

Significance
The major portion of hereditary breast cancer still remains unexplained, and many susceptibility loci are yet to be found. Exome sequencing of 24 high-risk familial BRCA1/2-negative breast cancer patients and further genotyping of a large sample set of breast/ovarian cancer cases and controls was used to discover previously unidentified susceptibility alleles and genes. A significant association of a FANCM nonsense mutation with breast cancer, especially triple-negative breast cancer, identifies FANCM as a breast cancer susceptibility gene. Identification of such risk alleles is expected to improve cancer risk assessment for breast cancer patients and families, and may lead to improvements in the prevention, early diagnosis, and treatment of cancer.
Keywords: breast cancer, DNA repair, FANCM, exome sequencing, triple-negative breast cancer
Abstract
Inherited predisposition to breast cancer is known to be caused by loss-of-function mutations in BRCA1, BRCA2, PALB2, CHEK2, and other genes involved in DNA repair. However, most families severely affected by breast cancer do not harbor mutations in any of these genes. In Finland, founder mutations have been observed in each of these genes, suggesting that the Finnish population may be an excellent resource for the identification of other such genes. To this end, we carried out exome sequencing of constitutional genomic DNA from 24 breast cancer patients from 11 Finnish breast cancer families. From all rare damaging variants, 22 variants in 21 DNA repair genes were genotyped in 3,166 breast cancer patients, 569 ovarian cancer patients, and 2,090 controls, all from the Helsinki or Tampere regions of Finland. In Fanconi anemia complementation gene M (FANCM), nonsense mutation c.5101C>T (p.Q1701X) was significantly more frequent among breast cancer patients than among controls [odds ratio (OR) = 1.86, 95% CI = 1.26–2.75; P = 0.0018], with particular enrichment among patients with triple-negative breast cancer (TNBC; OR = 3.56, 95% CI = 1.81–6.98, P = 0.0002). In the Helsinki and Tampere regions, respectively, carrier frequencies of FANCM p.Q1701X were 2.9% and 4.0% of breast cancer patients, 5.6% and 6.6% of TNBC patients, 2.2% of ovarian cancer patients (from Helsinki), and 1.4% and 2.5% of controls. These findings identify FANCM as a breast cancer susceptibility gene, mutations in which confer a particularly strong predisposition for TNBC.
Breast cancer is the most common cancer affecting women worldwide. It is also the principal cause of death from cancer among women globally, accounting for 14% of all cancer deaths (1). The etiology of breast cancer is multifactorial, and the risk depends on various factors like age, family history, and reproductive, hormonal, or dietary factors. The majority of breast cancers are sporadic, but approximately 15% of cases show familial aggregation (2, 3). Since the identification of the first breast and ovarian cancer susceptibility genes breast cancer 1 and 2 (BRCA1 and BRCA2, respectively) by linkage analysis and positional cloning, several breast cancer susceptibility genes and alleles with different levels of risk and prevalence in the population have been recognized. BRCA1 and BRCA2 mutation carriers have more than 10-fold increased risk of breast cancer compared with women in the general population, and mutations in TP53, PTEN, STK11, and CDH1 have also been associated with a high lifetime risk of breast cancer in the context of rare inherited cancer syndromes (4). In addition, rare variants in genes such as checkpoint kinase 2 (CHEK2), ataxia telangiectasia mutated (ATM), and BRCA1 interacting helicase BRIP1, that confer a two- to fourfold increased risk, and in partner and localizer of BRCA2 (PALB2), with even higher risk estimates, have been found with candidate gene approaches (5, 6), and an increasing number of common low-risk loci with modest odds ratios (ORs; as much as 1.26-fold increased risk for heterozygous carriers) have been identified by genome-wide association studies (7).
However, the major portion of hereditary breast cancer still remains unexplained, and many susceptibility loci are yet to be found. Exome sequencing combined with genotyping of the identified variants in case-control analysis is an effective method to recognize novel risk alleles, based on the assumption that disease-causing variants are rare and often accumulate in the protein-coding areas of the genome (8–10).
Since the discovery that proteins encoded by the BRCA1 and BRCA2 breast/ovarian cancer susceptibility genes are directly involved in homologous recombination repair of DNA double-strand breaks, it has been evident that other genes involved in DNA repair are attractive breast cancer susceptibility candidates (4). Biallelic mutations in ATM gene cause rare ataxia telangiectasia disease and are associated with an increased risk for breast cancer as a result of improper DNA damage response (11). Fanconi anemia (FA) is a rare genetic disorder caused by biallelic mutations in FA genes that also participate in DNA repair. At least 15 FA genes have been identified (12). Patients with heterozygous mutations in certain FA genes have an elevated risk for various cancers, and monoallelic mutations in at least four of these genes [BRCA2, BRIP1, PALB2, and RAD51 paralog C (RAD51C)] are associated with an increased risk of breast or ovarian cancer (12, 13). Recurrent founder mutations in several cancer susceptibility genes, including the BRCA2, PALB2, and RAD51C FA genes, have been identified in the Finnish population (14–16). The PALB2 and RAD51C founder mutations have been detected at 2% frequency in Finnish breast or ovarian cancer families (15–17), whereas, in other populations, mutations in these genes are rare and often unique for each family. Founder effects in the isolated populations such as Finland or Iceland may enrich certain mutations and thus explain a significant proportion of all mutations in certain genes (18, 19). This provides an advantage in the search for novel susceptibility genes and alleles.
In this study, we used exome sequencing to uncover previously unidentified recurrent breast or ovarian cancer predisposing variants in the Finnish population with a focus on DNA repair genes. Selected variants were further genotyped in a large case-control sample set. Our investigation revealed an association of a nonsense mutation (rs147021911) in an FA complementation gene, FANCM, with breast cancer, especially with triple-negative (TN) breast cancer (TNBC).
Results
Exome Sequencing and Identification of Candidate Variants.
Exome sequencing was performed on germ-line DNA samples of 24 BRCA1/2-negative patients from 11 breast cancer families. All families had at least three breast or ovarian cancer patients among first- or second-degree relatives. We identified a total of 80,918 variants in 80,867 nonsynonymous positions, with a mean read coverage of 101. More than 91% of the captured exome target region was covered by at least 10 reads in all samples.
Several filtering steps were applied to the exome sequencing data to prioritize variants for further validation and follow-up studies (Fig. S1). Variants with mean coverage <15 and common variants (minor allele frequency ≥ 1%) in the 1000 Genomes Project (20) or Exome Variant Server data [National Heart, Lung, and Blood Institute (NHLBI) Grand Opportunity (GO) Exome Sequencing Project; evs.gs.washington.edu/EVS/; January 2013] were excluded. Population-matched exome-sequenced controls (n = 144) allowed exclusion of variants and indels based on the frequency in the Finnish population. Annovar (21) was used for annotation of the variants. Protein truncating alterations including nonsense variations, frame-shift insertions/deletions, and splice-site variants were manually examined with Integrative Genomics Viewer (22) and RikuRator, an in-house visualization tool (created by Riku Katainen, University of Helsinki) to remove possible artifacts. Only variants predicted to be pathogenic were included in the study.
Given that the majority of the known breast cancer predisposition genes are involved in DNA double-strand break repair, we concentrated on variants identified in genes defined as DNA repair genes in the Gene Ontology project (via AmiGO browser) (23). A total of 22 variants were identified in 21 DNA repair genes, including 20 single-nucleotide variants (SNVs) and two indels (endonuclease NEIL1 c.314dupC and protein ligase and helicase SHPRH c.3577_3580delCTTA; Table S1). Both indels were confirmed by Sanger sequencing. Three variants showed some evidence of cosegregation with cancer in families. Specifically, an SHPRH deletion, a DNA glycosylase MPG splicing mutation, and an ATPase RUVBL1 missense variant were detected in both exome-sequenced members of the respective families. In addition, a single PAX interacting protein 1 (PAXIP1) missense variant was detected in two unrelated cases, raising the possibility that the variant is present at an increased frequency in Finnish breast cancer families. The rest of the variants were observed in only one case.
Genotyping Population-Matched Cases and Controls.
Associations between the selected variants and breast and ovarian cancer were evaluated in further studies. In phase I, the 22 variants were genotyped in a pilot set of 524 familial BRCA1/2-negative breast cancer patients from the Helsinki region of Finland to determine whether the variants were recurrent in Finnish families and thus relevant for genotyping in a larger set of samples. All patients had strong family history of breast cancer, with at least three breast or ovarian cancer patients in the family among first- or second-degree relatives (411 families with breast cancer only and 113 families with both breast and ovarian cancer). Genotyping of the SNVs was performed with Sequenom MassARRAY system using iPLEX Gold assays, genotyping of PAXIP1 c.803C>T and NEIL1 c.314dupC was performed with TaqMan real-time PCR, and genotyping of SHPRH c.3577_3580delCTTA was conducted by Sanger sequencing. After phase I, 13 SNVs and the SHPRH c.3577_3580delCTTA deletion were excluded from further analysis as no additional mutation carriers were found (Table S1). The Fanconi anemia complementation group A (FANCA) c.1682C>T missense variant was discarded because the iPLEX genotyping assay did not produce a reliable result.
In phase II, BRCA1 associated RING domain 1 (BARD1) c.2282G>A and PAXIP1 c.803C>T missense variants and the NEIL1 c.314dupC variant, which were not included in the iPLEX assay, were genotyped with TaqMan real-time PCR in population matched healthy female controls (n = 552) to define the mutation frequency in the general Finnish population. The frequencies of the missense variants BARD1 c.2282G>A (1.8% for controls, 2.2% for cases) and PAXIP1 c.803C>T (0.5% for controls, 1.2% for cases) were similar to the phase I familial cases or too rare for final statistical assessment in our datasets, and thus these variants were excluded from further analysis (Table S2). The NEIL1 c.314dupC mutation was present in one control and was selected for further genotyping in phase III. The remaining five SNVs were genotyped simultaneously in phase II and phase III samples by Sequenom MassARRAY.
In phase III, five SNVs were genotyped by Sequenom MassARRAY on an additional 233 familial BRCA1/2-negative breast cancer patients, 1,730 unselected breast cancer cases from the Helsinki region, and 679 unselected breast cancer patients (including 257 familial cases) from the Tampere region of southwestern Finland. For population controls, a total of 1,274 healthy females from Helsinki and 816 from the Tampere region were genotyped. The SNVs were also genotyped in a series of unselected ovarian cancer patients (n = 569) from the Helsinki region. The success rates for the assays varied between 97.9% and 100% after excluding samples with no acceptable results or low success rates. NEIL1 c.314dupC was genotyped with TaqMan real-time PCR in the familial and unselected breast cancer cases and population controls.
FANCA c.4228T>G was not observed in any of the phase III genotyped patients, RUVBL1 c.950G>A was detected in one additional breast and one ovarian cancer sample, and MPG c.40–1G>T, SLX4 structure-specific endonuclease subunit (SLX4) c.2484G>C, FANCM c.5101C>T, and NEIL1 c.314dupC were detected in 10 or more additional samples (Table S2).
FANCM c.5101C>T Associates with Breast Cancer Risk.
After combining results from the three phases of genotyping in two independent series of breast cancer cases and controls, a significant association with breast cancer was found for FANCM c.5101C>T nonsense mutation (14:45658326C>T, rs147021911, p.Q1701X). Approximately twofold increased mutation frequency was found among breast cancer cases (2.9%) compared with controls (1.4%) in the Helsinki sample set (OR = 2.06, 95% CI = 1.22–3.47; P = 0.006; Table 1). To identify the patient characteristics associated with the highest risk of disease, we divided the genotyped patients into subgroups according to family history of breast or ovarian cancer, estrogen receptor (ER) status, and TN subtype, i.e., ER-, progesterone receptor-, and HER2-negative tumors. The association was consistent among familial cases (OR = 2.21, 95% CI = 1.24–3.94; P = 0.006) and among unselected cases (OR = 2.01, 95% CI = 1.16–3.47; P = 0.011), with 3.2% carrier frequency among patients with strong family history of breast cancer and 2.9% among patients with only one affected first-degree relative. The frequencies were similar in breast cancer-only families (3.2%) and families with both breast and ovarian cancer (3.5%). Among the unselected ovarian cancer patients, a consistent but nonsignificant result was seen, with 2.2% mutation carrier frequency (OR = 1.56, 95% CI = 0.75–3.26; P = 0.235). When studied by subtype of breast cancer, the risk was somewhat higher in the ER-negative subgroup (OR = 2.38, 95% CI = 1.17–4.83; P = 0.013) than in the ER-positive subgroup (OR = 1.83, 95% CI = 1.05–3.17; P = 0.029), and the highest risk and most significant association was found with TN breast cancer (OR = 4.13, 95% CI = 1.76–9.67; P = 0.0004). Among the 143 TN cases in the Helsinki region, 24 had family history of breast cancer and two of them were mutation carriers (8.3%, OR = 6.33, 95% CI = 1.38–28.95; P = 0.047 vs. controls). Among the unselected TN patients, FANCM c.5101C>T associated with breast cancer (P = 0.0001; OR = 4.92, 95% CI = 2.01–12.07).
Table 1.
Frequency of the FANCM c.5101C>T nonsense mutation among unselected and familial breast cancer patients, unselected ovarian cancer patients, and population controls
| Study cohort | N | CC (%) | CT (%) | OR | 95% CI | P value |
| Helsinki | ||||||
| Controls | 1,271 | 1,253 (98.6) | 18 (1.4) | — | — | — |
| All BC | 2,405 | 2,336 (97.1) | 69 (2.9) | 2.06 | 1.22–3.47 | 0.0059 |
| Familial BC | 1,074 | 1,041 (96.9) | 33 (3.1) | 2.21 | 1.24–3.94 | 0.0061 |
| ≥3 affected | 524 | 507 (96.8) | 17 (3.2) | 2.33 | 1.19–4.57 | 0.0109 |
| BC only | 410 | 397 (96.8) | 13 (3.2) | 2.28 | 1.11–4.69 | 0.0217 |
| BC + OC | 114 | 110 (96.5) | 4 (3.5) | 2.53 | 0.84–7.61 | 0.1003 |
| 2 affected | 550 | 534 (97.1) | 16 (2.9) | 2.09 | 1.06–4.12 | 0.0307 |
| Unselected BC | 1,713 | 1,665 (97.2) | 48 (2.8) | 2.01 | 1.16–3.47 | 0.0109 |
| ER+ | 1,799 | 1,753 (97.4) | 46 (2.6) | 1.83 | 1.05–3.17 | 0.0293 |
| ER− | 423 | 409 (96.7) | 14 (3.3) | 2.38 | 1.17–4.83 | 0.0132 |
| TN | 143 | 135 (94.4) | 8 (5.6) | 4.13 | 1.76–9.67 | 0.0004 |
| Unselected OC | 548 | 536 (97.8) | 12 (2.2) | 1.56 | 0.75–3.26 | 0.2346 |
| Tampere | ||||||
| Controls | 809 | 789 (97.5) | 20 (2.5) | — | — | — |
| All BC | 674 | 647 (96.0) | 27 (4.0) | 1.65 | 0.91–2.96 | 0.0932 |
| Familial BC | 253 | 241 (95.3) | 12 (4.7) | 1.96 | 0.95–4.08 | 0.0651 |
| ER+ | 509 | 491 (96.5) | 18 (3.5) | 1.45 | 0.76–2.76 | 0.2610 |
| ER− | 124 | 117 (94.4) | 7 (5.6) | 2.36 | 0.98–5.70 | 0.0497 |
| TN | 61 | 57 (93.4) | 4 (6.6) | 2.77 | 0.92–8.37 | 0.0806 |
| Meta-analysis | ||||||
| All BC | 3,079 | — | — | 1.86 | 1.26–2.75 | 0.0018 |
| Familial | 1,327 | — | — | 2.11 | 1.34–3.32 | 0.0012 |
| ER+ | 2,308 | — | — | 1.66 | 1.09–2.52 | 0.0182 |
| ER− | 547 | — | — | 2.37 | 1.37–4.12 | 0.0021 |
| TN | 204 | — | — | 3.56 | 1.81–6.98 | 0.0002 |
BC, breast cancer; OC, ovarian cancer; ≥3 affected, families with ≥3 BC or OC cases among first- or second-degree relatives; 2 affected, families with two first-degree relatives with BC or OC. Two-sided P values are calculated with Pearson χ2 or Fisher exact test if the expected number of cell count is ≤5.
Consistent results were observed in the independent Tampere breast cancer case-control series (Table 1), with the highest OR among the TN cases (OR = 2.77, 95% CI = 0.92–8.37; P = 0.0806). Meta-analyses of the two series, combining the estimates from the Helsinki and Tampere datasets, confirmed the association among all of the genotyped breast cancer cases (N = 3,079; OR = 1.86, 95% CI = 1.26–2.75; P = 0.0018), among familial cases (OR = 2.11, 95% CI = 1.34–3.32; P = 0.0012), and the highest risk in the TN subgroup (OR = 3.56, 95% CI = 1.81–6.98; P = 0.0002). P < 0.0023 was considered significant after Bonferroni correction for the number of variants tested, and the P values remained significant after the multiple testing correction. In the meta-analysis, there was borderline-significant heterogeneity in the risks between ER-positive and TN breast cancer (Phet = 0.059), suggesting an increased risk especially for TNBC.
Altogether, 96 FANCM c.5101C>T mutation carriers were found among the breast cancer cases. In the Helsinki series, 17 carriers had a strong family history of breast cancer, 16 had one first-degree relative affected with breast or ovarian cancer, and 36 were sporadic breast cancer patients who did not fulfill the family history criteria. From the Tampere region, 12 familial and 15 sporadic cases carried the FANCM c.5101C>T mutation. The mean age of the FANCM c.5101C>T mutation carriers at breast cancer diagnosis was 55.2 y, whereas the mean age of the noncarriers was 56.4 y (P = 0.416). In both the Helsinki and Tampere series, the mutation was also detected in 38 female population controls.
Among the 548 unselected ovarian cancer cases, 12 carried the mutation. Age at ovarian cancer diagnosis was available for 11 mutation carriers and 525 noncarriers. The mean age was 53.7 y for the carriers and 55.4 y for the noncarriers (P = 0.569). Six of the tumors were of serous histology, two were mucinous, two were other subtypes, and histology was unknown for two.
To study the FANCM nonsense mutation in another strong founder population, we genotyped 965 unselected breast cancer patients, including 92 familial cases from Iceland, but no FANCM c.5101C>T mutations were detected in this sample set, suggesting the mutation is absent or extremely rare in Iceland.
To study the familial segregation of FANCM c.5101C>T, 45 individuals from eight mutation carrier families were genotyped. Among 11 female relatives with breast cancer, five carried the FANCM c.5101C>T mutation. Notably, all of these mutation carriers were first-degree relatives of the index patients, whereas all but one of the breast cancer cases with the WT genotype were more distant relatives. Among 16 healthy female relatives, seven carried the mutation (aged between 33 and 80 y). Most of the families showed incomplete segregation of the mutation, but, in one of the carrier families, all three sisters affected with breast cancer were mutation carriers (Fig. 1). Among eight genotyped relatives affected with other types of cancer, one man diagnosed with prostate cancer at the age of 48 y and one woman affected with an undefined cancer were mutation-positive. Among the relatives without available DNA samples, several other cancer types in addition to breast and ovarian cancer were also present, including prostate, pancreatic, skin, lung, colorectal, bone marrow, liver, and kidney cancer.
Fig. 1.
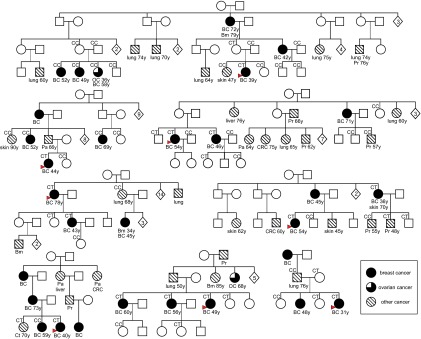
Pedigrees of eight FANCM c.5101C>T families. The FANCM c.5101C>T genotype is denoted above genotyped individuals. Cancer type and age at diagnosis are indicated below cancer patients. BC, breast cancer; Bm, bone marrow; Ct, connective tissue; Gb, glioblastoma; Mel, melanoma; OC, ovarian cancer; Pa, pancreatic cancer; Pr, prostate cancer.
To investigate whether the FANCM c.5101C>T causes nonsense-mediated mRNA decay, we performed allele-specific quantitative real time-PCR for WT and c.5101C>T mutant FANCM alleles. Equal proportions of the WT and mutant allele were detected in RNA from a c.5101C>T carrier, whereas only the WT allele was detected in controls (Fig. S2). These results indicate that the c.5101C>T allele was not subject to nonsense-mediated mRNA decay.
The other studied DNA repair gene variants did not show a statistically significant difference in frequencies between cases and controls (SI Results and Discussion and Table S2), although FANCA c.4228T>G was detected in two breast cancer cases and RUVBL1c.950G>A in three breast cancer cases and one ovarian cancer case, but not in any of the controls.
Discussion
We used exome sequencing of breast cancer families in the genetically homogenous Finnish population to identify recurrent alleles associated with cancer risk in previously unidentified breast and ovarian cancer susceptibility genes. By concentrating on DNA repair genes, 22 variants from our exome sequencing data were selected for further evaluation in large datasets of familial and unselected breast cancer patients and population controls from two independent case-control series in different regions of Finland as well as in a series of unselected ovarian cancer cases.
We found a c.5101C>T (rs147021911) variant encoding a premature FANCM stop codon (p.Q1701X) in 96 Finnish breast cancer patients and 12 Finnish ovarian cancer patients. Although FANCM c.5101C>T is very rare in the 1000 Genomes Project European population (minor allele frequency = 0.005), it is observed at a higher frequency in the Finnish population, and was here found at a twofold increased frequency among familial and unselected breast cancer cases from two studies relative to population controls. In the FANCM c.5101C>T mutation carrier families, additional genotyping of the available affected relatives did not demonstrate distinct segregation of the mutation. Instead, the mutation had incomplete segregation with the disease, consistent with FANCM c.5101C>T being a moderate-risk allele, which alone may not explain the clustering of the disease in the pedigrees in which, presumably, other unknown predisposition alleles may also segregate.
When considering breast tumor histopathology, FANCM c.5101C>T was observed more frequently in ER-negative than ER-positive breast cancer patients, and the strongest association was observed in TNBC patients, with ORs ranging from 2.77 to 4.13. This finding is consistent with enrichment for germ-line variants in DNA repair genes among TNBC cases. Specifically, mutations in the breast cancer predisposing genes BRCA1 and BRCA2 have been observed in as many as 15% of TNBC cases, and 70% of BRCA1-associated breast tumors exhibit TN characteristics. In addition, three known TNBC-specific low-risk loci (TERT, MERIT40, and MDM4) contain genes that participate in DNA repair and maintenance of genome stability (24, 25). Our findings suggest that FANCM is a breast cancer susceptibility gene, mutations in which confer a particularly strong predisposition for TNBC. The improved understanding of the etiology of the TN subtype of breast cancer may lead to identification of new targeted treatments or development of therapeutic agents for this form of breast cancer. Conversely, in the unselected ovarian cancer series, unambiguous association was not detected (OR = 1.56, 95% CI = 0.75–3.26; P = 0.235), although the OR and 95% CIs were consistent with an increased ovarian cancer risk. A larger sample set is needed for further investigation of the association of FANCM c.5101C>T mutation with ovarian cancer risk and to uncover potential associations with specific ovarian cancer subtypes.
We also genotyped the FANCM c.5101C>T nonsense mutation in 965 breast cancer patients from Iceland. The mutation was not found in this sample set and therefore may be very rare in the genetically isolated Icelandic population. However, other pathogenic, possibly population-specific FANCM mutations may confer an increased risk of breast cancer in this population. Furthermore, other cancer-predisposing FANCM mutations may also exist in the Finnish population, and further studies to evaluate the whole contribution of FANCM are warranted.
In the Finnish population, the CHEK2 moderate-risk allele c.1100delC is observed at a similar population-control frequency as the FANCM mutation in the Helsinki dataset, but with a higher frequency among familial breast cancer patients (5.5%), with approximately fourfold elevated risk (26). This is comparable to the FANCM mutation among the TN patients and specifically the TNBC patients with family history of breast cancer. CHEK2 is thought to be a risk modifier gene with multiplicative effects with other susceptibility alleles in breast cancer families. Thus, the breast cancer risk estimates for c.1100delC mutation carriers are influenced by family history. The lifetime risk of breast cancer for carriers of CHEK2 truncating mutations is estimated to range from 20% for a woman with no affected relative to 44% for a woman with a first- and a second-degree relative affected (27). Interestingly, the CHEK2 missense-mutation I157T is a lower-risk allele with approximately 1.5-fold elevated cancer risk among unselected and familial cases, whereas it associates specifically with approximately fourfold increased risk of lobular breast cancer (28).
FANCM is the most conserved protein within the FA pathway (29). The main function of this pathway is to activate a DNA damage response when encountering stalled replication forks in response to DNA damage. FANCM has a translocase and endonuclease activity, and its functions are essential for promoting branch migration of Holliday junctions and DNA repair structures at replication forks. It can suppress spontaneous sister chromatid exchanges and maintain chromosomal stability, suggesting a tumor-suppressor role in cells (29–31). The c.5101C>T variant results in truncation of the FANCM protein and would be predicted to cause nonsense-mediated mRNA decay. However, no nonsense-mediated mRNA decay of the c.5101C>T allele was detected, suggesting that the continued expression of the mutant allele and almost full-length protein levels may in part account for the moderate influence of the variant on risk. The FANCM c.5101C>T mutation is located at exon 20 causing a premature stop codon at Gln1701, which causes the loss of two protein domains (ERCC4 and RuvA domain 2-like) in the C terminus (Fig. 2). This may affect the DNA binding abilities of the FANCM protein during stalled replication fork processing and monoubiquitination of the repair complex proteins (32). The ERRC4 domain can contribute to DNA binding and it also has nuclease activity; however a full-length FANCM protein has stronger DNA binding abilities than its C-terminal domain. The other C-terminal domain RuvA domain 2-like including HtH motif strengthens the ability of FANCM to bind especially to Holliday junctions and participate in DNA repair. The two translocase domains in FANCM protein are located in the N terminus. It is likely that all these protein domains in FANCM act sequentially in replication fork processing (31, 33, 34) (Interpro; www.ebi.ac.uk/interpro/protein/Q8IYD8; July 2014).
Fig. 2.
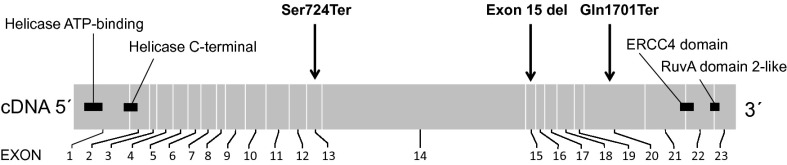
Drawing of FANCM indicating protein domains, the position of the c.5101C>T nonsense mutation, and positions of the mutations found from a single FA patient carrying FANCM and FANCA defects. Mutations and protein domains are mapped to the corresponding exons of FANCM.
Our findings are consistent with previous studies showing that fancm-deficient mice have increased cancer incidence (33). In addition, homozygous missense mutations in FANCM (c.5164C>T, p.P1722S) have been found in breast tumors (35), and four different FANCM SNPs have been associated with osteosarcoma risk (36). Also, in colorectal cancer (CRC) patients, an FANCM nonsense mutation (c.5791C>T, p.Arg1931Ter) has been identified (37), but further studies are needed to clarify the potential role of FANCM in CRC susceptibility. However, only one FA patient has been found to carry truncating FANCM mutation and this individual also carries biallelic mutations in the FANCA gene (33, 38). Furthermore, homozygous carriers of FANCM loss-of-function mutations c.5101C>T and c.5791C>T observed in the Finnish population do not show symptoms of FA (39). Thus, FANCM may have tumor suppressor activity, but, despite its role in the FA pathway, it may not contribute to FA.
Conclusions
In summary, the FANCM c.5101C>T nonsense mutation associates with breast cancer risk in the Finnish population. This is consistent with recent murine studies that indicate FANCM as a tumor-suppressor gene and increased cancer incidence in fancm-deficient mice. The highest mutation frequency and strongest association with breast cancer risk was observed among TNBC patients, further implicating DNA repair in the etiology of this aggressive form of breast cancer. Further studies in different populations, especially in familial cases, are essential for more precise estimation of breast cancer risks associated with this mutation. The discovery of variants such as FANCM c.5101C>T is essential for individualized breast cancer risk assessment and early diagnosis for breast cancer families.
Materials and Methods
Ethics Approval.
This study was approved by Helsinki University Central Hospital Ethics Committee (reference no. 272/13/03/03/2012), the Icelandic Data Protection Authority (reference no. 20010505239 and later amendments), and the National Bioethic Committee (reference no. 99/051-B1/B2 and later amendments).
Samples.
We selected 24 BRCA1/2-negative breast cancer patients from 11 Finnish breast cancer families (minimum of three breast or ovarian cancers in first- or second-degree relatives) for exome sequencing. We included two cases from nine families and three from two families. Selected exome variants were genotyped in additional breast and ovarian cancer patients and population controls in phases I–III, totaling 3,166 breast cancer cases and 2,090 healthy female population controls. Phase I consisted of 524 BRCA1/2-negative familial breast cancer patients from the Helsinki region of Finland. Phase II consisted of 552 population controls. Phase III consisted of 1,730 unselected breast cancer patients, 233 additional familial BRCA1/2-negative patients, and a total of 1,274 population controls (including the 552 controls from phase II) from the Helsinki region, as well as 679 unselected breast cancer patients (including 257 familial cases) and 816 population controls from the Tampere region of Finland. In addition, the selected substitutions were genotyped in an unselected series of ovarian cancer patients (n = 569) from the Helsinki region, and the identified FANCM nonsense mutation was also genotyped in an unselected series of Icelandic breast cancer patients, including 92 familial and 873 sporadic cases. The studied cohorts are described in SI Materials and Methods.
Exome Sequencing.
Exome sequencing and variant calling was performed at Genome Quebec Innovation Centre. The Agilent SureSelect Human All Exon 50-Mb kit was used to capture the exomic regions from 3 μg of genomic DNA. The sequencing was performed on Illumina HiSeq2000 sequencer with 100-bp paired-end reads. Details of the exome data analysis are given in SI Materials and Methods.
Variant Filtering.
We used several filtering criteria to select variants from exome sequencing data for further genotyping. Variants with mean read coverage less than 15 were excluded. Filtering of common variants (≥1% frequency) was performed using the 1000 Genomes Project (20), Exome Variant Server (NHLBI GO Exome Sequencing Project; evs.gs.washington.edu/EVS/; January 2013), and exome data from 144 Finnish noncancer control samples. The candidate genes and variants were annotated by using AmiGO (23) and Annovar (21), and genes participating in DNA repair were selected for further analysis. Only variants predicted to change protein sequence (frame-shift deletions and insertions, splicing alterations, and missense and nonsense SNVs) were considered. Of the missense variants, only those predicted to be pathogenic were included. After these filtering steps, we manually examined the remaining variants to visualize and verify their structure and position, and also to remove possible sequencing artifacts, with the use of the analysis and visualization program Interactive Genomics Viewer (22) and in-house visualization tool Rikurator (Riku Katainen, University of Helsinki). Indels that passed the filtering steps were confirmed by Sanger sequencing before genotyping (SI Materials and Methods).
Genotyping.
Genotyping of the selected 22 variants from 21 DNA repair genes was performed with Sequenom MassARRAY system, TaqMan real-time PCR, or Sanger sequencing (Table S1) as described in SI Materials and Methods. Variants detected in subsequent samples in phase I were selected for further genotyping. Those variants passing phase I that could be included on the Sequenom MassARRAY were simultaneously genotyped in phase II and phase III samples. Variants that were not included on the array because of technical reasons (BARD1 c.2282G>A, PAXIP1 c.803C>T, and NEIL1 c.314dupC) were first genotyped with TaqMan real-time PCR in population controls in phase II, and those that were detected frequently in controls were excluded from phase III. The phase III genotyping for the remaining variant (NEIL1 c.314dupC) was also executed with TaqMan real-time PCR. Additional relatives from the FANCM c.5101C>T mutation carrier families were genotyped by Sanger sequencing (Table S3). In addition, the FANCM mutation was also genotyped in 965 Icelandic breast cancer patients with TaqMan real-time PCR.
Nonsense-Mediated mRNA Decay.
Quantitative allele-specific FANCM RT-PCR was performed with RNA extracted from lymphoblastoid cells from a heterozygous carrier of the c.5101C>T variant and six noncarriers (SI Materials and Methods).
Statistical Analyses.
Two-sided P values were calculated by Pearson χ2 test or Fisher exact test if the expected number of cell count was five or less. For meta-analysis, we combined the estimates from Helsinki and Tampere with a fixed effects model by using the inverse variance-weighted method. Meta-analysis was performed by using R.3.0.1 environment (www.r-project.org/). For multiple testing correction, the Bonferroni method was used, and heterogeneity between ER-positive and TN subgroups was determined by two-sample z-test. The mean age of FANCM c.5101C>T mutation carriers and noncarriers was compared with Student paired t test.
Supplementary Material
Acknowledgments
We thank research nurses Irja Erkkilä and Virpi Palola and geneticist Salla Ranta for their help with collecting patient data and samples, the Finnish and the Icelandic Cancer Registries for cancer diagnostic data, Dr. Maija Wessman [Institute for Molecular Medicine Finland (FIMM) and Folkhälsan Research Center] for providing Finnish exome sequencing data, Riku Katainen from Lauri Aaltonen’s group at the University of Helsinki for RikuRator software, Evdis Gudmundsdottir for help in analyzing the FANCM c.5101C>T genotype data in Iceland, the staff at the Technology Centre of FIMM for SNV marker genotyping, and the staff at Genome Quebec Innovation Center for exome sequencing services. This study was supported by the Helsinki University Central Hospital Research Fund, Academy of Finland Grant 266528, the Sigrid Juselius Foundation, Nordic Cancer Union, Finnish Cancer Society, Alfred Kordelin Foundation, Orion-Farmos Research Foundation, Emil Aaltonen Foundation, and the Maud Kuistila Foundation. This work was also supported by the Breast Cancer Research Foundation (F.J.C.), National Institutes of Health Grants CA176785 and CA116167 (to F.J.C.), National Cancer Institute Specialized Program of Research Excellence Award in Breast Cancer (to Mayo Clinic) P50 CA116201 (to F.J.C.), Nordic Cancer Union (R.B.B. and E.S.F.), Research Fund of Landspitali University Hospital (R.B.B. and E.S.F.), and Gongum saman Research Fund (R.B.B. and E.S.F.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1407909111/-/DCSupplemental.
References
- 1.Jemal A, et al. Global cancer statistics. CA Cancer J Clin. 2011;61(2):69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Doyle C, et al. 2006 Nutrition, Physical Activity and Cancer Survivorship Advisory Committee American Cancer Society Nutrition and physical activity during and after cancer treatment: An American Cancer Society guide for informed choices. CA Cancer J Clin. 2006;56(6):323–353. doi: 10.3322/canjclin.56.6.323. [DOI] [PubMed] [Google Scholar]
- 3.Ferlay J, et al. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127(12):2893–2917. doi: 10.1002/ijc.25516. [DOI] [PubMed] [Google Scholar]
- 4.Foulkes WD. Inherited susceptibility to common cancers. N Engl J Med. 2008;359(20):2143–2153. doi: 10.1056/NEJMra0802968. [DOI] [PubMed] [Google Scholar]
- 5.Fletcher O, Houlston RS. Architecture of inherited susceptibility to common cancer. Nat Rev Cancer. 2010;10(5):353–361. doi: 10.1038/nrc2840. [DOI] [PubMed] [Google Scholar]
- 6.Antoniou AC, et al. Breast-cancer risk in families with mutations in PALB2. N Engl J Med. 2014;371(6):497–506. doi: 10.1056/NEJMoa1400382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Michailidou K, et al. Large-scale genotyping identifies 41 new loci associated with breast cancer risk. Nat Genet. 2013;45(4):353–361. doi: 10.1038/ng.2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Snape K, et al. Predisposition gene identification in common cancers by exome sequencing: Insights from familial breast cancer. Breast Cancer Res Treat. 2012;134(1):429–433. doi: 10.1007/s10549-012-2057-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bodmer W, Bonilla C. Common and rare variants in multifactorial susceptibility to common diseases. Nat Genet. 2008;40(6):695–701. doi: 10.1038/ng.f.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gibson G. Rare and common variants: Twenty arguments. Nat Rev Genet. 2011;13(2):135–145. doi: 10.1038/nrg3118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McKinnon PJ. ATM and the molecular pathogenesis of ataxia telangiectasia. Annu Rev Pathol. 2012;7:303–321. doi: 10.1146/annurev-pathol-011811-132509. [DOI] [PubMed] [Google Scholar]
- 12.Kee Y, D’Andrea AD. Molecular pathogenesis and clinical management of Fanconi anemia. J Clin Invest. 2012;122(11):3799–3806. doi: 10.1172/JCI58321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.D’Andrea AD. Susceptibility pathways in Fanconi’s anemia and breast cancer. N Engl J Med. 2010;362(20):1909–1919. doi: 10.1056/NEJMra0809889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sarantaus L, et al. Multiple founder effects and geographical clustering of BRCA1 and BRCA2 families in Finland. Eur J Hum Genet. 2000;8(10):757–763. doi: 10.1038/sj.ejhg.5200529. [DOI] [PubMed] [Google Scholar]
- 15.Erkko H, et al. A recurrent mutation in PALB2 in Finnish cancer families. Nature. 2007;446(7133):316–319. doi: 10.1038/nature05609. [DOI] [PubMed] [Google Scholar]
- 16.Pelttari LM, et al. RAD51C is a susceptibility gene for ovarian cancer. Hum Mol Genet. 2011;20(16):3278–3288. doi: 10.1093/hmg/ddr229. [DOI] [PubMed] [Google Scholar]
- 17.Heikkinen T, et al. The breast cancer susceptibility mutation PALB2 1592delT is associated with an aggressive tumor phenotype. Clin Cancer Res. 2009;15(9):3214–3222. doi: 10.1158/1078-0432.CCR-08-3128. [DOI] [PubMed] [Google Scholar]
- 18.Peltonen L, Jalanko A, Varilo T. Molecular genetics of the Finnish disease heritage. Hum Mol Genet. 1999;8(10):1913–1923. doi: 10.1093/hmg/8.10.1913. [DOI] [PubMed] [Google Scholar]
- 19.Gudmundsson J, et al. Frequent occurrence of BRCA2 linkage in Icelandic breast cancer families and segregation of a common BRCA2 haplotype. Am J Hum Genet. 1996;58(4):749–756. [PMC free article] [PubMed] [Google Scholar]
- 20.Abecasis GR, et al. 1000 Genomes Project Consortium An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491(7422):56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang K, Li M, Hakonarson H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Robinson JT, et al. Integrative genomics viewer. Nat Biotechnol. 2011;29(1):24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carbon S, et al. AmiGO Hub Web Presence Working Group AmiGO: Online access to ontology and annotation data. Bioinformatics. 2009;25(2):288–289. doi: 10.1093/bioinformatics/btn615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stevens KN, Vachon CM, Couch FJ. Genetic susceptibility to triple-negative breast cancer. Cancer Res. 2013;73(7):2025–2030. doi: 10.1158/0008-5472.CAN-12-1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Purrington KS, et al. GENICA Network Genome-wide association study identifies 25 known breast cancer susceptibility loci as risk factors for triple-negative breast cancer. Carcinogenesis. 2014;35(5):1012–1019. doi: 10.1093/carcin/bgt404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vahteristo P, et al. A CHEK2 genetic variant contributing to a substantial fraction of familial breast cancer. Am J Hum Genet. 2002;71(2):432–438. doi: 10.1086/341943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cybulski C, et al. Risk of breast cancer in women with a CHEK2 mutation with and without a family history of breast cancer. J Clin Oncol. 2011;29(28):3747–3752. doi: 10.1200/JCO.2010.34.0778. [DOI] [PubMed] [Google Scholar]
- 28.Liu C, Wang Y, Wang QS, Wang YJ. The CHEK2 I157T variant and breast cancer susceptibility: A systematic review and meta-analysis. Asian Pac J Cancer Prev. 2012;13(4):1355–1360. doi: 10.7314/apjcp.2012.13.4.1355. [DOI] [PubMed] [Google Scholar]
- 29.Blackford AN, et al. The DNA translocase activity of FANCM protects stalled replication forks. Hum Mol Genet. 2012;21(9):2005–2016. doi: 10.1093/hmg/dds013. [DOI] [PubMed] [Google Scholar]
- 30.Gari K, Décaillet C, Delannoy M, Wu L, Constantinou A. Remodeling of DNA replication structures by the branch point translocase FANCM. Proc Natl Acad Sci USA. 2008;105(42):16107–16112. doi: 10.1073/pnas.0804777105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Whitby MC. The FANCM family of DNA helicases/translocases. DNA Repair (Amst) 2010;9(3):224–236. doi: 10.1016/j.dnarep.2009.12.012. [DOI] [PubMed] [Google Scholar]
- 32.Meetei AR, et al. A human ortholog of archaeal DNA repair protein Hef is defective in Fanconi anemia complementation group M. Nat Genet. 2005;37(9):958–963. doi: 10.1038/ng1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bakker ST, et al. Fancm-deficient mice reveal unique features of Fanconi anemia complementation group M. Hum Mol Genet. 2009;18(18):3484–3495. doi: 10.1093/hmg/ddp297. [DOI] [PubMed] [Google Scholar]
- 34.Yang H, et al. Structural insights into the functions of the FANCM-FAAP24 complex in DNA repair. Nucleic Acids Res. 2013;41(22):10573–10583. doi: 10.1093/nar/gkt788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wood LD, et al. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318(5853):1108–1113. doi: 10.1126/science.1145720. [DOI] [PubMed] [Google Scholar]
- 36.Mirabello L, et al. A comprehensive candidate gene approach identifies genetic variation associated with osteosarcoma. BMC Cancer. 2011;11:209. doi: 10.1186/1471-2407-11-209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Smith CG, et al. Exome resequencing identifies potential tumor-suppressor genes that predispose to colorectal cancer. Hum Mutat. 2013;34(7):1026–1034. doi: 10.1002/humu.22333. [DOI] [PubMed] [Google Scholar]
- 38.Singh TR, et al. Impaired FANCD2 monoubiquitination and hypersensitivity to camptothecin uniquely characterize Fanconi anemia complementation group M. Blood. 2009;114(1):174–180. doi: 10.1182/blood-2009-02-207811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lim ET, et al. Distribution and medical impact of loss-of-function variants in the Finnish founder population. PLoS Genet. 2014;10(7):e1004494. doi: 10.1371/journal.pgen.1004494. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.