

Abstract
The nuclear IκB family protein IκBNS is expressed in T cells and plays an important role in Interferon (IFN)-γ and Interleukin (IL)-2 production. IκB-ζ, the most similar homolog of IκBNS, plays an important role in the generation of T helper (Th)17 cells in cooperation with RORγt, a master regulator of Th17 cells. Thus, IκB-ζ deficient mice are resistant to Th17-dependent experimental autoimmune encephalomyelitis (EAE). However, IκB-ζ deficient mice develop the autoimmune-like Sjögren syndrome with aging. Here we found that IκBNS-deficient (Nfkbid−/−) mice show resistance against developing Th17-dependent EAE. We found that Nfkbid−/− T cells have decreased expression of IL-17-related genes and RORγt in response to Transforming Growth Factor (TGF)-β1 and IL-6 stimulation. Thus, IκBNS plays a pivotal role in the generation of Th17 cells and in the control of Th17-dependent EAE.
Introduction
The transcriptional regulator IκBNS is a member of the nuclear IκB family, which also includes IκB-ζ and Bcl-3. IκBNS expression occurs in T cells and is rapidly induced upon T cell receptor (TCR) stimulation [1]. IκBNS has 7 ankyrin repeat domains that bind the p50 subunit of the DNA-binding protein nuclear factor-kappa B (NF-κB), but has no DNA-binding domain [2]. IκBNS interacts with NF-κB to control Interleukin (IL)-6 gene expression in macrophages [3], [4]. In T cells, IκBNS positively regulates IL-2 expression, a target of NF-κB [5]. Previously, Schmitz's group showed that IκBNS intrinsically induces Forkhead box P3 (Foxp3) positive regulatory T cells (Tregs) in vivo and in vitro [6]. Foxp3 is a master regulator of Tregs and can be induced by NF-κB activation [7].
TCR and Transforming Growth Factor (TGF)-β signaling are necessary for the generation of both Tregs and T helper (Th)17 cells [8]. IL-17 is a proinflammatory cytokine that plays an important role in autoimmune diseases and against bacterial infections [9]. The nuclear IκB family protein IκB-ζ can be induced in T cells in response to TGF-β1 and IL-6 stimulation and positively regulates Th17 generation in cooperation with RORγt [10]. Therefore, IκB-ζ-deficient mice are resistant to Th17-dependent experimental autoimmune encephalomyelitis (EAE), a model of multiple sclerosis. However, these mice have more effecter memory T cells and produce more Interferon (IFN)- γ in the periphery, leading to a Sjögren-like syndrome with age [11].
Here, we demonstrate that IκBNS-deficient (Nfkbid−/−) mice are resistant to Th17-dependent EAE. Further analysis revealed that the percentage of Th17 cells in the draining lymph nodes (LNs) of myelin oligodendrocyte glycoprotein (MOG)-immunized Nfkbid−/− mice was decreased relative to that of control mice. In addition, IκBNS-deficient T cells were less capable of generating Th17 cells in response to TGF-β1 and IL-6. Mechanistically, we found that IκBNS–deficient T cells show decreased RORγt induction in response to TGF-β1 and IL-6.
Materials and Methods
Mice
Nfkbid− /− mice (having a mixed C57/BL6 × BALB/c genetic background) were established as described previously [3]. All mice were maintained in specific pathogen-free conditions in the animal facilities of Tohoku University. All animal protocols were approved by the Institutional Committee for the Use and Care of Laboratory Animals of Tohoku University (2013MA029, 2013MA031 and 2013MA032).
Cells
CD4+CD25− T cells were prepared from mouse spleens using a CD4+CD25+ isolation kit II (Miltenyi Biotec; Bergisch Gladbach, Germany).
EAE induction and analysis of cytokine production
Nfkbid +/+ and Nfkbid −/− mice were injected subcutaneously (on the lower back) on day 0 with emulsions containing complete Freund's adjuvant (CFA; BD Difco™; Detroit, MI), 100 µg MOG peptide (MEVGWYRSPFSRVVHLYRNGK; MBL International Corporation, Nagoya, Japan), and 0.5 mg Mycobacterium tuberculosis H37RA (BD Difco™). In addition, these mice received 500 ng pertussis toxin (Sigma) by i.p. injection to boost immunological responses on day 0 and 2. These mice were observed until day 21 after immunization and clinical signs of EAE were scored according to a previously described protocol [12]. To study cytokine production, draining lymph node cells were derived and cultured for 72 h in the presence of 10 ng/ml MOG peptide.
In vitro T cell culture
Purified CD4+CD25− T cells were cultured in RPMI 1640 medium containing 10% heat-inactivated fetal calf serum, 100 units/mL penicillin and 100 µg/mL streptomycin at 37°C in 5% CO2. For cells in the Th0 condition, anti-CD3 (1 µg/mL) + anti-CD28 (1 µg/mL) stimulation was used. For cells in the Th17 condition, anti-CD3 (1 µg/mL) + anti-CD28 (1 µg/mL) with TGF-β1 (2 ng/mL) and IL-6 (50 ng/mL) stimulation was used [13].
Plasmids, antibodies, and cytokines
Expression vectors encoding FLAG-tagged mouse RORγt and IκB-ζ were constructed as described previously [10], [14]. Mouse IκBNS was inserted into a pcDNA3-FLAG vector at the EcoRI and BamHI sites. The mouse IL-17A promoter (−6647 to +1) was inserted into a pGL4.12 vector at the NheI and HindIII sites. pGL4 and pcDNA3 were obtained from Life Technologies (Rockville, MD), and phRL-TK was obtained from Promega Corp. (Madison, WI).
APC-conjugated anti-mouse CD62L (MEL-14), APC-conjugated anti-mouse IL-17A (TC11-18H10.1), PerCP/Cy5.5-conjugated anti-mouse CD8α (53-6.7), and Pacific Blue-conjugated anti-mouse CD4 (GK1.5) antibodies were purchased from BioLegend, Inc. (San Diego, CA). PE-conjugated anti-mouse IFN-γ (XMG1.7), purified anti-mouse CD3 (145-2C11), and purified anti-mouse CD28 (37.51) antibodies were obtained from eBioscience, Inc. (San Diego, CA). Rat anti-mouse Galectin-3 was obtained from Bay bioscience Co., Ltd (Hyogo, Japan). Biotinylated anti-rat IgG (E0468) was obtained from DAKO (Glostrup, Denmark). Recombinant human TGF-β1 and mouse IL-6 were obtained from PeproTech, Inc. (Rocky Hill, NJ).
Flow cytometric analysis
Cell surface antigens were stained in the dark at 4°C with antibodies diluted in PBS containing 0.5% BSA (FACS buffer). To study intracellular cytokine production, cells were stimulated with 250 nM ionomycin (BD Bioscience, San Jose, CA) and 50 nM phorbol 12-myristate 13-acetate (Sigma-Aldrich) in the presence of GolgiStop (BD Bioscience) for 4 h at 37°C in 5% CO2. Cells were fixed with 4% paraformaldehyde-PBS, permeabilized with 0.5% saponin in FACS buffer, and then stained in the dark at 4°C with the indicated antibodies [11]. Stained cells were analyzed with a Gallios™ flow cytometer (Beckman Coulter, Inc.; Brea, CA) and the data obtained were analyzed with FlowJo software (Tree Star, Inc.; Ashland, OR).
Enzyme-linked immunosorbent assays (ELISAs)
ELISA kits for IL-17A and IFN-γ (eBioscience) were used to quantify the respective cytokines in culture supernatants, according to the manufacturer's protocols.
Chromatin immunoprecipitation (ChIP)
Cultured CD4+ T cells were fixed in 1% formaldehyde, exposed to 0.2 M glycine to halt the fixation process, and washed in ice-cold PBS containing 0.5% BSA. Subsequently, cells were lysed by sonication in SDS lysis buffer containing 1% (wt/vol) SDS, 10 mM EDTA, and 50 mM Tris (pH 8.0). Cellular debris was removed by centrifugation. A ChIP assay was performed using antibodies against acetyl-histone H3 (Lys27) and normal rabbit IgG (Cell Signaling Technology; Danvers, MA) and Dynabeads Protein G (Life Technologies). Immunoprecipitated and input DNA was then analyzed by quantitative PCR using SYBR Premix EX Taq (Takara Bio). The sequences of primers used for quantitative PCR are as follows: 5′-GCTGCTGTTTCCTTGAGAGG-3′ and 5′-GCTGGATAAGGCAGGAACAG-3′ for conserved non-coding sequence (CNS) 1; 5′- CTGAGTTGGGGGCTGTGTAT -3′ and 5′-CATATCGAGGGTGTCGGACT-3′ for CNS 2; 5′-CAGCAGACACACATGCAAGA-3′ and 5′-CCTCAGGGGAGGGAATTAAG-3′ for CNS 3; 5′-CACCTCACACGAGGCACAAG-3′ and 5′-ATGTTTGCGCGTCCTGATC-3′ for the Il-17a promoter; and 5′-CACTTCCTGAAGGGGAATCA-3′ and 5′-GGGTGGGCTTAGAAGAGAGG-3′ for the Il-17f promoter.
Histology
Tissues were fixed by immersion in 10% formalin in phosphate-buffered saline and embedded in paraffin blocks. Three-micrometer-thick sections were stained with hematoxylin and eosin (HE staining) or luxol fast blue solution and cresyl violet solution (Klüver-Barrera staining), and then examined by light microscopy. Immunohistochemistry of galectin-3 was described previously [15]. Briefly, the paraffinized sections were antigen revealed by using a 0.01 M citrate buffer (pH 6.0) by the PascalR heat-induced target retrieval system (DAKO). Anti-galectin-3 antibody using at a dilution of 1∶100 in 2% BSA/PBS were added on the slides and incubated overnight at 4°C. Anti-galectin-3 antibody was detected with a biotinylated anti-Rat IgG (1∶200) for 30 min, followed by incubation with avidin-coupled peroxidase (Vectastain ABC kit, Vector Laboratories) for 30 min. The peroxidase binding sites were detected by staining with 3,3′-diaminobenzidine (DAB) in 50 mM Tris–EDTA buffer, pH 7.6.
Luciferase Assays
HEK 293 cells (1×105 cells) were transfected using the calcium phosphate-DNA coprecipitation method with IL-17A reporter and expression vectors (pcDNA3-RORγt, IκBNS, and IκB-ζ) with pRL-TK-Luc. Twenty-four hours after transfection, the medium was changed and the cells were incubated for a further 24 h. Luciferase activities were measured using the Dual-Luciferase Reporter Assay System, according to the manufacturer's instructions (Promega Corp., Madison, WI). Data shown are the mean ± SE of duplicate samples from a representative of at least 3 independent experiments.
Statistical Analysis
Paired data were evaluated using the Student's t test.
Results
Nfkbid −/− mice maintain immune homeostasis
A previous study showed that IκBNS plays an important role in the generation of Tregs [6]. However, Nfkbid −/− mice and bumble mutant mice (harboring a stop codon after exon 4 of the Nfkbid gene) appear healthy and do not exhibit an inflammatory phenotype in the periphery [16]. IκBNS shares greatest homology with IκB-ζ (43% identity at the gene level). We confirmed that Nfkbid −/− mice appear healthy and can live for up to 6 months without disease manifestation. At 8–12 weeks old, the percentage of effector/memory and naïve CD4+ cells in the peripheral LNs and spleen were comparable in Nfkbid +/+ and Nfkbid −/− mice (Fig. 1A and 1B). Although the overall percentages of IFN-γ- and IL-17A-producing CD4+ cells in the spleen and peripheral LNs were low (Fig. 1C and 1D), we were still able to demonstrate that the percent of IFN-γ-producing CD4+ cells in the peripheral LNs was lower in Nfkbid −/− mice compared to Nfkbid +/+ mice (Fig. 1D).
Figure 1. Characteristics of immune homeostasis in Nfkbiz −/− mice.
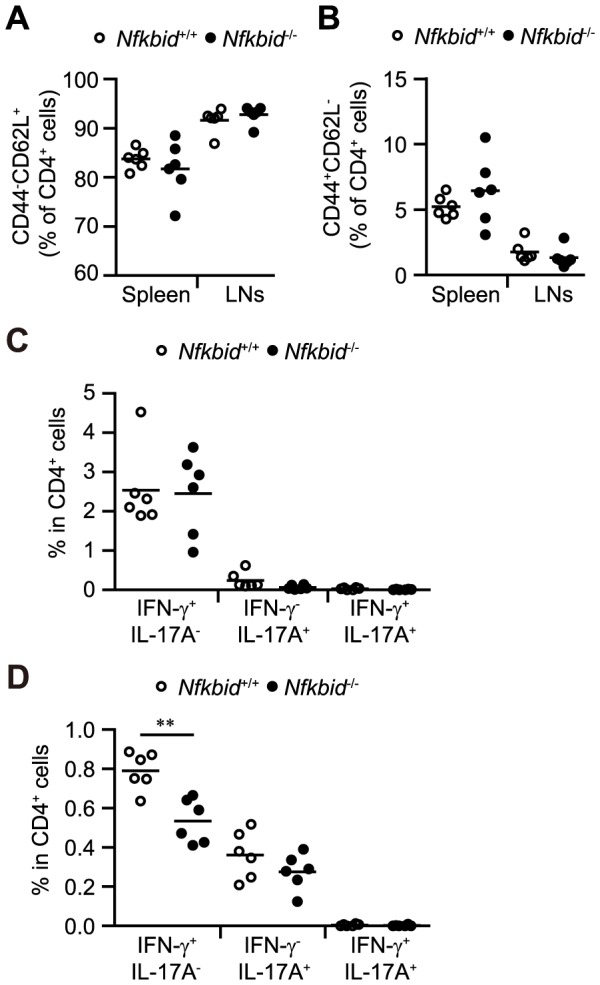
(A, B) Naive CD4+ cells in the spleen and lymph nodes (LNs) of 8–12 week old Nfkbid +/+ and Nfkbiz −/− mice. (C, D) Flow cytometric analysis of IFN-γ- and IL-17-producing CD4+ cells isolated from the spleen (C) and LNs (D) of Nfkbid +/+ and Nfkbiz −/− mice at 8–12 weeks of age. Paired data were evaluated using the Student's t test. **p<0.01.
Nfkbid −/− mice resist EAE development
Next, we generated classical Th17-dependent EAE models by immunizing mice with the MOG peptide [17], [18]. Ten to twelve days after MOG immunization, Nfkbid +/+ mice developed EAE, starting with paralysis of the tail, followed by paralysis in the front and hind limbs between days 18 and 21 (Fig. 2A). However, Nfkbid −/− mice showed significantly fewer clinical signs of diseases (Fig. 2A). Further, Nfkbid −/− mice have fewer IL-17A-producing CD4+ cells and reduced MOG antigen-specific IL-17A production in their draining LNs (Fig. 2B–D). We also confirmed that lymphocyte infiltration and demyelination occurred in the lumber section of spinal cords of Nfkbid +/+ EAE model mice, but not in those of Nfkbid −/− EAE models (Fig. 2E). Galectin-3, an activation maker of monocyte/macrophages/microglia [19], was observed as dark-brown staining in injured white matter of spinal cord in EAE model. We found that many galectin-3 positive cells in the lumber section of spinal cords of Nfkbid +/+ EAE model mice, but not Nfkbid −/− EAE model mice (Fig. 2E). In addition, adoptive transfer of CD4+ T cells from EAE models of Nfkbid −/− mice to naïve Nfkbid +/+ mice caused tail and hind limb paralysis, although these symptoms were less severe than those observed with adoptive transfer of CD4+ T cells from EAE models of Nfkbid +/+ mice (Fig. S1). Thus, T cells serve an intrinsic role in the resistance of EAE in Nfkbid −/− mice.
Figure 2. Experimental autoimmune encephalomyelitis (EAE) model in Nfkbiz −/− mice.

(A) Disease progression of EAE in Nfkbiz +/+ (n = 11–13) and Nfkbiz −/− mice (n = 9–11). (B–D) Analysis of mice 12 days after immunization. (B) Cytokine profile of CD4+ cells in draining LNs. (C, D) Measurement of IL-17A (C) and IFN-γ (D) supernatant concentrations by ELISAs (Nfkbiz +/+: n = 5; Nfkbiz −/−: n = 6), using cultured draining LNs incubated in the presence or absence of MOG peptide (10 ng/ml) for 72 h. Data shown represent mean ± S.E. Paired data were evaluated using the Student's t test. *p<0.05, **p<0.01. (E) Histology of spinal cord specimens in EAE models. Twelve days after MOG immunization, Nfkbiz +/+ and Nfkbiz −/− mice were sacrificed and their lumber section of spinal codes were collected. Three-micrometer-thick sections were stained with hematoxylin and eosin (HE), Klüver-Barrera staining (KB) or galectin-3 (Gal-3) immunohistochemistry. Serial sections were used for HE staining, KB staining and Gal-3 immunohistochemistry. Arrowheads in HE staining and KB staining indicate the demyelinated lesions. Data are representative of 3 independent experiments.
Reduced Th17 cell differentiation in Nfkbid −/− T cells
Next, we examined whether Nfkbid −/− T cells are capable of differentiating into Th17 cells. We found that Nfkbid −/− T cells show decreased expression of IL-17A (Fig. 3A and 3B) and Th17-related genes (Ccr6 and Il-17f; Fig. 3C and 3D) in response to TGF-β1 and IL-6 stimulation. In addition, detection of Acetylated histone H3 on Lysine 27 (H3K27Ac), a histone modification associated with open chromatin configurations, was reduced in the conserved non-coding sequence (CNS) 1, CNS 2, CNS 3, and Il-17f promoter regions in Nfkbid −/− T cells under Th17 conditions compared to that observed in Nfkbid +/+ T cells (Fig. 3E–F). These regions potentially regulate Il-17 gene expression [20]. Interestingly, the acetylation status of the CNS 1 region in Nfkbid −/− T cells did not change under Th17 conditions compared with Th0 conditions (Fig. 3F). A previous study indicated that the CNS 1 region plays an important role for both IL-17A and IL-17F expression [21]. Thus, Nfkbid −/− T cells were impaired in generating Th17 cells in response to TGF-β1 and IL-6 because of reduced histone H3 acetylation in the CNS 1 regions.
Figure 3. Nfkbiz −/− mouse T cells fail to generate Th17 cells in vitro.

(A–D) Expression of IL-17A protein or Il-17a mRNA (A, B) and of the Th17-related mRNAs Ccr6 and Il-17f (C, D) in CD4+ T cells from Nfkbiz +/+ and Nfkbiz −/− mice, cultured for 48 h under Th0 or Th17 conditions. (E) Diagram of Il-17a and Il-17f gene conservation. Red-arrows indicate the Il-17a promoter, the Il-17f promoter, and the CNS 1, CNS 2, and CNS 3 regions. (F) ChIP analysis of H3K27Ac. Cells were cultured under Th0 or Th17 conditions for 48 h. Data shown are from one experiment that was representative at three independent experiments. (A–D) Data shown represent mean ± S.E. (n = 3). Paired data were evaluated using the Student's t test. *p<0.05, **p<0.01.
IκBNS does not control il17a gene expression
IκB-ζ, a homolog of IκBNS, can be upregulated in T cells in response to TGF-β1 and IL-6 stimulation, directly binds to CNS1, and positively regulates IL-17A expression in cooperation with RORγt [22], [23]. Here, we found that IκBNS expression was comparable under Th0 and Th17 conditions (Fig. 4A). In addition, IκBNS expression had no effect on IL-17A reporter activity, even in the presence of RORγt (Fig. 4B). Thus, while IL-17A expression may be regulated by IκB-ζ, the homolog IκBNS does not transcriptionally control IL-17A.
Figure 4. IκBNS does not regulate Il-17a gene expression.
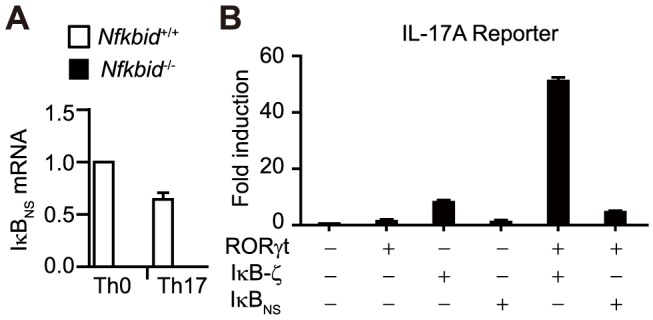
(A) Relative IκBNS mRNA expression levels in cultured CD4+ T cells under Th0 or Th17 conditions. IκBNS mRNA expression in CD4+ T cells cultured under Th0 conditions from control mice was set to a value of 1. Data shown represent mean ± S.E. (n = 3) (B) Il-17a promoter activity presented as the fold-increase over that observed in HEK293 cells transfected with the empty vector. Data shown are the mean ± S.D. of duplicate samples and are representative of three independent experiments.
Reduced RORγt expression in Nfkbid −/− T cells
IκBNS can be induced upon TCR stimulation and can control NF-κB transcriptional activity [6], [24]. In T cells, IκBNS deficiency leads to decreased production of IL-2 (a target of NF-κB) in response to TCR stimulation [5]. Thus, Nfkbid −/− T cells have a reduced ability to activate NF-κB in response to TCR stimulation. Rel (NF-κB subunit)-deficient T cells fail to generate Th17 cells since RORγt induction is diminished in response to TGF-β1 and IL-6 stimulation [25].
To understand the molecular mechanism underlying the control of IL-17A gene expression by IκBNS, we examined the expression of RORγt (a master regulator of Th17) and found that it was decreased in Nfkbid −/− T cells (Fig. 5). Thus, IκBNS controls NF-κB activation, which plays a pivotal role in RORγt expression and Th17 cell differentiation.
Figure 5. Nfkbiz −/− T cells show decreased RORγt expression.
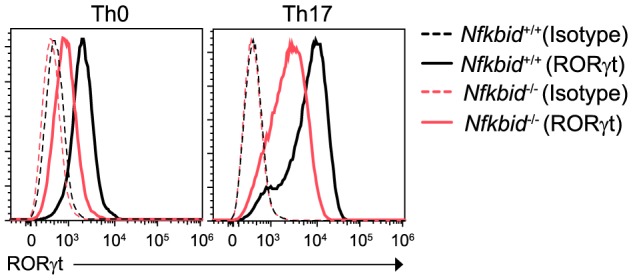
RORγt expression in CD4+ T cells from Nfkbiz +/+ and Nfkbiz −/− mice, cultured for 72 h under Th0 or Th17 conditions. Data are representative of three independent experiments.
Discussion
IκBNS, a member of the nuclear IκB family of proteins, is induced by TCR stimulation in thymocytes [1], [26]. Interestingly, Nfkbid −/− mice show a high sensitivity to lipopolysaccharide (LPS)-induced endotoxin shock and 4,4-dimethyl-4-silapentane-1-sulfonic acid-induced colitis because Nfkbid −/− DCs and macrophages produce large amounts of IL-6 in response to LPS stimulation [3]. It is well known that IL-6 positively regulates Th17 cell generation [27]. In addition, IκBNS may play a pivotal role in IL-10 production from regulatory DCs [28]. Thus, the intrinsic role of IκBNS in T cells may contribute to exacerbating Th17-dependent EAE. Although IκBNS is important for Foxp3+Tregs generation [6], Nfkbid −/− mice and bumble mutant mice appear healthy and do not exhibit an inflammatory phenotype in the periphery (Fig. 1) [16]. IκBNS may play a minor role in the immune suppression ability of Tregs.
IκB-ζ, a homolog of IκBNS, has a transcriptional activation domain and is important for Th17 cell differentiation [10]. IκBNS does not have a similar homologous transcriptional activation domain. In addition, IκB-ζ expression in T cells is dependent on TGF-β1 and IL-6 stimulation [10]. Thus, the regulation of IL-17A gene expression by IκB-ζ and IκBNS proceeds by different mechanisms.
Nfkbid −/− T cells show reduced proliferation and NF-κB activation in response to TCR [5]. However, a previous study showed that IL-17A promoter activity is dispensable for NF-κB activation [25]. Our ChIP data revealed that acetylation of histone H3 in the CNS 1 region does not change in Nfkbid −/− T cells in response to TGF-β1 + IL-6 (Fig. 3F). The CNS 1 region normally controls both IL-17A and IL-17F gene expression [21]. RORγt, a master regulator of Th17, has the ability to bind both CNS 1 and the Il-17a promoter region, and it positively regulates IL-17A gene expression [29]. Therefore, our results indicate that Nfkbid −/− T cells showed impaired Th17 cells differentiation because of a reduction in RORγt expression and histone H3 acetylation in the CNS 1 region. In conclusion, we show that IκBNS deficiency causes resistance to Th17-dependent autoimmune disease.
Supporting Information
Passive EAE model using adoptive T cell transfer. Collected draining LNs from the Nfkbiz +/+ and Nfkbiz −/− mice at day 12 after MOG immunizations. LN cells were re-stimulated by MOG (10 ng/ml) after 3 days in culture, and CD4+ T cells were isolated using the CD4+CD25+ Regulatory T cell Isolation Kit (Miltenyi Biotec). Nfkbiz +/+ mice (n = 3–4/group) were intravenously injected (5 × 105 CD4+ T cells/mouse) and EAE symptoms were scored for up to 12 days. In addition, these mice received 500 ng pertussis toxin (Sigma) by i.p. injection to boost their immunological responses on Days 0 and 2. Data shown represent mean + S.E. Paired data were evaluated using the Student's t test. *p <0.05.
(EPS)
Acknowledgments
We are grateful to Dr. Muta, Tatsushi (Tohoku University) for supervising this research and for helpful discussions. We are also grateful to Mr. Yasuyuki, Goto (Tohoku University) and Ms. Takahashi, Kyoko (Gifu University) for providing technical assistance.
Data Availability
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was supported in part by a Grant-in-Aid for Scientific Research on Innovative Areas (25118702) from the Ministry of Education, a Grant-in-Aid for Young Scientists (B) (24790458) from the Japan Society for the Promotion of Science, and by grants from the Takeda Science Foundation, the Uehara Memorial Foundation, the Novartis Foundation, and the Sumitomo Foundation to TMY. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Fiorini E, Schmitz I, Marissen WE, Osborn SL, Touma M, et al. (2002) Peptide-induced negative selection of thymocytes activates transcription of an NF-kappa B inhibitor. Molecular cell 9: 637–648. [DOI] [PubMed] [Google Scholar]
- 3. Kuwata H, Matsumoto M, Atarashi K, Morishita H, Hirotani T, et al. (2006) IkappaBNS inhibits induction of a subset of Toll-like receptor-dependent genes and limits inflammation. Immunity 24: 41–51. [DOI] [PubMed] [Google Scholar]
- 4. Hirotani T, Lee PY, Kuwata H, Yamamoto M, Matsumoto M, et al. (2005) The nuclear IkappaB protein IkappaBNS selectively inhibits lipopolysaccharide-induced IL-6 production in macrophages of the colonic lamina propria. Journal of immunology 174: 3650–3657. [DOI] [PubMed] [Google Scholar]
- 5. Touma M, Antonini V, Kumar M, Osborn SL, Bobenchik AM, et al. (2007) Functional role for I kappa BNS in T cell cytokine regulation as revealed by targeted gene disruption. Journal of immunology 179: 1681–1692. [DOI] [PubMed] [Google Scholar]
- 6. Schuster M, Glauben R, Plaza-Sirvent C, Schreiber L, Annemann M, et al. (2012) IkappaB(NS) protein mediates regulatory T cell development via induction of the Foxp3 transcription factor. Immunity 37: 998–1008. [DOI] [PubMed] [Google Scholar]
- 7. Ruan Q, Kameswaran V, Tone Y, Li L, Liou HC, et al. (2009) Development of Foxp3(+) regulatory t cells is driven by the c-Rel enhanceosome. Immunity 31: 932–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Maruyama T, Konkel JE, Zamarron BF, Chen W (2011) The molecular mechanisms of Foxp3 gene regulation. Seminars in immunology 23: 418–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Veldhoen M, Stockinger B (2006) TGFbeta1, a “Jack of all trades”: the link with pro-inflammatory IL-17-producing T cells. Trends in immunology 27: 358–361. [DOI] [PubMed] [Google Scholar]
- 10. Okamoto K, Iwai Y, Oh-Hora M, Yamamoto M, Morio T, et al. (2010) IkappaBzeta regulates T(H)17 development by cooperating with ROR nuclear receptors. Nature 464: 1381–1385. [DOI] [PubMed] [Google Scholar]
- 11. Okuma A, Hoshino K, Ohba T, Fukushi S, Aiba S, et al. (2013) Enhanced apoptosis by disruption of the STAT3-IkappaB-zeta signaling pathway in epithelial cells induces Sjogren's syndrome-like autoimmune disease. Immunity 38: 450–460. [DOI] [PubMed] [Google Scholar]
- 12. Perruche S, Zhang P, Liu Y, Saas P, Bluestone JA, et al. (2008) CD3-specific antibody-induced immune tolerance involves transforming growth factor-beta from phagocytes digesting apoptotic T cells. Nature medicine 14: 528–535. [DOI] [PubMed] [Google Scholar]
- 13. Maruyama T, Li J, Vaque JP, Konkel JE, Wang W, et al. (2011) Control of the differentiation of regulatory T cells and T(H)17 cells by the DNA-binding inhibitor Id3. Nat Immunol 12: 86–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yamazaki S, Muta T, Matsuo S, Takeshige K (2005) Stimulus-specific induction of a novel nuclear factor-kappaB regulator, IkappaB-zeta, via Toll/Interleukin-1 receptor is mediated by mRNA stabilization. The Journal of biological chemistry 280: 1678–1687. [DOI] [PubMed] [Google Scholar]
- 15. Satoh K, Niwa M, Goda W, Binh NH, Nakashima M, et al. (2011) Galectin-3 expression in delayed neuronal death of hippocampal CA1 following transient forebrain ischemia, and its inhibition by hypothermia. Brain research 1382: 266–274. [DOI] [PubMed] [Google Scholar]
- 16. Arnold CN, Pirie E, Dosenovic P, McInerney GM, Xia Y, et al. (2012) A forward genetic screen reveals roles for Nfkbid, Zeb1, and Ruvbl2 in humoral immunity. Proceedings of the National Academy of Sciences of the United States of America 109: 12286–12293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Oukka M (2007) Interplay between pathogenic Th17 and regulatory T cells. Annals of the rheumatic diseases 66 Suppl 3iii87–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kleinschek MA, Owyang AM, Joyce-Shaikh B, Langrish CL, Chen Y, et al. (2007) IL-25 regulates Th17 function in autoimmune inflammation. The Journal of experimental medicine 204: 161–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Reichert F, Rotshenker S (1999) Galectin-3/MAC-2 in experimental allergic encephalomyelitis. Experimental neurology 160: 508–514. [DOI] [PubMed] [Google Scholar]
- 20. Akimzhanov AM, Yang XO, Dong C (2007) Chromatin remodeling of interleukin-17 (IL-17)-IL-17F cytokine gene locus during inflammatory helper T cell differentiation. The Journal of biological chemistry 282: 5969–5972. [DOI] [PubMed] [Google Scholar]
- 21. Wang X, Zhang Y, Yang XO, Nurieva RI, Chang SH, et al. (2012) Transcription of Il17 and Il17f is controlled by conserved noncoding sequence 2. Immunity 36: 23–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhang F, Meng G, Strober W (2008) Interactions among the transcription factors Runx1, RORgammat and Foxp3 regulate the differentiation of interleukin 17-producing T cells. Nat Immunol 9: 1297–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ivanov, II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, et al. (2006) The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 126: 1121–1133. [DOI] [PubMed] [Google Scholar]
- 24. Ghosh S, Hayden MS (2008) New regulators of NF-kappaB in inflammation. Nature reviews Immunology 8: 837–848. [DOI] [PubMed] [Google Scholar]
- 25. Ruan Q, Kameswaran V, Zhang Y, Zheng S, Sun J, et al. (2011) The Th17 immune response is controlled by the Rel-RORgamma-RORgamma T transcriptional axis. The Journal of experimental medicine 208: 2321–2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yamamoto M, Takeda K (2008) Role of nuclear IkappaB proteins in the regulation of host immune responses. Journal of infection and chemotherapy: official journal of the Japan Society of Chemotherapy 14: 265–269. [DOI] [PubMed] [Google Scholar]
- 27. Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, et al. (2006) Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 441: 235–238. [DOI] [PubMed] [Google Scholar]
- 28. Fujita S, Seino K, Sato K, Sato Y, Eizumi K, et al. (2006) Regulatory dendritic cells act as regulators of acute lethal systemic inflammatory response. Blood 107: 3656–3664. [DOI] [PubMed] [Google Scholar]
- 29. Hermann-Kleiter N, Meisel M, Fresser F, Thuille N, Muller M, et al. (2012) Nuclear orphan receptor NR2F6 directly antagonizes NFAT and RORgammat binding to the Il17a promoter. Journal of autoimmunity 39: 428–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Passive EAE model using adoptive T cell transfer. Collected draining LNs from the Nfkbiz +/+ and Nfkbiz −/− mice at day 12 after MOG immunizations. LN cells were re-stimulated by MOG (10 ng/ml) after 3 days in culture, and CD4+ T cells were isolated using the CD4+CD25+ Regulatory T cell Isolation Kit (Miltenyi Biotec). Nfkbiz +/+ mice (n = 3–4/group) were intravenously injected (5 × 105 CD4+ T cells/mouse) and EAE symptoms were scored for up to 12 days. In addition, these mice received 500 ng pertussis toxin (Sigma) by i.p. injection to boost their immunological responses on Days 0 and 2. Data shown represent mean + S.E. Paired data were evaluated using the Student's t test. *p <0.05.
(EPS)
Data Availability Statement
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its Supporting Information files.