

Abstract
Obesity is a common and complex health problem, which impacts crucial organs; it is also considered an independent risk factor for chronic kidney disease. Few studies have analyzed the consequence of obesity in the renal proximal convoluted tubules, which are the major tubules involved in reabsorptive processes. For optimal performance of the kidney, energy is primarily provided by mitochondria. Melatonin, an indoleamine and antioxidant, has been identified in mitochondria, and there is considerable evidence regarding its essential role in the prevention of oxidative mitochondrial damage. In this study we evaluated the mechanism(s) of mitochondrial alterations in an animal model of obesity (ob/ob mice) and describe the beneficial effects of melatonin treatment on mitochondrial morphology and dynamics as influenced by mitofusin-2 and the intrinsic apoptotic cascade. Melatonin dissolved in 1% ethanol was added to the drinking water from postnatal week 5–13; the calculated dose of melatonin intake was 100 mg/kg body weight/day. Compared to control mice, obesity-related morphological alterations were apparent in the proximal tubules which contained round mitochondria with irregular, short cristae and cells with elevated apoptotic index. Melatonin supplementation in obese mice changed mitochondria shape and cristae organization of proximal tubules, enhanced mitofusin-2 expression, which in turn modulated the progression of the mitochondria-driven intrinsic apoptotic pathway. These changes possibly aid in reducing renal failure. The melatonin-mediated changes indicate its potential protective use against renal morphological damage and dysfunction associated with obesity and metabolic disease.
Introduction
Obesity is considered an independent risk factor for chronic kidney disease, when associated with type 2 diabetes and hypertension [1], [2] and also in nondiabetic people [3], [4]. This condition is characterized by an excessive metabolic demand in adipose tissue and concomitant diabetic nephropathy [5]. Several lines of evidence highlight that excessive glomerular filtration with an increase in Bowman’s space is associated with an elevated glomerular hydrostatic pressure and that elevated proximal tubular sodium reabsorption plays a central role in the pathogenesis of glomerular hyperfiltration [6]. Although nephropathy in obese subjects is both common and complex and it is poorly understood. Moreover, few data are available on morphological changes that develop during proximal tubule damage [7]. Experimental data in animal models suggest that, in the diabetic kidney, the metabolic energy necessary to remove toxic deposits and to perform hyperfiltration by proximal tubules is deficient. This energy is provided by mitochondria and is essential for tubular-glomerular feedback [8].
Research has shown that mitochondrial damage and their functional disruption is due to increased generation of reactive oxygen species (ROS) and to mutations in proteins involved in the fusion-fission machinery. These represent an important pathogenic mechanism in renal diseases. Indeed, the morphology of mitochondria is related to the balance between fission and fusion [9]. Fission results in the formation of solitary organelles that may be easily removed by mitophagy or may be activated by the pro-apoptotic Bcl2 family machinery resulting in cytochrome c release and caspase activation and apoptosis. In contrast, the mitochondrial fusion machinery is associated with three GTPase dynamin-like proteins, named optic atrophy 1 (Opa1) and mitofusins (Mfn) 1 and 2 [10], [11]. In particular, Mfn2 is a transmembrane protein linked to the structural bridge between mitochondria and endoplasmic reticulum [12] and even through it may not be necessary for the juxtaposition of the organelles [13] it is related to calcium flux in metabolically active tissues. Mfn2, is closely associated to the development of diabetes, but its specific roles in the diabetic kidney remain unknown [5], [14].
Melatonin (N-acetyl-5-methoxytryptamine), a widely produced indoleamine and antioxidant [15]–[17], has been successfully used to limit metabolic alterations and mitochondrial dysfunctions in obese and in aged animals [18]–[22]. Emerging evidence has outlined the crucial role of melatonin in the prevention of oxidative mitochondrial damage due to its direct influence on free radical removal, cardiolipin peroxidation and cytochrome c release from mitochondria [23]–[25]. Moreover in metabolic diseases, such as obesity, inflammatory cytokines stimulate the generation of mitochondrial superoxide anion radicals and induce oxidative/nitrosative stress that causes defective aggregation of respiratory chain complexes. Melatonin neutralizes these changes and reduces radical mediated mitochondrial dysfunction [20], [26], [27].
Evidence has also shown an intimate interplay between misshaped mitochondria and altered mitochondrial bioenergetics. A mitochondrial reduction in membrane potential and fragmentation may be related to Mfn2 expression during obesity. These observations along with a reduction in the amplitude of the nocturnal pineal melatonin peak suggest that lower melatonin levels may be related to the mitochondrial deficiencies [10], [28], [29]. Thus, in the current study we carefully analyzed the mitochondrial structure in the proximal convoluted tubules of obese mice.
The aims of this study were to clarify mitochondrial alterations by investigating the roles of Mfn2 protein associated with convoluted tubule modifications in obese mice and to determine if melatonin would ameliorate or attenuate mitochondria dysfunction and the mitochondrial deformities. We also evaluated whether melatonin affects mitochondria-Mfn2-driven intrinsic apoptosis by an immunohistochemical analysis of specific markers such as cytochrome c, Bax and the caspase cascade (caspase 3 and caspase 9).
Materials and Methods
Animals and experimental protocols
Forty male mice, 20 lean and 20 obese (B6.V-Lepob/OlaHsd), 4 weeks old were purchased from Harlan Laboratories S.r.l. (Udine, Italy) and divided into four groups (n = 10 mice/group) as follows: a) lean mice without treatment (lean); b) lean mice treated with melatonin for 8 weeks (lean+mel); c) obese mice without treatment (ob/ob); d) obese mice treated with melatonin for 8 weeks (ob/ob+mel). Melatonin (kindly provided by Chronolife S.r.l., Roma, Italy) was dissolved in 1% ethanol and diluted in the drinking water to yield a calculated final dose of 100 mg/kg body weight/day from postnatal week 5 to 13. The feeder jars were refilled daily with normal rodent chow, obtained from Harlan Laboratories S.r.l. (Udine, Italy) and the mean daily food intake were calculated by two observers blinded to the treatment. This study was carried out in strict accordance with the protocols of the Italian Ministry of Health and complied with “Guiding Principles in the Use of Animals in Toxicology” which were adopted by the Society of Toxicology in 1989. Protocols were approved by the Animal Care and Use Committee of the University of Brescia, Italy. Additional details about experimental procedures have been reported previously [18]. At the conclusion of postnatal week 13, all animals were killed by cervical dislocation and blood samples and kidneys were collected and processed for the serum analyses, the histological and immunofluorescence assays and for ultrastructural analyses.
Analysis of serum and urine
Creatinine concentration in serum was determined using an enzymatic assay (Biovision, Milpitas, California, US), according to the manufacturers’ instructions.
Urine was collected in metabolic cages before death and 8-epiprostaglandin-F2α (8-epi-PGF2α) was calculated using an EIA kit (8-isoprostane; Cayman Chemical, Ann Arbor, MI, USA), according to the manufacturers’ instructions.
Histopathology
One kidney was processed for histopathological staining and immunofluorescence assays. The tissue was fixed in 4% buffered paraformaldehyde for 24 hours, dehydrated in progressive ethanol solutions, xylene and embedded in paraffin wax, following the standard procedures. Subsequently, 7 µm-thick paraffin sections were cut stained using a Periodic acid-Schiff PAS solution to assess glomerular and tubular changes and mesangial proliferation [30]. On PAS stained sections at 400x magnification, two observers performed, in a blinded fashion, morphometric computerized analysis to evaluate glomerular area, using a light microscope (Olympus, Germany) equipped with an image analyzer (Image Pro Plus, Italy). In particular, 80 glomeruli, where vascular pole was evident, were examined in kidneys of each experimental group, as previously described [31].
Transmission electron microscopy
The second kidney of each mouse was treated for ultrastructural analysis according to Rezzani et al. [32]. Briefly renal tissue was fixed by immersion in 2.5% glutaraldehyde in cacodylate buffer 0.1 M (pH 7.4) for 3 hours at +4°C and postfixed in 2% osmium tetroxide in cacodylate buffer for 1 hour at +4°C. Dehydration process was performed in increasing ethanol concentrations and propylene oxide followed by Araldite-Epon resin embedding. Semithin sections (1 µm-thick) were collected at an UltraCut E ultramicrotome stained by toluidine blue and observed at a light microscope (Olympus, Germany) to assess the presence of glomeruli and cortical tubules. Subsequently, from representative blocks, 70–80 nm-thick ultrathin sections were obtained using a diamond knife, collected on formvar coated grids, double stained with uranyl acetate and lead citrate and observed under a transmission electron microscope (Tecnai G2 Spirit) at 80 kV. Morphometric computerized analysis of the area of 100 mitochondria for each experimental group was blindly analyzed at 26.000x magnification by two observers blinded of the treatment.
Immunofluorescence and immunohistochemistry assay
Alternate kidney sections were deparaffinized, rehydrated and incubated in 3% hydrogen peroxide, blocked with 1% bovine serum albumin (BSA) for 1 hour at room temperature, and then incubated overnight at 4°C with the following primary antibodies: mouse monoclonal antibody against mitofusin 2 (diluted 1∶600; H00009927-M01 Abnova, Taipei City, Taiwan); mouse monoclonal antibody against cytochrome c (diluted 1∶200; sc-13156 Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA); rabbit polyclonal antibody against Bax (diluted 1∶400; sc-526 Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA); mouse monoclonal antibody against caspase 3 (diluted 1∶400; sc-70497 Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA) and rabbit polyclonal antibody against caspase 9 (diluted 1∶400; sc- 7885 Abcam, Cambridge, United Kingdom). After rinsing with phosphate buffered saline (PBS) the sections were labeled using goat anti-rabbit Alexa Fluor 546 (A11035) and goat anti-mouse or goat anti-rabbit Alexa Fluor 488 (A11029 and A11034 respectively) conjugated secondary antibodies (1∶200; Invitrogen, Paisley, United Kingdom). Finally, the sections were counter-stained with 4′,6-diamidino-2-phenylindole (DAPI), mounted and observed with a confocal microscope (LSM 510 Zeiss, Munich, Germany) [33].
For the immuohistochemical analysis, the sections were incubated in 3% hydrogen peroxide for 30 minutes, to inactivate the endogenous peroxidase activity. Then, after incubation in 1% BSA for 1 hour at room temperature, were incubated in mouse monoclonal antibody against mitofusin 2 (diluted 1∶200; H00009927-M01 Abnova, Taipei City, Taiwan) for 2 hours at 37°C. The sections were then sequentially incubated in anti-mouse biotinylated immunoglobulin and in avidin-biotin peroxidase complex. The reaction products were visualized using 0.33% hydrogen peroxide and 0.05% 3,3′-diaminobenzidine tetrahydrochloride as chromogen. The sections were finally counter-stained with haematoxylin, mounted and observed with a light microscope (Olympus, Germany).
The immunofluorescence and immunohistochemical controls were performed by omitting the primary antibody and in the presence of isotype matched IgGs.
Staining intensity of all the immunofluorescence assays was evaluated by two observers blinded to the treatments. The observers, using transmitted light, identified convoluted proximal tubules and then quantified the immunoposotivity. The evaluations were assumed to be correct if the values were not significantly different. In case of dispute concerning interpretation the case was reconsidered to reach a unanimous agreement.
In particular, the immunopositivity for mitofusin 2, cytochrome c, Bax, caspase 3 and caspase 9 were calculated, using an image analyzer (Image Pro Plus, Milan, Italy), for standardized areas, measuring 20 random fields with the same area for each experimental animals.
Statistical analyses
The data were pooled to represent a mean value±standard deviation and statistical significance of differences among the experimental groups was evaluated by analysis of variance corrected by Bonferroni test with significance set at p<0.05. The levels of immunopositivity are expressed as arbitrary unit (AU).
Results
All animals of each experimental group survived and melatonin supplementation in drinking water for 8 weeks was well tolerated. The food intake of the ob/ob animals was not significantly different from that of the lean group. Body and kidney weight for each animal was measured; weight parameters in ob/ob group were significantly higher compared those in control mice. We observed that melatonin supplementation to ob/ob mice did not influence significantly body or kidney weights, although the kidney/body weight ratio indicated proportional changes in the different experimental groups (Table 1).
Table 1. Food intake, body and kidney weights.
| lean | lean plus melatonin | ob/ob | ob/ob plus melatonin | |
| Food intake (g/day) | 4.45.±0.2 | 4.37±0.12 | 4.32±0.11 | 4.39±0.15 |
| Body weight (g) | 31.2±0.23 | 30.5±0.12 | 54.3±0.37 | 49.7±0.26 |
| Kidney weight (g) | 0.322±0.03 | 0.335±0.04 | 0.428±0.02 | 0.397±0.03 |
| Kidney/Body weight ratio (mg/g) | 10.27±0.18 | 10.86±0.22 | 8.21±0.16 | 7.65±0.18 |
Already at 13 weeks of age the obese mice showed an elevated value of serum creatinine respect to lean control mice, with or without melatonin treatment; this alteration of creatinine levels reflect loss of renal function. Moreover, renal oxidative stress, determined by urinary 8-isoprostane assay, was increased in obese mice compared to lean mice, with or without melatonin treatment Interestingly, melatonin treatment induced a significative reduction in serum creatinine and in urinary 8-isoprostane in the obese mice (Table 2).
Table 2. Creatinine and renal oxidative damage.
| lean | lean plus melatonin | ob/ob | ob/ob plus melatonin | |
| Creatinine (mg/dL) | 0.17±0.2 | 0.16±0.1 | 0.5±0.1 | 0.23±0.1 |
| 8-isoprostane (pg/µmol) | 465±7 | 489±14 | 1153±25 | 576±18 |
We next assessed renal morphology and proximal tubules ultrastructure. Normal features of glomeruli and cortical proximal renal tubules in lean mice, with or without melatonin treatment, were observed; in particular, proximal and distal tubular epithelium was regular with a PAS-positive brush border and central nuclei and showed a large lumen (Figure 1a, b). However, ob/ob mice showed irregular glomerular tufts with abundant mesangial matrix and a wide subcapsular space, an enlarged tubular epithelium, often devoid of a brush border, and a lumen filled with proteic aggregates in both proximal and distal tubules (Figure 1c). We observed that the renal histological architecture was partially restored after melatonin supplementation and that melatonin prevented major changes in proximal tubules where abnormal PAS-positive deposits disappeared and the brush border epithelium was preserved (Figure 1d). Moreover, quantitative data on total glomerular area, including both the glomerular tuft and subpodocytic space, documented that in ob/ob mice the total area decreased compared to that in lean mice, with or without melatonin treatment; in ob/ob mice that received melatonin, the total area was increased (about 15%) with respect to ob/ob non-treated group (p<0.05) (Figure 1e). The ultrastructural evaluation performed on cortical proximal tubules showed; in the lean group treated and untreated with melatonin, normal proximal tubular epithelial cells with regular apical microvilli and many mitochondria associated with basal membrane invaginations (Figure 2a, c). The mitochondria, in particular, appeared elongated with well-preserved regular cristae and a homogeneous inner matrix (Figure 2b, d). In contrast, proximal tubules of ob/ob mice presented apoptotic or necrotic nuclei and cytoplasm occupied by scattered round mitochondria (Figure 2e), that appeared hydropic, with irregular and peripherally-located short cristae (Figure 2f). After melatonin supplementation to the obese animals, mitochondria appeared elongated with more regular cristae (Figure 2g, h). Mitochondrial area was lower in ob/ob group with respect to that in lean mice and this was significantly increased after melatonin supplementation (Figure 2i).
Figure 1. Histopathology.

Photomicrographs showing PAS staining in the kidney of lean (a), lean mice treated with melatonin (b), ob/ob (c) and ob/ob treated with melatonin (d) mice. The graph summarizes the quantitative analyses of glomerular area for each experimental group (e). *p<0.05 vs lean, #p<0.05 vs lean plus melatonin and + vs ob/ob. (G) glomerulus; (P) proximal tubule and (D) distal tubule. Bar equals 20 µm.
Figure 2. Transmission electron microscopy.

Ultrastructural photomicrographs showing cortical proximal tubules and mitochondria of lean (a, b), lean mice treated with melatonin (c, d), ob/ob (e, f) and ob/ob treated with melatonin mice (g, h). The graph summarizes the quantitative analyses of mitochondrial area of each experimental groups (i). (M) mitochondria and (N) nucleus. Bar equals 1 µm. *p<0.05 vs lean, #p<0.05 vs lean plus melatonin and + vs ob/ob.
The serum and urine assays and the morphological analyses did not reveal significantly differences between melatonin treated and non-melatonin treated control mice; thus, in immunofluorescence observations, they are considered without distinction (defined as lean mice). Immunofluorescent Mfn2 assay (green staining) showed that this mitochondrial marker was present as an intense basolateral signal in cortical tubules of lean mice (Figure 3a, e), whereas it was almost absent in renal proximal tubules of ob/ob mice (Figure 3b, f). After melatonin treatment, Mfn2 was partially preserved with a weak/moderate signal (Figure 3c). No expression or a very weak signal of Mfn2 was detected at glomerular level of each experimental group.
Figure 3. Mitofusin 2 immunofluorescence assay.

Photomicrographs showing kidney mitofusin 2 immunostaining of lean (a, e), ob/ob (b, f) and ob/ob treated with melatonin (c) mice and relative quantitative analyses of immunopositivity (d). Bar equals 20 µm. *p<0.05 vs lean. Nuclei were stained with 4,6 diamidino-2-phenilindole. Photomicrographs of mitofusin 2 staining showing a cortical proximal tubule immunopositive (e) and an other one immunonegative (f).
Cytochrome c immunofluorescence staining (identified in green) was almost undetectable in lean mice (Figure 4a) and became evident, with a moderate/weak signal, in ob/ob mice (Figure 4b). No expression was detected at glomerular level of mice of either group. Moreover, also Bax immunofluorescence (green staining) showed absence or very weak expression in lean mice (Figure 4e) and a moderate/weak expression in cortical proximal tubules of ob/ob mice (Figure 4f). No expression was detected at the glomerula. Regarding caspases, their localization and expression (green staining for caspase 3 and red signal for caspase 9) were undetectable/very weak in lean mice (Figure 4i), whereas, in ob/ob mice both caspases became clearly expressed (moderate signal) in cortical tubules, but sometimes scattered in different sites. In particular, caspase 3 was prevalently localized in the proximal tubules, while caspase 9 was observed in the distal tubules (Figure 4l).
Figure 4. Apoptotic markers immunofluorescence assay.

Photomicrographs of immunofluorescence analyses of kidney cytochrome c (green staining) of lean (a), ob/ob (b) and ob/ob treated with melatonin (c) mice. Immunofluorescence analyses of Bax (green staining) in kidney of lean (e), ob/ob (f) and ob/ob treated with melatonin (g) mice. Double immunofluorescence of caspase 3 (green staining) and caspase 9 (red staining) in kidney of lean (i), ob/ob (l) and ob/ob treated with melatonin (m) mice. Nuclei were stained with 4,6 diamidino-2-phenilindole. Bar equals 20 µm. The graphs summarize the quantitative analysis, respectively, of cytochrome c (d), Bax (h) and both caspase 3 and caspase 9 (n). *p<0.05 vs lean and #p<0.05 vs ob/ob.
Moreover, we observed, after melatonin supplementation, that cytochrome c and also Bax were greatly attenuated (very weak/absent signal) due to melatonin treatment (Figure 4c, g) and also expression of both caspases decreased significantly in the proximal tubules (Figure 4m). Relative immunopositivity quantifications of each experimental group are plotted in figure 4d, h and n.
Discussion
In the present study we showed that mitochondria of proximal tubules are morphologically altered in obese mice. Mnf2 expression was undetectable; while, staining of cytochrome c, Bax and capsases was evident in obese animals. Melatonin in the drinking water modified mitochondrial morphology and distribution in proximal tubules and greatly reduced specific markers of the intrinsic apoptotic pathway in kidneys of obese mice.
The obese mice developed an elevated serum creatinine already in the earliest phases of the pathogenesis, demonstrating that the onset of nephropathy is rapid and progressive. With regard to mitochondrial morphology, our findings are in agreement with several studies performed using muscles, liver and heart of obese animals [34], [35]. In our study, mitochondria of obese mice were roundish when compared to the elongated shape of mitochondria in control conditions. During ultrastructural analysis, cortical proximal tubules were found to have, in lean mice, elongated mitochondria, surrounded by a double membrane and with regular lamellar cristae. In contrast, in ob/ob mice, proximal tubular mitochondria were round with a few short cristae and with osmiophilic granules in the inner matrix. These findings, supported also by the observed increased in kidney 8-isoprostane, suggest that the mitochondria of the proximal convoluted tubules are significantly damaged in ob/ob mice, as a consequence of renal oxidative stress [36], [37].
To best define the mitochondrial morphological alteration in obesity, we observed a correlation between Mfn2 expression and the cytoarchitecture of mitochondria and their cristae in proximal tubules [5], [38]–[41]. Indeed, mitochondrial organization is commonly affected by a reduction in Mf2 in many organs including liver, heart and skeletal muscle, where this pleiotropic protein is associated with resistance to oxidative and mitochondrial DNA damage [41]–[43].
Recent studies indicate that, in healthy cells, Mfn2 is associated with the Bcl2 family proteins including Bax and Bak. This association in the cytosol allows maintenance of mitochondrial morphology during mitochondriogenesis [44]. During apoptosis, however, Bax translocates to the outer mitochondrial membrane (OMM), changes its conformation by becoming oligomeric, and alters its interaction with Mfn2 [45], [46]. Thus, here we analyzed the renal localization of Bax, and downstream pro-apoptotic markers including cytochrome c and caspases in ob/ob mice. While in control animals these signals were lacking, in the obese mice intense Bax and cytochrome c stainings were observed. It is known that Bax, upon oxidative damage, translocates to the OMM where it forms pore channels that allowed the flux of pro-apoptotic proteins from the inner mitochondrial membrane (IMM) to the cytosol, e.g. cytochrome c, Smac/Diablo, and the apoptosis inducing factor [47]. Moreover, in addition to opening the OMM pore, Bax remodels the cristae by interacting with specific proteins resident in the IMM [48], [49], in advance of the apoptosis cascade [50].
Regarding mitochondria-Mfn2-driven intrinsic apoptotic pathway, we detected the prevalence of caspase 3 in the proximal tubules and the caspase 9 signal in the distal tubules of ob/ob mice. To explain these observations, we noted that, among caspases, there are different roles with early initiator members, e.g. caspase 9, and later effector signals, such as caspase 3. When Bax in mitochondria triggers cytochrome c release, the apoptosome complex activates caspase 9 which further stimulates other downstream caspases, such as caspase 3. Moreover, the different distribution of the caspases as we observed may be due to the greater susceptibility of the proximal tubules to oxidative injury [51], [52] and resistance of the distal tubules to these processes which may adopt also an anaerobic metabolism [53].
Interestingly, melatonin restored creatinine levels, kidney oxidative stress and all damage linked to proximal tubules and especially mitochondria structure and Mfn2 expression. Melatonin is a potent antioxidant [17], [26], [54] and its actions on the mitochondrial alterations strongly indicates that the modification on these organelles were related to ROS production [55] as also occurs during aging [56]. These results are supported also by other studies showing similar morphological changes in muscle mitochondria of streptozotocin-treated mice and effects of N-acetylcysteine, another antioxidant [36]. In obese mice melatonin clearly attenuated mitochondria-driven apoptosis; this observation is in line with that of Radogna et al. [57], who showed a direct antagonistic effect of melatonin on Bax activation at the mitochondrial level during an apoptotic challenge. Moreover, we showed that melatonin strongly limited the caspase cascade expression in obese kidneys consistent with its anti-apoptotic activities observed by Molpeceres et al. (2007) [58].
Our study would stimulate the attention of scientists on the role of melatonin in the kidney of obese mice, specifically in terms of its ability to remodel mitochondrial structure and to restore Mfn2 in cortical proximal tubules. Via these crucial events, melatonin may negatively influence mitochondrial Bax sensitization and consequently cytochrome c efflux, thereby abrogating the caspase-dependent intrinsic mitochondrial apoptotic program (Figure 5).
Figure 5. Melatonin remodels mitochondria in obese mice kidneys.
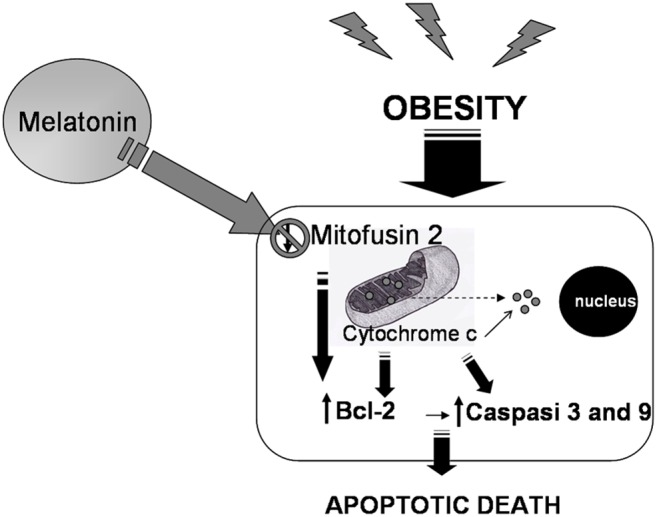
A schematic representation of the apoptotic pathway induced by obesity mediated alterations in the kidney conditioned by upstream mitofusin 2 and the inhibiting action of melatonin on the mitochondria-driven intrinsic apoptotic cascade.
Acknowledgments
Sincere thanks to Professor Raymond Coleman for his linguistic improvement, to Dr. Francesca Bonomini for her technical support on confocal microscopy and to Miss Stefania Castrezzati for her technical support on transmission electron microscopy.
Data Availability
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper.
Funding Statement
The authors have no support or funding to report.
References
- 1. Muntner P, He J, Chen J, Fonseca V, Whelton PK (2004) The metabolic syndrome and chronic kidney disease in US adults. Ann Int Med 140: 167–174. [DOI] [PubMed] [Google Scholar]
- 2. Wickman C, Kramer H (2013) Obesity and kidney Disease: Potential Mechanisms. Seminars Nephrol 33: 14–22. [DOI] [PubMed] [Google Scholar]
- 3. Kurella M, Lo JC, Chertow GM (2005) Metabolic syndrome and the risk for chronic kidney disease among nondiabetic adults. J Am Soc Nephrol 16: 2134–2140. [DOI] [PubMed] [Google Scholar]
- 4. Leoncini G, Viazzi F, Agabiti Rosei E, Ambrosioni E, Costa FV, et al. (2012) Metabolic sindrome and chronic kidney disease in high-risk Italian hypertensive patients: the I-DEMAND study. J Nephrol 25: 63–74. [DOI] [PubMed] [Google Scholar]
- 5. Tang WX, Wu WH, Zeng XX, Bo H, Huang SM (2012) Early protective effect of mitofusin 2 overexpression in STZ-induced diabetic rat kidney. Endocrine 41: 236–247. [DOI] [PubMed] [Google Scholar]
- 6. Tobar A, Ori Y, Benchetrit S, Milo G, Herman-Edelstein M, et al. (2013) Proximal tubular hypertrophy and enlarged glomerular and proximal tubular urinary space in obese subjects with proteinuria. PlosOne 8: e75547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kelly KJ, Wu P, Patterson CE, Temm C, Dominguez JH (2008) LOX-1 and inflammation: a new mechanism for renal injury in obesity and diabetes. Am J Physiol Renal Physiol 294: F1136–F1145. [DOI] [PubMed] [Google Scholar]
- 8. Zhai XY, Birn H, Jensen KB, Thomsen JS, Andreasen A, et al. (2003) Digital three dimensional reconstruction and ultrastructure of the mouse proximal tubule. J Am Soc Nephrol 14: 611–619. [DOI] [PubMed] [Google Scholar]
- 9. Westermann B (2010) Mitochondrial fusion and fission in cell life and death. Nat Rev Mol Cell Biol 11: 872–884. [DOI] [PubMed] [Google Scholar]
- 10. Galloway C, Yoon Y (2012) What comes first, misshape or dysfunction? The view from metabolic excess. J Gen Physiol 139: 455–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Suend, Norris K, Youle R (2008) Mitochondrial dynamics and apoptosis. Genes Dev 22: 1577–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. de Brito O, Scorrano L (2008) Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 456: 605–610. [DOI] [PubMed] [Google Scholar]
- 13. Cosson P, Marchetti A, Ravazzola M, Orci L (2012) Mitofusin 2–independent juxtaposition of endoplasmic reticulum and mitochondria: an ultrastructural study. PlosOne 7: e46293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sebastián D, Hernández-Alvarez MI, Segalés J, Sorianello E, Muñoz JP, et al. (2012) Mitofusin 2 (Mfn2) links mitochondrial and endoplasmic reticulum function with insulin signaling and is essential for normal glucose homeostasis. Proc Natl Acad Sci USA 109: 5523–5528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Acuña-Castroviejo D, Escames G, Venegas C, Díaz-Casado ME, Lima-Cabello E, et al.. (2014) Extrapineal melatonin: sources, regulation, and potential functions. Cell Mol Life Sci. [Epub ahead of print]. [DOI] [PMC free article] [PubMed]
- 16. Galano A, Tan DX, Reiter RJ (2011) Melatonin as a natural ally against oxidative stress: a physiochemical examination. J Pineal Res 51: 1–16. [DOI] [PubMed] [Google Scholar]
- 17. Galano A, Tan DX, Reiter RJ (2013) On the free radical scavenging activities of melatonin’s metabolites, AFMK and AMK. J Pineal Res 54: 245–257. [DOI] [PubMed] [Google Scholar]
- 18. Favero G, Lonati C, Giugno L, Castrezzati S, Rodella LF, et al. (2013) Obesity-related dysfunction of the aorta and prevention by melatonin in ob/ob mice. Acta Histochem 115: 783–788. [DOI] [PubMed] [Google Scholar]
- 19. Hu ZP, Fang XL, Fang N, Wang XB, Qian HY, et al. (2013) Melatonin ameliorates vascular endothelial dysfunction, inflammation and atherosclerosis by suppressing the TLR4/NF-kB system in high-fat-fed rabbits. J Pineal Res 55: 388–398. [DOI] [PubMed] [Google Scholar]
- 20. Solís-Muñoz P, Solís-Herruzo JA, Fernández-Moreira D, Gómez-Izquierdo E, García-Consuegra I, et al. (2011) Melatonin improves mitochondrial respiratory chain activity and liver morphology in ob/ob mice. J Pineal Res 51: 113–123. [DOI] [PubMed] [Google Scholar]
- 21. Mendes C, Lopes AM, do Amaral FG, Peliciari-Garcia RA, Turati Ade O, et al. (2013) Alterations of the aging animal to exercise: role of daily supplementation with melatonin. J Pineal Res 55: 229–239. [DOI] [PubMed] [Google Scholar]
- 22. Jiménez-Aranda A, Fernández-Vázquez G, Campos D, Tassi M, Velasco-Perez L, et al. (2013) Melatonin induces browning of inguinal white adipose tissue in Zucker diabetic fatty rats. J Pineal Res 55: 416–423. [DOI] [PubMed] [Google Scholar]
- 23. Reiter R, Paredes S, Korkmaz A, Jou MJ, Tan DX (2008) Melatonin combats molecular terrorism at the mitochondrial level. Interdiscipl Toxicol 1: 137–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Paradies G, Petrosillo G, Padies V, Reiter RJ, Ruggiero FM (2010) Melatonin, cardiolipin and mitochondria bioenergetics in health and diseases. J Pineal Res 48: 297–310. [DOI] [PubMed] [Google Scholar]
- 25. Hsiao CW, Peng TI, Peng AC, Reiter RJ, Tanaka M, et al. (2013) Long term Aβ exposure augments in mCa2+ independent in mROS-mediated depletion of cardiolipin for the shift of a lethal transient mitochondrial permeability transition to its permanent mode in NARP cybrids: a protective targeting of melatonin. J Pineal Res 54: 107–125. [DOI] [PubMed] [Google Scholar]
- 26. Tan DX, Manchester LC, Liu X, Rosales-Corral SA, Acuna-Castroviejo D, et al. (2013) Mitochondria and chloroplasts as the original sites of melatonin synthesis: a hypothesis related to melatonin’s primary function and evolution in eukaryotes. J Pineal Res 54: 127–138. [DOI] [PubMed] [Google Scholar]
- 27. Lanoix D, Lacasse A, Reiter R, Vaillancourt C (2013) Melatonin: the watchdog of villous trophoblast homeostasis against hypoxia/reoxygenation-induced oxidative stress and apoptosis. Mol Cell Endocrinol 381: 35–45. [DOI] [PubMed] [Google Scholar]
- 28. Cano P, Jiménez-Ortega V, Larrad A, Reyes Toso CF, Cardinali DP, et al. (2008) Effect of a high-fat diet on 24-h pattern of circulating levels of prolactin, luteinizing hormone, testosterone, corticosterone, thyroid-stimulating hormone and glucose, and pineal melatonin content, in rats. Endocrine 33: 118–25. [DOI] [PubMed] [Google Scholar]
- 29. Raskind MA, Burke BL, Crites NJ, Tapp AM, Rasmussen DD (2007) Olanzapine-induced weight gain and increased visceral adiposity is blocked by melatonin replacement therapy in rats. Neuropsychopharmacology 32: 284–288. [DOI] [PubMed] [Google Scholar]
- 30. Stacchiotti A, Ricci F, Rezzani R, Li Volti G, Borsani E, et al. (2006) Tubular stress proteins and nitric oxide synthase expression in rat kidney exposed to mercuric chloride and melatonin. J Histochem Cytochem 54: 1149–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Stacchiotti A, Li Volti G, Lavazza A, Rezzani R, Rodella LF (2009) Schisandrin B stimulates a cytoprotective response in rat liver exposed to mercuric chloride. Food Chem Toxicol 47: 2834–2840. [DOI] [PubMed] [Google Scholar]
- 32. Rezzani R, Favero G, Stacchiotti A, Rodella LF (2013) Endothelial and vascular smooth muscle cell dysfunction mediated by cyclophylin A and the atheroprotective effects of melatonin. Life Sci 92: 875–882. [DOI] [PubMed] [Google Scholar]
- 33. Rodella L, Favero G, Rossini C, Foglio E, Bonomini F, et al. (2013) Aging and vascular dysfunction: beneficial melatonin effects. Age (Dordr) 35: 103–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Garcia-Ruiz I, Rodriguez-Juan C, Diaz-Sanjuan T, Martínez MA, Muñoz-Yagüe T, et al. (2007) Effects of rosiglitazone on the liver histology and mitochondrial function in ob/ob mice. Hepatology 46: 414–423. [DOI] [PubMed] [Google Scholar]
- 35. Dong F, Zhang X, Yang X, Esberg LB, Yang H, et al. (2006) Impaired cardiac contractile function in ventricular myocytes from leptin-deficient ob/ob obese mice. J Endocrinol 188: 25–36. [DOI] [PubMed] [Google Scholar]
- 36. Bonnard C, Durand A, Peyrol S, Esberg LB, Yang H, et al. (2008) Mitochondrial dysfunction results from oxidative stress in the skeletal muscle of diet-induced insulin-resistant mice. J Clin Invest 118: 789–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Quigley JE, Elmarakby AA, Knight SF, Manhiani MM, Stepp DW, et al. (2009) Obesity induced renal oxidative stress contributes to renal injury in salt-sensitive hypertension. Clin Exp Pharmacol Physiol 36: 724–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zorzano A, Liesa M, Palacin M (2009) Role of mitochondrial dynamics proteins in the pathophysiology of obesity and type 2 diabetes. Int J Biochem Cell Biol 41: 1846–1854. [DOI] [PubMed] [Google Scholar]
- 39. Zhan M, Brooks C, Liu F, Sun L, Dong Z (2013) Mitochondrial dynamics: regulatory mechanisms and emerging role in renal pathophysiology. Kidney Int 83: 568–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cheng Z, Almeida F (2014) Mitochondrial alteration in type 2 diabetes and obesity. An epigenetic link. Cell Cycle 13: 890–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Liesa M, Palacin M, Zorzano A (2009) Mitochondrial dynamics in mammalian health and disease. Physiol Rev 89: 799–845. [DOI] [PubMed] [Google Scholar]
- 42. Chen H, Vermulst M, Wang Y, Chomyn A, Prolla TA, et al. (2010) Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell 141: 280–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Jheng H, Tsai P, Guo S, Kuo LH, Chang CS, et al. (2012) Mitochondrial fission contributes to mitochondrial dysfunction and insulin resistance in skeletal muscle. Mol Cell Biol 32: 309–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Karbowski M, Norris K, Cleland M, Jeong SY, Youle RJ (2006) Role of Bax and Bak in mitochondrial morphogenesis. Nature 443: 658–662. [DOI] [PubMed] [Google Scholar]
- 45. Brooks C, Wei Q, Feng L, Dong G, Tao Y, et al. (2007) Bak regulates mitochondrial morphology and pathology during apoptosis by interacting with mitofusins. Proc Natl Acad Sci 104: 11649–11654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gall J, Wang Z, Liesa M, Molina A, Havasi A, et al. (2013) Role of mitofusin 2 in the renal stress response. PlosOne 7: e31074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Brady N, Elmore S, Van Beek J, Krab K, Courtoy PJ, et al. (2004) Coordinated behavior of mitochondria in both space and time: a reactive oxygen species-activated wave of mitochondrial depolarization. Biophys J 87: 2022–2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yamaguchi R, Lartigue I, Perkins G, Scott RT, Dixit A, et al. (2008) Opa1-mediated cristae opening is Bax/Bak and BH3 dependent, required for apoptosis, and independent of Bak oligomerization. Mol Cell 31: 557–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ghibelli L, Diederich M (2010) Multistep and multitask Bax activation. Mitochondrion 10: 604–613. [DOI] [PubMed] [Google Scholar]
- 50. Ulivieri C (2010) Cell death: Insights into the ultrastructure of mitochondria. Tissue Cell 42: 339–347. [DOI] [PubMed] [Google Scholar]
- 51. D’Elia J, Roshan B, Maski M, Weinrauch LA (2009) Manifestation of renal disease in obesity:pathophysiology of obesity-related dysfunction of the kidney. Int J Nephrol Renovasc Dis 2: 39–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Brezniceanu M, Liu F, Wei C, Chénier I, Godin N, et al. (2008) Attenuation of interstitial fibrosis and tubular apoptosis in db/db transgenic mice overexpressing catalase in renal proximal tubular cells. Diabetes 57: 451–459. [DOI] [PubMed] [Google Scholar]
- 53. Hall A, Unwin R, Parker N, Duchen M (2009) Multiphoton imaging reveals differences in mitochondrial function between nephron segments. J Am Soc Nephrol 20: 1293–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tan DX, Chen LD, Poeggeler B (1993) Melatonin: a potent endogenous hydroxyl radical scavenger. Endocr J 1: 57–60. [Google Scholar]
- 55. Martin-Cano F, Camello-Almaraz C, Acuňa-Castroviejo D, Pozo MJ, Camello PJ (2014) Age-related changes in mitochondrial function of mouse colonic smooth muscle: beneficial effects of melatonin. J Pineal Res 56: 163–174. [DOI] [PubMed] [Google Scholar]
- 56. Runkel ED, Baumeister R, Schulze E (2014) Mitochondrial stress: Balancing friend and foe. Exp Gerontol 56C: 194–201. [DOI] [PubMed] [Google Scholar]
- 57. Radogna F, Cristofanon S, Paternoster L, D'Alessio M, De Nicola M, et al. (2008) Melatonin antagonizes the intrinsic pathway of apoptosis via mitochondrial targeting of Bcl-2. J Pineal Res 44: 316–325. [DOI] [PubMed] [Google Scholar]
- 58. Molpeceres V, Mauriz J, Garcia-Mediavilla M, González P, Barrio JP, et al. (2007) Melatonin is able to reduce the apoptotic liver changes induced by aging via inhibition of the intrinsec pathway of apoptosis. J Gerontol 62: 687–695. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper.