs) concurrently), seamless heterogeneous cluster usage (i.e., campus grids and cloud providers), and support for arbitrary MD codes such as GROMACS, while ensuring that all statistics are unbiased. We applied AWE-WQ to a 34 residue protein which simulated 1.5 ms over 8 months with peak aggregate performance of 1000 ns/h. Comparison was done with a 200 μs simulation collected on a GPU over a similar timespan. The folding and unfolded rates were of comparable accuracy.
Introduction
Proteins are complex molecules of fundamental importance in biological processes. Numerical simulation using molecular dynamics (MD) has proven to be a powerful tool to predict many important properties such as the native state of the protein or its free energy.1,2 In this paper, we will focus on methods, based on MD, to calculate reaction rates, which are defined as transition rate between metastable states or conformations of the protein. As a byproduct of our analysis, we will also calculate the main mechanisms of the reaction, i.e., the transition pathways. For example, a cartoon model of the free energy for a biomolecule is shown in Figure 1. It illustrates schematically how MD trajectories explore the conformational space. The left region represents the reactant states (R), and the right the product states (P). Trajectories spend most of their time in R or P with infrequent transitions due to the energy barrier that separates the two states.
Figure 1.
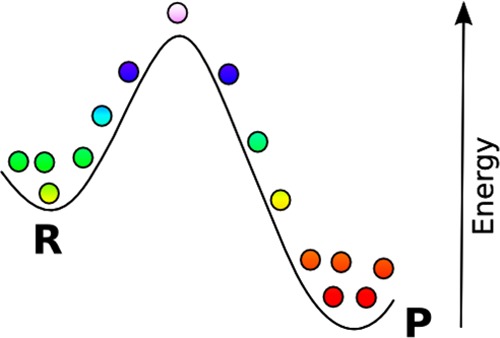
Molecular dynamics sampling of a one-dimensional free-energy surface between reactant (R) and product (P) states. By periodically recording the molecular conformation, energy, and other observables, various relevant properties of the protein can be computed.
In 1977 McCammon et al. applied MD to the bovine pancreatic trypsin inhibitor (BPTI).3 While the system was simple (vacuum with a crude force field), the simulation nonetheless contributed to shifting the view of proteins as rigid “brass models.” Since this initial simulation, MD has been used to study a wide variety of topics, such as identification of integral motions such as hinge bending modes,4 tRNA flexibility,5 and the study of E. coli chaperone GroEL.6 The application of MD to larger and more complicated molecules impacts development of force-field parameters such as the CHARMM7,8 and AMBER families,9 and solvent models (generaly categorized as implicit10 or explicitly defined11−13). Recently, MD has been used to study an HIV capsid14 and a complete satellite tobacco mosaic virus,15 as well as screening designed proteins.16
MD simulation of protein is a notoriously difficult computational task. In fact, a large portion of the major supercomputers’ time is currently dedicated to this type of simulation. The main difficulty is that the time scales of interest are typically in the millisecond (10–3 s) while a typical time step in MD is on the order of the femtosecond (10–15 s). Therefore, a brute force simulation would require on the order of 1000 billion time steps, which is impractical given the current hardware.
High-performance computing infrastructures allow development of efficient parallel algorithms to speed up simulations.17−19 Specialized hardware, such as MDGRAPE20,21 and Anton,22,23 provide orders-of-magnitude speedup over traditional high-power computing (HPC) simulations. Similarly, work with graphical processing units (GPUs) have shown the that GPUs achieved greater performance over a single cntral processing unit (CPU),24−27 allowing a single GPU to simulate biological systems with comparable performance to a cluster.27,28
Most approaches to date attempted to accelerate a single (or a few) very long simulations. Due to the sequential nature of MD simulations, this is clearly a challenging task, which limited scalability. Force-calculations are a major bottleneck for MD. As such, much work has gone into the development of fast and efficient algorithms to dedicate large resources to long simulations of molecules. For instance, NAMD and AMBER were among the first to achieve scaling to hundreds of nodes and microsecond-time scales using parallel implementations.29,30 Improvements to constraint algorithms,31,32 particle interactions,33−35 and memory access patterns17,36 have resulted in significant performance improvements over the decades.
At the other end, a recent class of methods is attempting to predict equilibrium properties, such as free energy, reaction rate and reaction pathways, from a large number of short trajectories. Since the trajectories are largely independent, this approach is naturally scalable. In fact, in this paper, we will demonstrate large-scale simulations using parallel computer grids, showing that scalability and performance can be achieved even on slow networks or when using geographically remote compute nodes.
This class of approaches have been applied with great success by several groups, using a method called the Markov state model (MSM).37−40 This method partitions the conformational space into cells or states. Then a stochastic transition matrix is computed. The entry (i ,j) in the matrix stores the probability of a trajectory to go from cell i to cell j after some lag time τ. From the eigenvalues, eigenvectors, and other properties of the matrix, we can extract all relevant time scales and kinetics of interest.
This type of approach requires running a large number of trajectories of length τ and as such is easily parallelizable. The accuracy of this method is independent of the presence of energy barriers or of the metastability of regions in conformational space. Instead it relies on efficient local sampling to predict time scales that can be orders of magnitude longer than the aggregate simulation time.
One limitation of MSM is that its accuracy depends on the choice of τ. In fact, getting converged rates is very difficult. At short lag times τ, a significant bias is present in the prediction, that is even in the presence of infinite sampling the predicted rate is incorrect. This can be addressed by increasing the lag time but this has two limitations. First, this leads to an increase in required simulation time. Second, this typically leads to a larger variance. In practice, it is difficult to obtain converged results with respect to the lag time and sampling size.
Once a MSM has been computed, one can obtain the implicit time scales from the diagonalization of the stochastic transition matrix. These implicit time scales are highly sensitive to the time lag, or the time between samples used to construct the transition matrix: at short time lags implicit time scales have a bias that makes them appear much faster than they really are. At large time lags the statistical error dominates, and it is hard to find a compromise that balances these two errors.
In this paper, we considered a variant method, called the weighted ensemble (WE) approach which has a higher computational cost but convergence is more robust and easier to monitor.41−43 WE also avoids the systematic biases found in MSM.44 The WE method was introduced by Huber and Kim41 to address the problem of capturing infrequent reaction events under Brownian dyanamics. Further studies have applied it to simulated annealing (SI) obtaining higher success rate (order of magnitude) for finding the global optimum as compared to traditional SI,45 as well as in the investigation of super oxide dismutase and monoclonal antibody NC6.8. Zuckerman et al. have extended WE and describe several key properties:46 (i) WE produces unbiased results for both Markovian and non-Markovian dynamics, indicating that the method is much more general than previously understood and (ii) the method depends on the number of cells, which may be updated dynamically, allowing the system to discover unknown cells without loss of accuracy in the calculation of reaction rates. Applications of WE to toy models and alanine dipeptid1e,42 coarse-grained models of adenylate cyclase,43 and the sodium symport Mhp2 transporter protein47 have yielded promising results.
In WE a large number of parallel walkers (MD trajectories) are used. Later, we will explain in more details the algorithm, but the basic process is to start a large number of trajectories from regions in a partitioning of the free-energy surface. After a small number of MD integration steps we consider the population of walkers in each cell. Then, using a statistically unbiased procedure, we are able to either remove walkers from overpopulated cells or, on the contrary, repopulate cells that are becoming depleted. This is done through an appropriate subselection (killing walkers) or duplication process. This ensures that when the simulation restarts the number of walkers in each cell is the same, and is equal to some target number. Then, we resume the simulation and integrate forward in time each walker using MD. This process is repeated until the quantities of interest, such as the reaction rates, which are computed through an appropriate postprocessing procedure, are converged.
This method avoids the slow time scales found in MD by ensuring that all cells are equally populated. This means for example that regions near the transition barrier between two metastable regions are sampled as often as minimum free energy basins. This method can be interpreted as a way of enhancing the sampling of conformational space. We note that its efficiency still depends on an appropriate choice of macro-states or cells, although the fact that these cells form a simple partition and can be constructed in a variety of ways (e.g., they can be simple Voronoi cells) gives a lot of flexibility to the method. Some of the advantages of WE is that it is easy to setup, is inherently massively parallel and scalable, and is unbiased. For example, unlike MSM, WE is guaranteed to converge to the exact result with enough samples.
Notwithstanding these properties, AWE, like MSM and other related methods, remains computationally expensive due to the large number of walkers that need to be run. This paper will present a software infrastructure we developed to facilitate running such calculations with accelerated convergence. This is achieved by an appropriate selection of the initial conditions as described in43,44 hence the method is known as accelerated weighted ensemble (AWE), an extension of WE where the initial weights have been approximated to the steady state weights to accelerate the convergence. We compute them from a transition matrix obtained from the simulation, based on cells as they come from clustering, perhaps using MSMBuilder. AWE also uses a simpler and more accurate method for resampling and generating new walker populations through splitting and merging of walkers.44
AWE allows one to eliminate the bias introduced in MSM by generating the statistically exact distribution of walkers inside each cell. MSM uses the equilibrium distribution for the walkers in each cell, which causes a bias. Instead, AWE uses an out-of-equilibrium distribution corresponding to walkers flowing from reactants to product states (e.g., folding/unfolding) and vice versa, thereby producing exact statistics.44
We focused on the following main goals when designing the software:
-
1.
Allow using any MD code in a black-box manner, without having to make intrusive changes inside the code. This is important since many MD codes are available and people are often required to use a particular MD code because of some special required capability. For instance, the following MD codes are usable with our framework: AMBER,48 CHARMM,49 GROMACS,17,50 LAMMPS,51 NAMD,18 MDynaMix,52 Orac,53,54 GROMOS,55 Tinker,56 Desmond,57 DL_POLY.58
-
2.
Dynamically scale to available resources as they are added and removed as availability or funding allows and robustly handle resource failure. Details are discussed later, but the main idea is to support, for example, starting AWE using a dedicated cluster locally then adding cloud infrustructure such as Amazon EC2.
-
3.
Use a scripting language for quick prototyping, interactive simulations, and user-friendly codes. We based our software on Python for this purpose. The use of this simple scripting interface also means that it is not difficult to reuse our framework for other methods that require running a large number of short trajectories with some appropriate postprocessing, e.g., MSM and other related methods.
In this paper, we present a distributed computing implementation of AWE, based on WorkQueue (WQ), that is capable of using computing resources with different architectural properties (e.g., GPU, CPU), as well as different job execution semantics such as dedicated (HPC grid) or cycle-scavenging (Condor) resources. At peak performance, for our validation, AWE-WQ simulated an aggregate 1.5 ms, with a peak performance of 1000 ns/h, showing that a large amount of resources can be efficiently managed by the system.
Similar software packages include the Copernicus framework developed by Pronk et al.,59 Adaptive Markov State Models (adaptive MSMs) by Bowman et al.,60 and the Weighted Ensemble Simulation Toolkit with Parallelization and Analysis (WESTPA) developed by Zwier et al.61 Copernicus is a framework for running ensemble molecular dynamics that allows multiple resources to be used, supports multiple project types, and adaptive sampling. Adaptive MSMs iteratively build MSM models to determine the contribution of each state to the slowest kinetic process. Based on this information further simulations are run from states based proportionally to their contribution to the uncertainty. WESTPA is a software framework for running WE simulations. Currently under development by Lillian Chong and Dan Zuckermann, WESTPA has been used to study several systems, such as molecular association62 and the sodium symporter Mhp1.47
The fault-tolerance model, intelligent task scheduling, and caching distinguish AWE-WQ. These features allow the program to dynamically handle resource addition and removal, automatically handle worker failures, recover from machine failures, support clusters of heterogeneous computers, and minimize data transfer.
We first describe the algorithm, design challenges, and implementation in the Design and Implementation section and provide a brief overview of usage. Second, in the Results section, we demonstrate that AWE-WQ meets the criteria described in the Design and Implementation section as well as provide a validation of the implementation on a nontrivial protein. Finally, we provide public access to the software and the data sets used for the results as well as describe the current limitations and future directions in Availability and Future Directions section.
Design and Implementation
Accelerated Weighted Ensemble Algorithm
The WE approach proceeds as follows and illustrated in Figure 2.
-
1.
Partition state-space into C cells.
-
2.
Determine W number of simulations (walkers) to maintain in each cell.
-
3.
Parallel step (walk): run the N = W × C short simulations, assign each of the N final conformations to a cell.
-
4.
Synchronization barrier (resample): once all the N simulations are assigned, merge and split the walkers to maintain W walkers in each cell and update the associated weights.
-
5.
Go to step 3 unless weights converged.
Figure 2.
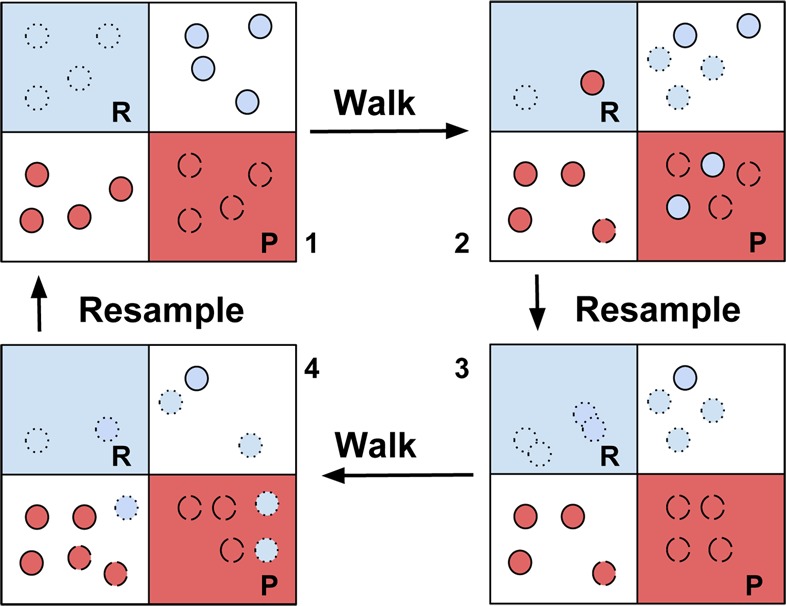
Illustration of WE algorithm. Each region represents a cell on the energy surface, the circles the walking simulations, the colors the association with the reactant (R) or product (P) regions.
The splitting and merging technique17 used in AWE generally works as follows. Given a population of walkers with weights wi, we first calculate a mean weight Wm = Σi(wi/ntargetwalkers), where ntargetwalkers is the desired number of walkers that we are trying to achieve. Then, if a walker has a weight greater than Wm, it is split into an integer number of walkers of weight Wm, with a remainder that is reinserted into the list of walkers to process. Walkers that have a weight less than Wm are merged in a statistically exact way to create walkers with a weight greater or equal to Wm. By repeating this process of splitting and merging walkers, we are able to generate a population of walkers with weights exactly equal to Wm, in a statistically exact manner. This is different from the procedure in the work of Huber and Kim41 that generates walkers with weights between Wm/2 and 2Wm but not exactly equal to the target weight Wm.
The choice of method for defining cells is orthogonal to WE. As long as a cell definition exists, in cells.dat currently, AWE-WQ is able to use it. This allows one to define cells arbitrarily to examine the effect of a cell definition method. We based the cells on the MSM construction in order to accelerate the convergence of cell weights. As explained previously, the main reason to use WE is to remove the bias present in MSM models due to the lag time and the (incorrect) distribution of walkers inside each cell.
Upon reaching steady-state the weight of each cell converges to its probability, allowing this procedure to calculate free energy. The resampling procedure allows even low-probability cells to be accurately sampled.
Calculation of transition rates requires a further modification: Define two sets of states, R and P, associated with the reactant and product regions (Figure 2). Each walker is assigned a color, such as blue for R and red for P, corresponding to the previously visited region. At each step in the simulation whenever a blue walker enters P, its color changes to red, and vice versa. The rate from R to P is then directly obtained by computing the flux of blue particles that change color, and similarly for the P to R rate. If extended to multiple colors and sets multiple kinetic rates can be determined. In practical terms, the R and P regions may correspond to the unfolded and folding regions and the fluxes to the folding and unfolding rates. By providing an initial approximation of the free energy the convergence to steady state can be accelerated.
From an implementation standpoint, the method requires the following three steps (as illustrated in Figure 3): (1) Most of the work is done in parallel as a large number of independent short MD trajectories. (2) The parallel barrier: wait for all MD steps to complete then collect walker assignments and final positions, split and merge walkers, and update weights. (3) Go to step 1 if needed.
Figure 3.
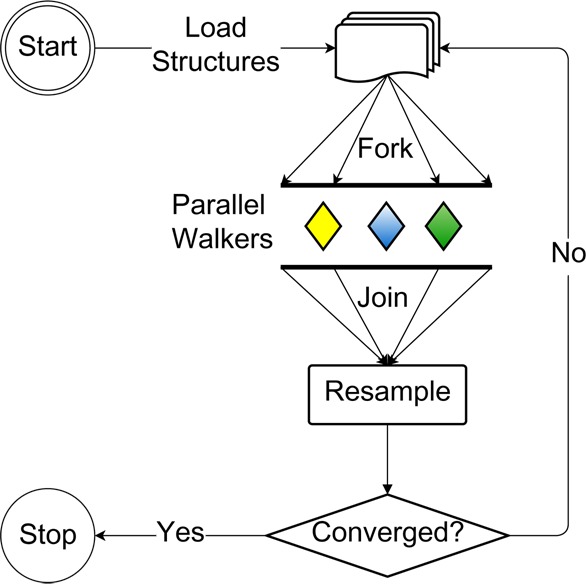
AWE-WQ flowchart of the major steps. On startup molecule conformations are loaded into the walker datastructures. Each walker is then, in parallel, executed and assigned to a cell. The join waits for all walkers to finish, before the resample procedure is applied. If the system has converged then the program halts, others walkers are rerun.
Usage
In order to use AWE-WQ one must define the cells from which to run the walkers. The general protocol, illustrated in Figure 4, is as follows: (1) sample the search space, (2) determine cell definitions, (3) prepare input files, (4) run AWE-WQ, (5) monitor progress.
Figure 4.

Diagram of the workflow to run AWE-WQ with associated programs and descriptions. First a sampling must be run with some MD software such as GROMACS, CHARMM, AMBER, etc. Next, define the cells using some clustering procedure. In this case, the MSMBuilder package provides some infrastructure. Prepare the input to AWE-WQ by converting the files and defining metastable states. Run AWE-WQ by starting the master process and allocating workers with various resources. Finally, continuously monitor the status and progress of AWE-WQ.
Sampling
First
some sampling must be done to explore
state space. This can be accomplished with programs such as GROMACS,
CHARMM, AMBER, NAMD, or using infrastructure such as Folding@home.
For this instance, sampling of alanine dipeptide has been run previously,
which can be extracted:![]()
Determine Cell Definitions
To start,
we have collected
MD data and stored them in the XTC directory. The directory structure
is XTC/RUN#/frame0.xtc, where # is the trajectory number. The sampling data is imported into MSMBuilder
using the ConvertDataToHDF.py command, paramterized with a PDB file
containing the system coordinates and the path to the MD data:![]()
The next step defines the cells. Conformations are clustered
with
a hybrid k-centers/k-medoids algorithm
using the RMSD between atoms as the distance metric. The AtomIndices.dat defines the atoms to consider when computing the distance between
conformations. Using a subset (such as all non-hydrogens) prevents
too fine a granularity from overfitting the data. Finally, we will
cluster the data into 100 groups.![]()
By inspecting the implied time scales (not shown)
we build a Markov
state model at lagtime 10.![]()
AWE-WQ Input Preparation
Extract
a given number of
conformations from each of the cells defined above (SaveStructures.py)
and import the cell definitions from MSMBuilder format to AWE-WQ format
(awe-import-gens) and setup the initial files for the AWE-WQ run (awe-prepare).
Regions of metastable states need to then be determined, which are
ultimately given as a list of the cells belonging to each region (e.g.,
“folded” and “unfolded”) and scripts are
given to do so using RMSD to a reference structure (awe-calc-gens-rmsd,
awe-define-regions). We plan to maintain 10 simulations in the cells,
so we need to extract conformations from the states using MSMBuilder.![]()
In order to run AWE-WQ
we must then import the cell
definitions which were written by MSMBuilder to Gens.lh5.![]()
In order
to compute the fluxes we need specify regions of metastable
states. AWE-WQ provides some commands to assist with this process: awe-calc-gens-rmsd and awe-define-regions. We use awe-calc-gens-rmsd to compute the
RMSD of each cell to some reference conformation such as the native
state.
By
plotting the distribution of values we can classify conformations
with RMSD ≤ 2.3 Å as folded and those
with RMSD ≥ 2.5 Å as unfolded. The two
output files folded.dat and unfolded.dat now contain the integer indices
of the states belonging to these regions.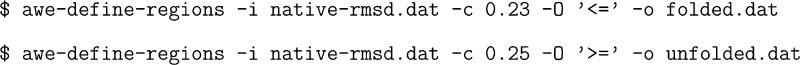
We can now prepare for AWE-WQ by checking dependencies and populating the directory with other necessary files by running awe-prepare.
Running AWE-WQ
There are two components to consider when running AWE-WQ: the master process and the resources. The master is the driver of the algorithm, managing task definitions, scheduling, processing, and the resampling procedure. In order to run the walkers, resources must be allocated.
Master
Start the AWE-WQ process
on a machine. This
process loads the initial conformations (walkers) for each cell, and
is in charge of scheduling each task, processing the result, and the
resampling procedure. This runs AWE-WQ maintaining 10 walkers in 100
cells, whose definition is provided in cells.dat with initial weights in Data/Populations.dat. The coordinates for the walkers are found in the Walkers directory.
The metastable regions are provided in folded.dat and unfolded.dat as a list of cell id numbers
belonging to each region. Finally, we give a name to the master (“awe-wq”)
to that workers can easily locate the host and port.
Workers
Resources
can be allocated either directly
using work_queue_worker or managed automatically
using work_queue_pool. Using work_queue_worker also allows the worker to operate as a “Foreman”,
enabling the hierarchical distribution of tasks. The work_queue_pool program maintains workers for the master based on need and is in
charge of submission to various backends such as the local machine,
Condor, SGE, PBS, etc. Since the master has started we can start some
workers locally.![]()
Monitoring AWE Progress
Use work_queue_status to get the current resources runtime status (number of workers, number of tasks waiting/completed, etc.). By using awe-plot-wq-stats the plot of the resource usage over the runtime of the program can be obtained.
Additionally, the current status of the master,
such as workers busy and tasks complete can be viewed using the work_queue_status command.
The awe-flux command allows the convergence of the calculated flux to be monitored. Once convergence within a determined threshold is obtained the program may be halted.
Additionally, other analyses are appropriate. For instance, the energy surface for alanine dipeptide can be visualized as a function of its dihedral angles. As such, we can plot, as shown in Figure 5, the cell coordinates and the initial estimation of the weights as well as computed weights after several iterations of AWE-WQ.
Figure 5.
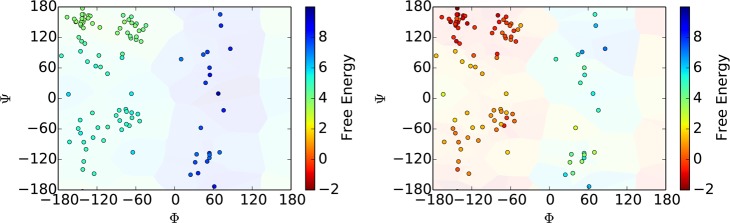
Cell weights before and after running AWE-WQ.
Design and Implementation
Since the AWE method follows a pattern of fan-out followed by fan-in we implement using a master/worker paradigm. AWE-WQ is designed to (1) support off-the-shelf MD backend software, (2) dynamically scale according to resource availability, (3) support heterogeneous runtime environments, and (4) be robust in the face of system failure. Application of AWE to a nontrivial biological system may have on the order of 10 000 simulations per iteration. On one CPU, if each walker requires 30 min to simulate, 7 months would be required to run a single iteration of AWE. Since walkers are short, assuming 30 min per walker, 7 months would be required to run 10 000 walkers for each AWE iteration using a single processor. With 1000 CPUs, the expected time drops to 5 h, assuming uniform performance. Additionally, the synchronization barrier allows straggling workers to slow down the entire application. Since we expect to need hundreds of iterations to converge, the challenge is to implement AWE such that it scales to 1000+ processors and can be sustained for months with no major slowdown due to stragglers.
Support of à la Carte MD Software
Rather than reimplement the core MD algorithms which are present in multiple software packages (i.e., GROMACS, AMBER, CHARMM, etc.) we decided to provide flexibility of MD backend. This flexibility presented several challenges by imposing a requirement of specifying the steps needed to run the backend, the transfer of files, the execution environment, and the interaction with the other steps of the procedure. The approach taken in the following: a walker is described as a task which consists of a program to run and the input and output files. Once specified, this task is scheduled to run on an available worker, which is a process that accepts any input files, executes the task’s command, and returns the results upon request.
We used GROMACS 4.5 for the MD backend. The awe-prepare script sets up files and scripts in the local directory necessary to run awe-wq. One of these scripts, execute-task.sh, executes the commands on the worker. In order to use a different MD backend execute-task.sh needs to define how to run the MD, and awe-wq needs to specify the files to send to the worker.
Dynamically Using Available Resources
Our solution to the worker fault-tolerance problem implies the elasticity—the ability to dynamically scale to available resources—of the program. If a task fails during execution, it is rescheduled to run on the next available worker. As resources are added or removed, tasks are started or reschedule, respectively. This allows a project to be started with the campus cluster, then EC2 machines run until budget exhaustion, then continued with only the cluster. Managing the resource pools are the interactions between the master, the Catalog Server (CS), and WorkQueue Pool (WQP) processes. The CS allows workers to dynamically determine the location of the master by mapping a project name (e.g., awe-wq) to a hostname and port (e.g., fah.crc.nd.edu:1024). The master registers this information when started, which allows it to be restarted in the event of machine failure without requiring the resources to be reallocated. In addition, the CS store information about the current resource requirements of a master, such as the number of tasks waiting to be scheduled. A WQP can thus use this information to automatically start workers based on the runtime needs. This is further enabled by the asynchronous model, which treats tasks as independent, arbitrarily executable entities. This would not have been possible using an MPI-based implementation.
Heterogeneous Runtime Environments
Additionally, AWE-WQ can use heterogeneous resources, allowing systems with different properties and (potentially) operational semantics to be used. For instance, this supports using GPU-based as well as CPU-based MD backend codes. This is accomplished by extending the task abstraction to dynamically resolve file location upon worker connection rather than statically. For example, an input file can be specified for a task as binaries/$OS/$ARCH/mdrun. When a worker connects to the master it reports the operating system and architecture of the remote node in the $OS and $ARCH variables, which may then be expanded to send binaries/Linux/i686/mdrun and binaries/Darwin/x86_64/mdrun to to workers running on a 32-bit Linux and 64-bit Mac OS machine, respectively. Finally, these variables can be overridden by the user to provide additional granularity.
Fault-tolerance
AWE-WQ is implemented with the assumption that machine failures are not only possible but in fact common, which is the opposite of the paradigm underlying MPI infrastructures. As such two broad categories of failures need to be accounted for: failure of the worker or master processes. In the first case, worker failure are handled transparently and tasks rescheduled to the next available worker.
Failure of the master node would normally result in the termination with an inability to resume. This is overcome by using a transactional logging mechanism: a combination of persistent and volatile logs. Each result from a walker is added to a log and after each AWE iteration the state of the system is checkpointed to disk and the log cleared. If the process fails, then the state is initialized from the checkpoint and the log used to replay and missed results from that iteration. This way only uncompleted walkers need to be restarted.
Intelligent Scheduling
Several components of task scheduling affect the scalability. The first is knowledge of a worker’s past task execution times. This allows tasks to be scheduled preferentially to the most performant machines. The second involves task replication to remove the effect of straggling workers. By monitoring the state of the workers—either busy or idle (wi)—and the number of tasks waiting to be assigned a worker, task replication can be engaged when wi > 0. Once a task returns, all its replicates are canceled, results are accepted in a first-come-first-serve fashion. By combining replication with preferential scheduling, workers are continuously used and slow workers allowed to contribute results until the fast machines are available, which then run the replicated tasks.
Event Model
The inner loop of AWE-WQ is an event-loop with asynchronous task execution. Each iteration begins by translating the internal data model of the walkers into task, which are submitted to the WQ scheduler. While tasks are executing, the program is idle until a result is returned. Each result creates an event that must be handled, either by translation or resubmission. Handlers are lightweight: read the final coordinates and cell assignment into the data models. Since task executions are asynchronous and results are processed serially, the event model enforces load balancing by spreading out the calls to the event handlers.
Caching Allows Scaling
Increasing scalability is further accomplished by minimizing the communication overhead and maximizing the parallel work. Ideally, the master process will spend most of its time waiting. By categorizing files as those common to all tasks (common) or specific to the current one (unique), the common files can be cached on the workers. Since we expect the number of walkers to be larger than resource availability the caching mechanism shifts overhead to depend on resources rather than problem size. For instance, using a GROMACS backend each task requires 34 MB in input files, binaries, and miscellaneous files.
In the case of the WW domain simulation, each task required 34 MB of GROMACS-related files as overhead and 100 KB of task specific data. Assuming 10 000 tasks/iteration and 500 iterations, the total amount of data transferred is 34 (MB/task) × 10 000 (tasks/iteration) × 500 iterations × 2–20 (TB/MB) = 162 TB. By caching input files, data transferred depends only on the number of new workers seen, rather than total number of tasks, which may be orders of magnitude greater. In this case, assuming 1000 workers are connected the total data transferred becomes 34 MB × 1000 × 2–10 (GB/MB) = 33 GB.
Results
We have run AWE-WQ for several months where 3.5 million tasks have executed over 600 compute years in a heterogeneous environment simulating over 1.5 ms of time. In this section we describe the application the AWE algorithm to a nontrivial biological system.
Heterogeneous Resource Usage
Available resources consisted of the following: Notre Dame HPC grid with 6000 cores shared among campus users; Notre Dame Condor pool with variable (usually around 1200) cores and flocking capabilities with Purdue University and the University of Wisconsin–Madison; Stanford University’s Institute for Computational and Mathematical Engineering (ICME) cluster with 200 CPUs and 100 NVIDIA GPUs, and cloud virtual machines via Amazon EC2 and Microsoft Azure.
Figure 6 displays the distribution of task execution times. The multimodal distribution reflects the differences of performance within each pool. The GPU cluster executes tasks within minutes, while the ND HPC grid and EC2 machines are the best performant CPU-based pools. The range of performance of Condor machines is unsurprising and illustrative of the cycle-scavenging nature of the platform. The Azure machines were the least performant resources, partly due to the necessity of running the tasks via Cygwin.
Figure 6.
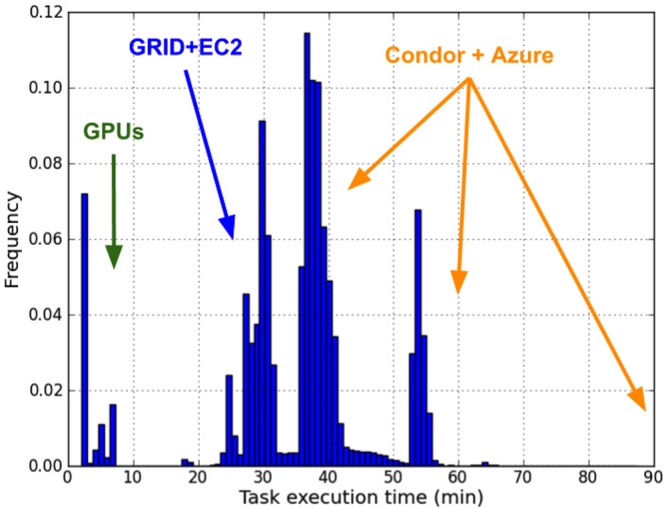
Distribution of task execution times indicate several peaks associated with the different underlying resources being used.
Elasticity and Scalability
Figure 7 demonstrates the fault-tolerance elasticity and scalability of the application as an expanded view of the number of busy resources over the runtime—the inset shows the entire run. Unexpected failures of the master (months 4, 6) initiated development and integration of features, bug fixes, and monitoring capabilities. Due to power outages and infrastructure upgrades the master process restarted multiple times. In spite of these failures over 250 iterations were completed without data loss due to the transactional logging mechanism. The periodically persisting state additionally allowed us to integrate new features, such as task replication, and bug fixes over the course of the project’s execution.
Figure 7.
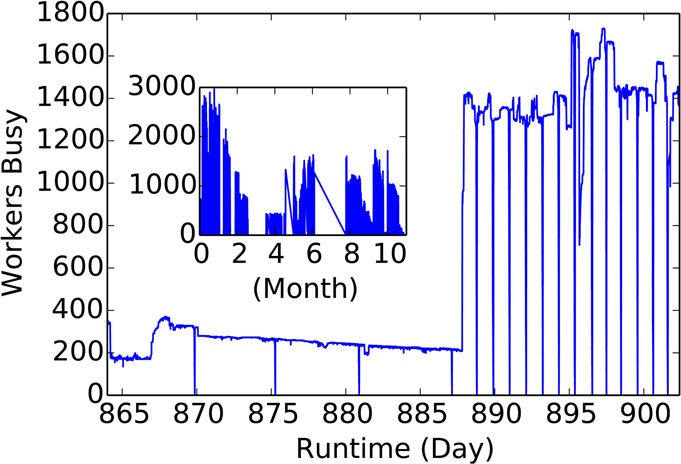
Expanded view of the entire run (inset) demonstrating elasticity, scalability, and fault-tolerance. In the inset, long gaps (such as the sudden failures at month 4 and 6) in activity indicate times when major development was done on the code (features added, bugs fixed). Despite the troubles experienced by the master node, calcuation of the results resumed once the code was deployed. Plotting the number of busy workers over many days shows the ability of the software to adapt to a changing environment. The periodic dips indicate the global synchronization barrier, each requiring 12 000 tasks to complete. A large increase after day 885 occurred when many nodes were suddenly available and were automatically incorporated.
Focusing on the region between days 865 and 900 shows a steady decrease in the number of busy resources until a new pool of workers became available and AWE-WQ automatically started using them. It must be emphasized as well that minimal human input was needed to take advantage of the additional resources: due to the use of the Catalog Server all that was required was to start a WQ Pool process for the additional resources.
The periodic dips, every 5 days in the first half and every ≈20 h for the second, correspond to the global synchronization barrier, each period requiring 12 000 tasks to complete. By scheduling the tasks based on resource availability and progression through the AWE algorithm, resources are fully utilized and the long tail effect from straggling workers is eliminated. Due to the use of the caching mechanism communication overhead is reduced: while the entire task payload (common and unique files) is 34 MB it is only for the first task a worker executes that his is transferred. Each subsequent task only requires 100 KB of data to be transferred (both in- and outbound traffic).
WW Domain Protein
The WW Domain is a 34 residue protein domain with two conserved tryptophan (W) residues and comprised of two antiparallel β-sheets as shown in Figure 8. We studied a mutant which is known to fold in 13 μs.63 Simulation parameters were the Amber96 force field with generalized Born implicit solvent, a viscosity parameter (γ) of 1 ps–1, temperature of 395 K (close to the optimal folding temperature64), saving conformations every 1 ns. The simulation was run for 200 μs during which multiple folding and unfolding events were observed. The conformations sampled by the simulation were then clustered using the α and β carbons using a cutoff of 3 Å. In order to reduce the time spent sampling the unfolded space we established that conformations with an RMSD to the folded structure less than 3 and 6 Å, and greater as folded, intermediate, and unfolded, respectively. Using these definitions the number of unfolded states was reduced, for a total of 601 cells. The AWE-WQ parameters were thus 601 cells, 20 walkers per cell, and walker length of 500 ps using the same MD parameters as the source data. GROMACS 4.5 was used for the MD backend.
Figure 8.
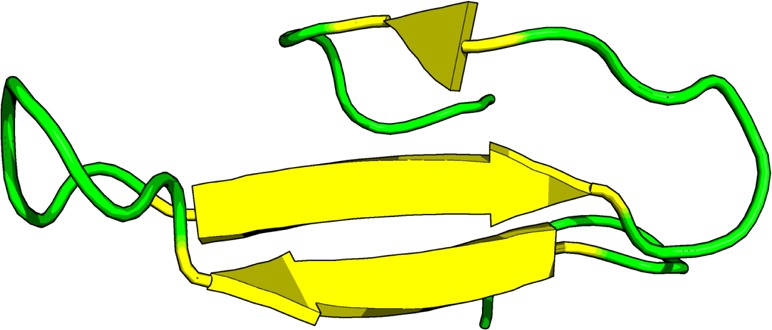
WW domain forming three antiparallel β-sheets separated by two flexible loop regions.
WW Results
Over the course of many months AWE-WQ completed 345 iterations, with an aggregate 2.3 ms of time simulated using 3.6 million tasks, and a peak performance of 1000 ns/h. Analysis indicates that the AWE-WQ results converge within acceptable error tolerance. Table 1 displays the forward and backward rate estimations computed using AWE-WQ and the 200 μs simulation. The AWE estimation lies within the confidence interval—with smaller statistical error—than the brute force simulation.
Table 1. Forward and Backward Reaction Rates As Computed from AWE-WQ and the Brute-Force Simulation.
| method | forward (μs–1) | backward (μs–1) |
|---|---|---|
| AWE-WQ | 1.5 ± 0.3 | 2.5 ± 0.5 |
| brute-force | 1.8 ± 1.0 | 2.0 ± 0.9 |
Figure 9 illustrates that forward and backward fluxes took 30 iterations to converge to the confidence interval built from brute force simulation and are stabilized within the interval afterward. The resampling procedure of AWE introduces no bias in the estimation of transition rates while exhibiting smaller statistical error, showing an improvement over the brute force method.
Figure 9.
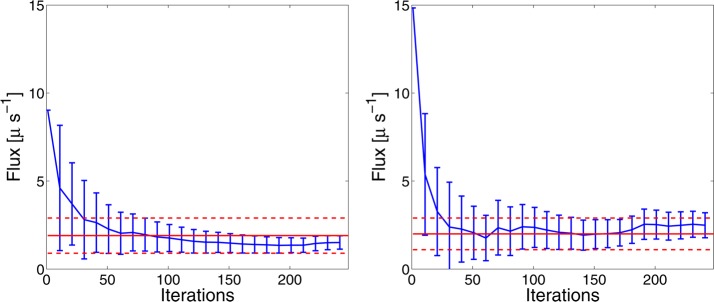
Average forward (top) and backward (bottom) flux from AWE (blue curve) with error bar of one standard deviation estimated by the block averaging method, compared with confidence interval built from the brute-force simulation. The red solid line represents the mean flux from the brute-force simulation, and the dashed lines are mean flux plus/minus one standard deviation.
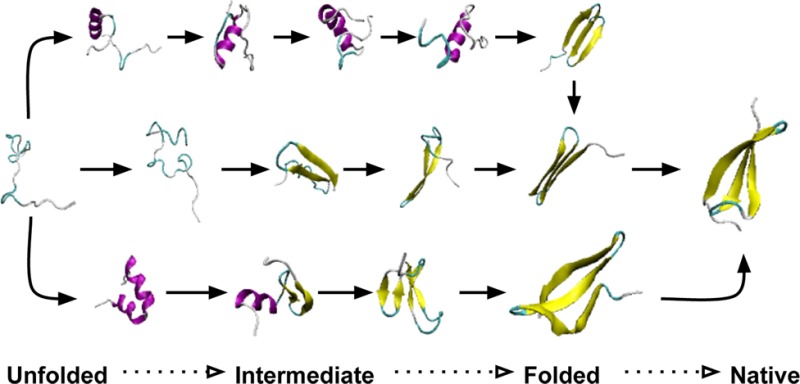
Transitions among the states are examined with a time lag of τ = Δt = 0.5 ns. Three significant pathways from extended to folded state are extracted from the network and shown in Figure 10. Starting from an unfolded conformation, two pathways involve multiple helical intermediate structures before β-sheet formation occurs via rearrangement. The third pathway occurs as a rapid collapse before the formation of Loop 2. Loop 1 is then able to form, after which the molecule undergoes further refinement to arrive at the native state.
Figure 10.

First three significant pathways from extended to folded conformations. Two pathways (top, middle) involve helix formation as an intermediary while the third pathway (bottom) consists of a collapse then refinement.
Conclusions
AWE-WQ meets our criteria: allowing the use of off-the-shelf components for MD backends, pooling of heterogeneous resources, scalability to over 1000 workers, and ability to cope with failures of both worker and master processes. The WW domain was used to validate AWE-WQ by applying the method to a long MD simulation in which multiple folding events are observed. The forward and backward reaction rates computed by AWE-WQ are within the range and have smaller error, of the rates calculated from the reference simulation.
Availability and Future Directions
Availability
AWE-WQ is available under the GPLv2 license. Documentation and data sets are available online.65 The source repository for AWE-WQ can be found online on Github.66 Build dependencies for AWE-WQ include a Python interpreter, the GROMACS xtc library,67 and the GNU Scientific Library.68
Future Directions
We are working on improvements to the AWE and WorkQueue components of AWE-WQ.
The current version requires initial sampling for cell definition. These cell are static for the AWE-WQ run. Thus, AWE-WQ is limited by the sampling techniques applied and is unable to discover new regions of state-space. Furthermore, a uniform partitioning scheme is used, which potentially leads to overrepresentation of uninteresting regions (such as unfolded protein). Reducing the number states for computational efficiency risks reduction in granularity in the regions that benefit the most (e.g., transition regions) and reduces accuracy. Goals for future versions of AWE-WQ are planned to allow for cell discovery and improved cell partitioning methods.
With regards to infrastructure, the next challenges to overcome are the continuous usage of 5000 and 10 000 workers. Recent development to the WorkQueue infrustructure have demonstrated that using a hierarchical topology can reduce communication overhead on the master by 96%.69 Additionally, support for other MD backend software, as well as explicit solvent models, is under development.
Acknowledgments
The authors thank the Notre Dame Center for Research Computing, the Stanford Institute for Computation and Mathematical Engineering, and the Folding@home donors for use of compute resources. Funding has been provided by NSF CCF-1018570, NIH 1R01 GM101935-01, NIH 7R01 AI039071. This work was supported in part by National Science Foundation grant OCI-1148330. Eric Darve was supported by the Department of Energy grant DE-FG02-11ER26061.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Li A.; Dagget V. Characterization of the Transition State of Protein Unfolding by Use of Molecular Dynamics: Chymotrypsin Inhibitor 2. Proc. Natl. Acad. Sci. U.S.A. 1994, 91, 10430–10434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li A.; Daggett V. Identification and Characterization of the Unfolding Transition State of Chymotrypsin Inhibitor 2 by Molecular Dynamics Simulations. J. Mol. Biol. 1996, 257, 412–429. [DOI] [PubMed] [Google Scholar]
- McCammon J. A.; Gelin B. R.; Karplus M. Dynamics of Folded Proteins. Nature 1977, 267, 585–590. [DOI] [PubMed] [Google Scholar]
- Colonna-Cesari F.; Perahia D.; Karplus M.; Eklund H.; Brädén C. I.; Tapia O. Interdomain Motion in Liver Alcohol Dehydrogenase. Structural and Energetic Analysis of the Hinge Bending Mode. J. Biol. Chem. 1986, 261, 15273–15280. [PubMed] [Google Scholar]
- Harvey S.; Prabhakaran M.; Mao B.; McCammon J. Phenylalanine Transfer RNA: Molecular Dynamics Simulation. Science 1984, 223, 1189–1191. [DOI] [PubMed] [Google Scholar]
- Xu Z.; Horwich A. L.; Sigler P. B. The Crystal Structure of the Asymmetric GroELGroES-(ADP)7 Chaperonin Complex. Nature 1997, 388, 741–750. [DOI] [PubMed] [Google Scholar]
- MacKerell A. D.; Bashford D.; Bellott M.; Dunbrack R. L.; Evanseck J. D.; Field M. J.; Fischer S.; Gao J.; Guo H.; Ha S.; Joseph-McCarthy D.; Kuchnir L.; Kuczera K.; Lau F. T. K.; Mattos C.; Michnick S.; Ngo T.; Nguyen D. T.; Prodhom B.; Reiher W. E.; Roux B.; Schlenkrich M.; Smith J. C.; Stote R.; Straub J.; Watanabe M.; Wiórkiewicz-Kuczera J.; Yin D.; Karplus M. All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [DOI] [PubMed] [Google Scholar]
- Huang J.; MacKerell A. D. CHARMM36 All-atom Additive Protein Force Field: Validation Based on Comparison to NMR Data. J. Comput. Chem. 2013, 34, 2135–2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornak V.; Abel R.; Okur A.; Strockbine B.; Roitberg A.; Simmerling C. Comparison of Multiple Amber Force Fields and Development of Improved Protein Backbone Parameters. Proteins: Struc., Funct., and Bioinformatics 2006, 65, 712–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.; Brooks C. L. I.; Khandogin J. Recent Advances in Implicit Solvent-based Methods for Biomolecular Simulations. Curr. Opin. Struct. Biol. 2008, 18, 140–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen W. L.; Chandrasekhar J.; Madura J. D.; Impey R. W.; Klein M. L. Comparison of Simple Potential Functions for Simulating LiquidWater. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar]
- JORGENSEN W. L. Quantum and Statistical Mechanical Studies of Liquids 0.10. Transferable Intermolecular Potential Functions for Water, Alcohols, and Ethers – Application to Liquid Water. J. Am. Chem. Soc. 1981, 103, 335–340. [Google Scholar]
- Berendsen H. J. C.; Grigera J. R.; Straatsma T. P. The Missing Term in Effective Pair Potentials. J. Comput. Chem. 1987, 91, 6269–6271. [Google Scholar]
- Zhao G.; Perilla J. R.; Yufenyuy E. L.; Meng X.; Chen B.; Ning J.; Ahn J.; Gronenborn A. M.; Schulten K.; Aiken C.; Zhang P. Mature HIV-1 Capsid Structure by Cryo-electron Microscopy and All-atom Molecular Dynamics. Nature 2013, 497, 643–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freddolino P. L.; Arkhipov A. S.; Larson S. B.; McPherson A.; Schulten K. Molecular Dynamics Simulations of the Complete Satellite Tobacco Mosaic Virus. Structure 2006, 14, 437–449. [DOI] [PubMed] [Google Scholar]
- Kiss G.; Pande V. S.; Houk K. N. Molecular Dynamics Simulations for the Ranking, Evaluation, and Refinement of Computationally Designed Proteins. Methods in Protein Design 2013, 523, 145–170. [DOI] [PubMed] [Google Scholar]
- Hess B.; Kutzner C.; van der Spoel D.; Lindahl E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [DOI] [PubMed] [Google Scholar]
- Phillips J. C.; Braun R.; Wang W.; Gumbart J.; Tajkhorshid E.; Villa E.; Chipot C.; Skeel R. D.; Kal e L.; Schulten K. Scalable Molecular Dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks B. R.; Bruccoleri R. E.; Olafson B. D.; States D. J.; Swaminathan S.; Karplus M. CHARMM: a Program for Macromolecular Energy, Minimization, and Dynamics Calculations. J. Comput. Chem. 1983, 4, 187–217. [Google Scholar]
- Fukushige T.; Taiji M.; Makino J.; Ebisuzaki T.; Sugimoto D. A Highly Parallelized Special-purpose Computer for Many-body Simulations with an Arbitrary Central Force: MD-GRAPE. Astrophysics 1996, 468, 51–61. [Google Scholar]
- Sumanth J. V.; Swanson D. R.; Jiang H. Performance and Cost Effectiveness of a Cluster ofWorkstations and MD-GRAPE 2 for MD Simulations. J. Parallel Distr. Com. 2003, 244–249. [Google Scholar]
- Shaw D. E.; Deneroff M. M.; Dror R. O.; Kuskin J. S.; Larson R. H.; Salmon J. K.; Young C.; Batson B.; Bowers K. J.; Chao J. C.; Eastwood M. P.; Gagliardo J.; Grossman J. P.; Ho C. R.; Ierardi D. J.; Kolossvary I.; Klepeis J. L.; Layman T.; Mcleavey C.; Moraes M. A.; Mueller R.; Priest E. C.; Shan Y.; Spengler J.; Theobald M.; Towles B.; Wang S. C. Anton, a Special-purpose Machine for Molecular Dynamics Simulation. Communications of the ACM 2008, 51, 91–97. [Google Scholar]
- Shaw D. E.Anton: a Special-purpose Machine That Achieves a Hundred-fold Speedup in Biomolecular Simulations Proceedings of the 22nd international symposium on High-performance parallel and distributed computing, New York, June 17–21, 2013; pp 129–130.
- Liu W.; Schmidt B.; Voss G.; Mueller-Wittig W. Accelerating Molecular Dynamics Simulations Using Graphics Processing Units with CUDA. Comput. Phys. Commun. 2008, 179, 634–641. [Google Scholar]
- Phillips J. C.; Stone J. E.; Schulten K. Adapting a Message-driven Parallel Application to GPU-accelerated Clusters. 2008 SC - International Conference for High Performance Computing. Networking, Storage and Analysis 2008, 1–9. [Google Scholar]
- Luttmann E.; Ensign D. L.; Vaidyanathan V.; Houston M.; Rimon N.; Øland J.; Jayachandran G.; Friedrichs M.; Pande V. S. Accelerating Molecular Dynamic Simulation on the Cell Processor and Playstation 3. J. Comput. Chem. 2009, 30, 268–274. [DOI] [PubMed] [Google Scholar]
- Friedrichs M. S.; Eastman P.; Vaidyanathan V.; Houston M.; Legrand S.; Beberg A. L.; Ensign D. L.; Bruns C. M.; Pande V. S. Accelerating Molecular Dynamic Simulation on Graphics Processing Units. J. Comput. Chem. 2009, 30, 864–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson J. A.; Lorenz C. D.; Travesset A. General Purpose Molecular Dynamics Simulations Fully Implemented on Graphics Processing Units. J. Chem. Phys. 2008, 227, 5342–5359. [Google Scholar]
- Nelson M. T.; Humphrey W.; Gursoy A.; Dalke A.; Kale L. V.; Skeel R. D.; Schulten K. NAMD: a Parallel, Object-Oriented Molecular Dynamics Program. International Journal of High Performance Computing Applications 1996, 10, 251–268. [Google Scholar]
- Duan Y.; Kollman P. A. Pathways to a Protein Folding Intermediate Observed in a 1-microsecond Simulation in Aqueous Solution. Science 1998, 282, 740–744. [DOI] [PubMed] [Google Scholar]
- Ryckaert J.-P.; Ciccotti G.; Berendsen H. J. C. Numerical Integration of the Cartesian Equations of Motion of a System with Constraints: Molecular Dynamics of N-alkanes. J. Chem. Phys. 1977, 23, 327–341. [Google Scholar]
- Hess B. P-LINCS: a Parallel Linear Constraint Solver for Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 116–122. [DOI] [PubMed] [Google Scholar]
- Liem S. Y.; Brown D.; Clarke J. H. R.Molecular Dynamics Simulations on Distributed Memory Machines. Comput. Phys. Commun. 1991, 67, 261–267. [Google Scholar]
- Bowers K. J.; Dror R. O.; Shaw D. E. Overview of Neutral Territory Methods for the Parallel Evaluation of Pairwise Particle Interactions. J. Phys.: Conf. Ser. 2005, 16, 300–304. [Google Scholar]
- Bowers K. J.; Dror R. O.; Shaw D. E. The Midpoint Method for Parallelization of Particle Simulations. J. Chem. Phys. 2006, 124, 184109. [DOI] [PubMed] [Google Scholar]
- Meloni S.; Rosati M.; Colombo L. Efficient Particle Labeling in Atomistic Simulations. J. Chem. Phys. 2007, 126, 121102. [DOI] [PubMed] [Google Scholar]
- Shirts M. R.; Pande V. S. Mathematical Analysis of Coupled Parallel Simulations. Phys. Rev. Lett. 2001, 86, 4983–4987. [DOI] [PubMed] [Google Scholar]
- Pande V. S.; Beauchamp K.; Bowman G. R. Everything You Wanted to Know about Markov State Models but Were Afraid to Ask. Methods 2010, 52, 99–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singhal N.; Pande V. S. Error Analysis and Efficient Sampling in Markov State Models for Molecular Dynamics. J. Chem. Phys. 2005, 123, 204909. [DOI] [PubMed] [Google Scholar]
- Swope W. C.; Pitera J. W.; Suits F. Describing Protein Folding Kinetics by Molecular Dynamics Simulations. 1. Theory. J. Phys. Chem. B 2004, 108, 6571–6581. [Google Scholar]
- Huber G. A.; Kim S. Weighted-ensemble Brownian Dynamics Simulations for Protein Association Reactions. Biophys. J. 1996, 70, 97–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatt D.; Zhang B. W.; Zuckerman D. M. Steady-state Simulations Using Weighted Ensemble Path Sampling. J. Chem. Phys. 2010, 133, 014110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatt D.; Zuckerman D. M. Heterogeneous Path Ensembles for Conformational Transitions in Semiatomistic Models of Adenylate Kinase. J. Chem. Theory Comput. 2010, 6, 3527–3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darve E.; Ryu E.. Innovations in Biomolecular Modeling and Simulations; The Royal Society of Chemistry, 2012; Vol. 1, Chapter 7, pp 138–206. [Google Scholar]
- Huber G. A.; McCammon J. A. Weighted-ensemble Simulated Annealing: Faster Optimization on Hierarchical Energy Surfaces. Phys. Rev. E 1997, 55, 4822–4825. [Google Scholar]
- Zhang B. W.; Jasnow D.; Zuckerman D. M. The ”weighted Ensemble” Path Sampling Method Is Statistically Exact for a Broad Class of Stochastic Processes and Binning Procedures. J. Chem. Phys. 2010, 132, 054107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adelman J. L.; Dale A. L.; Zwier M. C.; Bhatt D.; Chong L. T.; Zuckerman D. M.; Grabe M. Simulations of the Alternating Access Mechanism of the Sodium Symporter Mhp1. Biophys. J. 2011, 101, 2399–2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case D. A.; Darden T. A.; Cheatham T. E. III; Simmerling C. L.; Wang J.; Duke R. E.; Luo R.; Walker R. C.; Zhang W.; Merz K. M.; Roberts B.; Hayik S.; Roitberg A.; Seabra G.; Swails J.; Goetz A. W.; Kolossvary I.; Wong K. F.; Paesani F.; Vanicek J.; Wolf R. M.; Liu J.; Wu X.; Brozell S. R.; Steinbrecher T.; Gohlke H. l.; Cai Q.; Ye X.; Hsieh M. J.; Cui G.; Roe D. R.; Mathews D. H.; Seetin M. G.; Salomon-Ferrer R.; Sagui C.; Babin V.; Luchko T.; Gusarov S.; Kovalenko A.; Kollman P. a.. AMBER 12; University of California, San Fransisco, 2012,.
- Brooks B. R.; Brooks C. L. III; Mackerell A. D. Jr.; Nilsson L.; Petrella R. J.; Roux B.; Won Y.; Archontis G.; Bartels C.; Boresch S.; Caflisch A.; Caves L.; Cui Q.; Dinner A. R.; Feig M.; Fischer S.; Gao J.; Hodoscek M.; Im W.; Kuczera K.; Lazaridis T.; Ma J.; Ovchinnikov V.; Paci E.; Pastor R. W.; Post C. B.; Pu J. Z.; Schaefer M.; Tidor B.; Venable R. M.; Woodcock H. L.; Wu X.; Yang W.; York D. M.; Karplus M. CHARMM: the Biomolecular Simulation Program. J. Comput. Chem. 2009, 30, 1545–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pronk S.; Ṕall S.; Schulz R.; Larsson P.; Bjelkmar P.; Apostolov R.; Shirts M. R.; Smith J. C.; Kasson P. M.; van der Spoel D.; Hess B.; Lindahl E. GROMACS 4.5: a High-throughput and Highly Parallel Open Source Molecular Simulation Toolkit. Bioinformatics 2013, 29, 845–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plimpton S. Fast Parallel Algorithms for Short-Range Molecular-Dynamics. J. Chem. Phys. 1995, 117, 1–19. [Google Scholar]
- Lyubartsev A. P.; Laaksonen A. M. Dyna Mix–a Scalable Portable Parallel MD Simulation Package for Arbitrary Molecular Mixtures. Comput. Phys. Commun. 2000, 128, 565–589. [Google Scholar]
- Procacci P.; Darden T. A.; Paci E.; Marchi M. ORAC: a Molecular Dynamics Program to Simulate Complex Molecular Systems with Realistic Electrostatic Interactions. J. Comput. Chem. 1997, 18, 1848–1862. [Google Scholar]
- Marsili S.; Signorini G. F.; Chelli R.; Marchi M.; Procacci P. ORAC: a Molecular Dynamics Simulation Program to Explore Free Energy Surfaces in Biomolecular Systems at the Atomistic Level. J. Comput. Chem. 2010, 31, 1106–1116. [DOI] [PubMed] [Google Scholar]
- Christen M.; Hunenberger P. H.; Bakowies D.; Baron R.; Burgi R.; Geerke D. P.; Heinz T. N.; Kastenholz M. A.; Krautler V.; Oostenbrink C.; Peter C.; Trzesniak D.; Van Gunsteren W. F. The GROMOS Software for Biomolecular Simulation: GROMOS05. J. Comput. Chem. 2005, 26, 1719–1751. [DOI] [PubMed] [Google Scholar]
- Pappu R. V.; Hart R. K.; Ponder J. W. Analysis and Application of Potential Energy Smoothing and Search Methods for Global Optimization. J. Phys. Chem. B 1998, 102, 9725–9742. [Google Scholar]
- Bowers K.; Chow E.; Xu H.; Dror R.; Eastwood M.; Gregersen B.; Klepeis J.; Kolossvary I.; Moraes M.; Sacerdoti F.; Salmon J.; Shan Y.; Shaw D. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. ACM/IEEE Supercomputing. 2006, 43–43. [Google Scholar]
- Todorov I. T.; Smith W. DL POLY 3: the CCP5 National UK Code for Molecular-Dynamics Simulations. Philos. Trans. R. Soc., A 2004, 362, 1835–1852. [DOI] [PubMed] [Google Scholar]
- Pronk S.; Larsson P.; Pouya I.; Bowman G. R.; Haque I. S.; Beauchamp K.; Hess B.; Pande V. S.; Kasson P. M.; Lindahl E. Copernicus: a New Paradigm for Parallel Adaptive Molecular Dynamics. IEEE/ACM Supercomputing 2011, 1–10. [Google Scholar]
- Bowman G. R.; Ensign D. L.; Pande V. S. Enhanced Modeling Via Network Theory: Adaptive Sampling of Markov State Models. J. Chem. Theory Comput. 2010, 6, 787–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwier M.; Adelman J.; Rego N.; Kaus J.; Wong K.; Pratt A.; Wang D.; Lettieri S.; Suarez E.; Grabe M.; Zuckerman D.; Chong L.. WESTPA: a Portable, Highly Scalable Software Package for Weighted Ensemble Simulation and Analysis, in preparation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwier M. C.; Kaus J. W.; Chong L. T. Efficient Explicit-Solvent Molecular Dynamics Simulations of Molecular Association Kinetics: Methane/Methane, Na +/Cl, Methane/Benzene, and K +/18-Crown-6 Ether. J. Chem. Theory Comput. 2011, 7, 1189–1197. [DOI] [PubMed] [Google Scholar]
- Liu F.; Du D.; Fuller A. a.; Davoren J. E.; Wipf P.; Kelly J. W.; Gruebele M. An Experimental Survey of the Transition between Two-state and Downhill Protein Folding Scenarios. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 2369–2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw D. E.; Maragakis P.; Lindorff-Larsen K.; Piana S.; Dror R. O.; Eastwood M. P.; Bank J. A.; Jumper J. M.; Salmon J. K.; Shan Y.; Wriggers W. Atomic-Level Characterization of the Structural Dynamics of Proteins. Science 2010, 330, 341–346. [DOI] [PubMed] [Google Scholar]
- Cooperative Computing Lab Software. www.nd.edu/∼ccl/software/awe.
- AWE-WQ Source Code Control. www.github.com/cooperative-computing-lab/awe.
- GROMACS XTC Library.www.gromacs.org/Developer_Zone/Programming_Guide/XTC_Library.
- Galassi M., Davies J., Theiler J., Gough B., Jungman G., Alken P., Booth M., Rossi F.GNU Scientific Library Reference Manual; Network Theory Ltd., 2009 [Google Scholar]
- Albrecht M.; Rajan D.; Thain D.. MakingWork Queue Cluster-friendly for Data Intensive Scientific Applications. IEEE International Conference on Cluster Computing (CLUSTER), Indianapolis, IN, Sep 23–27, 2013; pp 1–8.