

Sulfonylureas suppress the stimulatory effect of Mg-nucleotides on recombinant β-cell (Kir6.2/SUR1) but not cardiac (Kir6.2/SUR2A) KATP channels.
Abstract
Sulfonylureas, which stimulate insulin secretion from pancreatic β-cells, are widely used to treat both type 2 diabetes and neonatal diabetes. These drugs mediate their effects by binding to the sulfonylurea receptor subunit (SUR) of the ATP-sensitive K+ (KATP) channel and inducing channel closure. The mechanism of channel inhibition is unusually complex. First, sulfonylureas act as partial antagonists of channel activity, and second, their effect is modulated by MgADP. We analyzed the molecular basis of the interactions between the sulfonylurea gliclazide and Mg-nucleotides on β-cell and cardiac types of KATP channel (Kir6.2/SUR1 and Kir6.2/SUR2A, respectively) heterologously expressed in Xenopus laevis oocytes. The SUR2A-Y1206S mutation was used to confer gliclazide sensitivity on SUR2A. We found that both MgATP and MgADP increased gliclazide inhibition of Kir6.2/SUR1 channels and reduced inhibition of Kir6.2/SUR2A-Y1206S. The latter effect can be attributed to stabilization of the cardiac channel open state by Mg-nucleotides. Using a Kir6.2 mutation that renders the KATP channel insensitive to nucleotide inhibition (Kir6.2-G334D), we showed that gliclazide abolishes the stimulatory effects of MgADP and MgATP on β-cell KATP channels. Detailed analysis suggests that the drug both reduces nucleotide binding to SUR1 and impairs the efficacy with which nucleotide binding is translated into pore opening. Mutation of one (or both) of the Walker A lysines in the catalytic site of the nucleotide-binding domains of SUR1 may have a similar effect to gliclazide on MgADP binding and transduction, but it does not appear to impair MgATP binding. Our results have implications for the therapeutic use of sulfonylureas.
INTRODUCTION
Sulfonylureas are potent stimulators of insulin secretion that have been used for many years to treat type 2 diabetes and, more recently, neonatal diabetes (Gribble and Reimann, 2003; Pearson et al., 2006). They act by binding to ATP-sensitive K+ (KATP) channels in pancreatic β-cells and causing them to close. This results in a membrane depolarization that opens voltage-gated calcium channels, thereby increasing intracellular calcium and triggering insulin release (Ashcroft and Rorsman, 2013).
KATP channels are composed of four pore-forming Kir6.2 subunits and four regulatory, sulfonylurea receptor (SUR) subunits (Shyng and Nichols, 1997). There are three main types of sulfonylurea receptor: SUR1, which forms the KATP channel in endocrine cells and brain, SUR2A, which is found in heart and skeletal muscle, and SUR2B, which comprises the smooth muscle KATP channel (Aguilar-Bryan et al., 1995; Inagaki et al., 1996). Sulfonylureas bind to their eponymous receptor with high affinity and induce pore closure. High-affinity inhibition is not complete, however, but reaches a maximum of ∼50–80%, producing a pedestal in the concentration-response curve (Gribble et al., 1997a). Single-channel recordings reveal the pedestal arises because KATP channels with bound sulfonylurea are still able to open, albeit with lower open probability (Barrett-Jolley and Davies, 1997). Thus, sulfonylureas act as partial antagonists of the KATP channel. At higher concentrations, sulfonylureas also produce a low-affinity inhibition that is independent of SUR and probably involves a binding site on Kir6.2 (Gribble et al., 1997a).
The binding site for sulfonylureas has not been fully mapped, but there is evidence it involves residues in the intracellular loop between transmembrane domains (TMs) 5 and 6 (Vila-Carriles et al., 2007) and a residue in the intracellular loop between TMs 15 and 16 (S1237 in SUR1; Ashfield et al., 1999). Mutation of S1237 in SUR1 to tyrosine abolishes the ability of tolbutamide and nateglinide to block Kir6.2/SUR1 channels (Ashfield et al., 1999; Hansen et al., 2002). In SUR2A the equivalent residue is a tyrosine, which accounts for the inability of these drugs to block Kir6.2/SUR2 channels. Residues in the N terminus of Kir6.2 are also involved in binding of both the sulfonylurea glibenclamide and the glinide repaglinide (Hansen et al., 2005; Vila-Carriles et al., 2007; Kühner et al., 2012). Thus, the sulfonylurea-binding site involves multiple regions of the protein (Winkler et al., 2007). How drug binding is transduced into closure of the Kir6.2 pore is unknown.
KATP channel activity is also regulated by cell metabolism, via changes in intracellular adenine nucleotides (Fig. 1, A and B). Binding of ATP (or ADP) to Kir6.2 results in channel closure (Tucker et al., 1997). Conversely, interaction of MgATP or MgADP with the two nucleotide-binding sites (NBSs [NBS1 and NBS2]) of SUR stimulates channel activity (Nichols et al., 1996; Gribble et al., 1997b, 1998a). It is believed this is mediated by occupancy of NBS2 by MgADP and that MgATP must be first hydrolyzed to MgADP (Zingman et al., 2001). Glucose metabolism leads to an increase in (Mg)ATP and a concomitant fall in MgADP, thereby inhibiting KATP channel activity and stimulating insulin secretion (Ashcroft et al., 1984).
Figure 1.

Nucleotide and sulfonylurea interactions with SUR. (A–D) Schematic showing interactions of nucleotides (A and B) and of nucleotides plus sulfonylureas (C and D) with SUR1 (A and C) and SUR2A (B and D). Minus signs indicate inhibitory effects; plus signs indicate interactions that stimulate channel activity.
The stimulatory effect of Mg-nucleotides on KATP channel activity involves at least three processes: an increase in the number of functional channels (N) in the plasma membrane, stabilization of the open state of active channels (which increases the single-channel open probability, PO), and a reduction in the inhibitory effect of nucleotides at Kir6.2 (which produces an increase in the IC50 for nucleotide inhibition; Bokvist et al., 1991; Hopkins et al., 1992; Nichols et al., 1996; Shyng et al., 1997; Ribalet et al., 2000; Abraham et al., 2002; Proks et al., 2010). All of these effects decline slowly after patch excision, a phenomenon which has been termed DAMN (decline in activation by Mg-nucleotides; Proks et al., 2010).
Sulfonylureas and nucleotides interact with one another in a complex way to regulate KATP channel activity. For example, MgATP and MgADP reduce glibenclamide binding to both SUR1 and SUR2A (Hambrock et al., 2002; Löffler-Walz et al., 2002). Conversely, in the presence of 100 µM intracellular MgADP, sulfonylurea inhibition of β-cell KATP (Kir6.2/SUR1) channels is enhanced (Zünkler et al., 1988; Gribble et al., 1997a, 1998b; Dabrowski et al., 2001; Proks et al., 2002), whereas that of cardiac KATP (Kir6.2/SUR2A) channels is reduced (Venkatesh et al., 1991; Gribble et al., 1998b). Chimera experiments suggest that these different responses of Kir6.2/SUR1 and Kir6.2/SUR2A channels involve TMs 8–11 of SUR (Reimann et al., 2003). It is therefore tempting to speculate that these domains may be involved in transducing nucleotide binding to SUR1 into channel activation; however, this has not been explicitly demonstrated.
The reduction in the maximal extent of high-affinity sulfonylurea inhibition of Kir6.2/SUR2A channels in the presence of MgADP is likely to be a consequence of the stabilization of the open state of the channel by the Mg-nucleotide (Fig. 1 D), as single-channel studies have demonstrated both MgADP (Li et al., 2002) and MgUDP (Alekseev et al., 1998) stabilize the open state. Similarly, maximal sulfonylurea inhibition is reduced when the channel open state is stabilized by PIP2 (Koster et al., 1999; Krauter et al., 2001) or by mutations in KATP channel subunits (Trapp et al., 1998; Koster et al., 1999). Because MgADP promotes channel opening, it also increases the IC50 for sulfonylurea inhibition of cardiac KATP channels (Venkatesh et al., 1991).
Similar effects would be expected for Kir6.2/SUR1 channels. However, in contrast to Kir6.2/SUR2A channels, an increase in the maximal extent of high-affinity sulfonylurea inhibition is actually observed in the presence of MgADP (Gribble et al., 1997a). It has been proposed that this occurs because sulfonylureas abolish the stimulatory effects of MgADP on Kir6.2/SUR1 channels and thus reveal the full extent of nucleotide inhibition at Kir6.2 (which is normally partially masked by the stimulatory effects of nucleotides). This produces an apparent enhancement of sulfonylurea inhibition (Fig. 1 C). However, this idea has not been tested directly, and whether sulfonylureas also influence MgATP activation is unknown. Furthermore, in almost all cases, only a single drug and nucleotide concentration were examined and the concentration dependence of these effects is unknown. Nevertheless, a mechanistic understanding of drug-nucleotide interactions is important because the different response of SUR isoforms likely contributes to the fact that (at the cellular level) these drugs are more potent on β-cell KATP channels than cardiac KATP channels (Lawrence et al., 2001).
In this paper, we provide a detailed quantitative analysis of the interactions between sulfonylureas and nucleotides on Kir6.2/SUR1 and Kir6.2/SUR2A channels. In most experiments, we used the sulfonylurea gliclazide because this drug has the advantage that its effects on Kir6.2/SUR1 are readily reversible, which enables multiple applications of the drug to the same patch (Gribble and Ashcroft, 1999) and correction for the effects of rundown and DAMN (Proks et al., 2010). Furthermore, in inside-out patches, the gliclazide concentration-inhibition relationship exhibits a clearly defined separation between inhibition at the high-affinity (IC50 ∼ 50 nM) site on SUR and the low-affinity (IC50 ∼ 3 mM) site on Kir6.2 (Gribble and Ashcroft, 1999). Because therapeutic concentrations of gliclazide in the plasma of patients with type 2 diabetes are typically around 10 µM (Abdelmoneim et al., 2012), only the effects of gliclazide at the high-affinity site are clinically relevant, and we therefore restricted our analysis to gliclazide concentrations of 100 µM or less. Our results shed new light on the mechanistic basis of the complex interaction between sulfonylureas and Mg-nucleotides on the activity of cardiac and β-cell KATP channels.
MATERIALS AND METHODS
Molecular biology
All experiments were performed on recombinant KATP channels heterologously expressed in Xenopus laevis oocytes. We used human Kir6.2 (GenBank accession no. NM_000525 with E23 and I377), rat SUR1 (GenBank accession no. L40624), and rat SUR2A (GenBank accession no. D83598); we used rodent rather than human SUR because in our hands it expressed larger currents. Site-directed mutagenesis of SUR1, SUR2A, or Kir6.2, preparation of mRNA, and isolation of Xenopus oocytes was performed as described previously (Proks et al., 2005). Oocytes were injected with 0.8 ng wild-type or mutant Kir6.2 mRNA and ∼4 ng SUR mRNA, and currents were recorded 1–3 d after injection.
Electrophysiology
Macroscopic currents were recorded as previously described (Proks et al., 2005) from giant inside-out patches at −60 mV and 20–22°C, filtered at 5 kHz, and digitized at 20 kHz. The pipette solution contained (mM) 140 KCl, 1.2 MgCl2, 2.6 CaCl2, and 10 HEPES (pH 7.4 with KOH). The intracellular (bath) solution contained (mM) 107 KCl, 1 CaCl2, 2 MgCl2, 10 EGTA, 10 HEPES (pH 7.2 with KOH), and MgATP or MgADP as indicated. The Mg-free intracellular solution contained (mM) 107 KCl, 1 K2SO4, 10 EGTA, 10 HEPES (pH 7.2 with KOH), and K2ATP or K2ADP as indicated. A 100 mM stock solution of gliclazide and glibenclamide in DMSO was prepared daily and diluted as required.
KATP channels in excised patches, whether from pancreatic β-cells, mammalian cell lines, or Xenopus oocytes, undergo both fast and slow rundown. Fast rundown is virtually complete within the first minute, and the mean open probability (PO) stabilizes at a similar value in all cases; it is independent of Mg2+ or mutation of the Walker A (WA) lysines in the NBSs of SUR1 (Table S1). Nucleotide sensitivity of KATP channels was assessed when fast rundown was complete. Slow rundown was compensated by averaging the current values in control solution before and after nucleotide/drug application. Because inhibition of Kir6.2/SUR1 channels by glibenclamide was poorly reversible on the time scale of our experiments, we estimated the effect of the drug on Kir6.2/SUR1 currents as follows. A single exponential was fit to the time course of the current decline before drug application. The control current amplitude (IC) was then estimated by extrapolating the exponential to the time at which the current in the presence of glibenclamide reached a steady-state (I). Currents were then plotted as I/IC.
Channel activation was expressed as (ING − IG)/(IMAX − I0), where ING is the steady-state current in the presence of both drug and test nucleotide concentration, IG is the current in the presence of gliclazide before nucleotide application, IMAX is the steady-state KATP current in the absence of drug and presence of a maximal stimulatory concentration of nucleotide (10 mM for MgATP and 1 mM for MgADP), and I0 is the current in the absence of both drug and nucleotide (Fig. 2). When measuring channel activation, both rundown and the decline in the stimulatory effect of Mg-nucleotides after patch excision were compensated as described previously (Proks et al., 2010). At MgADP concentrations of 10 mM or greater, a slowly developing inhibition made measurements unreliable: we therefore restricted our analysis to MgADP concentrations of 3 mM or below. The time course of Mg-nucleotide deactivation (i.e., the decrease in KATP current after nucleotide removal) was well fit with a single exponential both in the presence and absence of sulfonylurea. A similar finding in the absence of the drug was described previously (Proks et al., 2010).
Figure 2.

Method of quantifying interactions between Mg-nucleotides and gliclazide. Representative record showing activation of Kir6.2-G334D/SUR1 by MgADP (1 or 3 mM) in the presence and absence of 30 µM gliclazide (as indicated). IMAX,1 and IMAX,2 are the steady-state KATP currents in the presence of a maximal stimulatory concentration of 1 mM MgADP before and after gliclazide application, respectively; I0,1 and I0,2 are the currents in the control (drug and nucleotide free) solution measured immediately before nucleotide application (I0,1) or after (I0,2) gliclazide application; IG is the current in the presence of gliclazide alone; and ING is the steady-state current in the presence of both drug and the test nucleotide concentration. The dotted line indicates the zero current level.
The relationship between ADP concentration and KATP current inhibition in Fig. 4 and between sulfonylurea concentration and KATP current inhibition in Figs. 3, 5, and 6, was fit with
| (1) |
where IX is the steady-state KATP current in the presence of the test nucleotide or drug concentration [X], IC is the current in nucleotide (or drug)-free solution obtained by averaging the current before and after application, IC50 is the nucleotide (drug) concentration at which the inhibition is half maximal, h is the Hill coefficient, and a is the fraction of KATP current remaining at gliclazide concentrations that saturate the high-affinity binding site (for ADP, a = 0). The factor L equals 1 except for data in Fig. 6 (closed symbols), where it reflects the extent of channel activation by Mg-nucleotides in drug-free solution.
Figure 4.

Effect of sulfonylureas on (Mg)ADP modulation of SUR1- and SUR2A-containing channels. (A and B) Concentration-response relationships for ADP modulation of Kir6.2/SUR1 channels in the absence (A) or presence (B) of 2 mM Mg2+ and the absence (open symbols) or presence (closed symbols) of 30 µM gliclazide. (A) The lines are the best fit of Eq. 1 to the mean data: IC50 = 64 µM, h = 0.81 (open squares; n = 6); IC50 = 57 µM, h = 0.95 (closed squares; n = 6). (B) The lines are the best fit to the mean data of Eq. 4 with IC50 = 224 µM, h1 = 1.3 and EC50 = 11 µM, h2 = 1.3 and a = 2.7 (open circles; n = 12) or of Eq. 1 with IC50 = 64 µM, h = 0.91 (closed circles; n = 6). (C and D) Concentration-response relationships for ADP modulation of Kir6.2/SUR2A-YS channels in the absence (C) and presence of 2 mM Mg2+ (D) and in the presence (closed symbols; n = 6) or absence (open symbols; n = 6) of 30 µM gliclazide. (C) The lines are the best fit of Eq. 1 to the mean data: IC50 = 94 µM, h = 1.1 (open squares; n = 6); IC50 = 76 µM, h = 1.2 (closed squares; n = 6). (D) The solid line is the best fit to the mean data of Eq. 4 with IC50 = 240 µM, h1 = 1.2 and EC50 = 19 µM, h2 = 1.5 and a = 1.3. The dotted line is the concentration-inhibition curve for Kir6.2/SUR2A-YS channels in the absence of Mg2+ and gliclazide (C, open squares). (E and F) Concentration-response relationships for ADP modulation of Kir6.2/SUR2A channels in the absence (E) and presence of 2 mM Mg2+ (F) and in the absence (open symbols) or presence (closed symbols) of 1 µM glibenclamide. (E) The lines are the best fit of Eq. 1 to the mean data: IC50 = 90 µM, h = 0.86 (open squares; n = 6); IC50 = 70 µM, h = 0.95 (closed squares; n = 6). (F) Both open and closed circles are the mean of seven experiments. The solid line is the best fit to the mean data of Eq. 4 with IC50 = 270 µM, h1 = 1.4 and EC50 = 18 µM, h2 = 1.3 and a = 1.0. The dotted line is the concentration-inhibition curve for Kir6.2/SUR2A channels in the absence of Mg2+ and glibenclamide (E, open squares). Mean ± SEM.
Figure 3.

Sulfonylurea inhibition of Kir6.2/SUR1- and Kir6.2/SUR2A-containing channels. (A and B) Representative KATP currents (top) and concentration-response relationships (bottom) for glibenclamide inhibition of Kir6.2/SUR1 (A; n = 6) and Kir6.2/SUR2A (B; n = 6) channels. The lines are the best fit of Eq. 1 to the mean data: (A) IC50 = 2.8 nM, h = 0.93, a = 0.32; (B) IC50 = 13 nM, h = 0.94, a = 0.32. (C and D) KATP currents (top) and concentration-response relationships (bottom) for gliclazide block of Kir6.2/SUR1 (C; n = 6) and Kir6.2/SUR2A-YS (D; n = 5) channels. The lines are the best fit of Eq. 1 to the mean data: (C) IC50 = 72 nM, h = 1.2, a = 0.42; (D) IC50 = 1.3 µM, h = 1.1, a = 0.65. Mean ± SEM.
Figure 5.

Effect of MgADP and MgATP on sulfonylurea inhibition of SUR1- and SUR2A-containing channels. (A–F) Currents in the presence of sulfonylurea (I) expressed as a fraction of that in drug-free solution (Ic). (A and B) Concentration-response relationships for gliclazide inhibition of Kir6.2/SUR1 (A) and Kir6.2/SUR2A-YS (B) channels in the presence and absence (same data as in Fig. 3) of 100 µM MgADP. The lines are the best fit of Eq. 1 to the mean data: IC50 = 72 nM, h = 1.2, a = 0.42 (A, open circles; n = 6); IC50 = 187 nM, h = 1.1, a = 0.07 (A, closed circles; n = 6); IC50 = 1.3 µM, h = 1.1, a = 0.65 (B, open circles; n = 5); IC50 = 1.6 µM, h = 1.2, a = 0.85 (B, closed circles; n = 5). (C and D) Concentration-response relationships for glibenclamide inhibition of Kir6.2/SUR1 (C) and Kir6.2/SUR2A (D) channels in the presence and absence (data from Fig. 3) of 100 µM MgADP. The lines are the best fit of Eq. 1 to the mean data: IC50 = 2.8 nM, h = 0.93, a = 0.32 (C, open circles; n = 6); IC50 = 3.7 nM, h = 1.2, a = 0.04 (C, closed circles; n = 6); IC50 = 13 nM, h = 0.94, a = 0.32 (D, open circles; n = 5); IC50 = 30 nM, h = 0.75, a = 0.68 (D, closed circles; n = 5). (E and F) Concentration-response relationships for gliclazide inhibition of Kir6.2-G334D/SUR1 (E) and Kir6.2-G334D/SUR2A-YS (F) channels in the presence and absence of 1 mM MgATP. The lines are the best fit of Eq. 1 to the mean data: IC50 = 70 nM, h = 1.0, a = 0.39 (E, open squares; n = 6); IC50 = 210 nM, h = 1.0, a = 0.21 (E, closed squares; n = 6); IC50 = 1.3 µM, h = 1.2, a = 0.64 (F, open squares; n = 5); IC50 = 2.5 µM, h = 1.0, a = 0.91 (F, closed squares; n = 5). Mean ± SEM.
Figure 6.

Effect of MgADP and MgATP on gliclazide inhibition of Kir6.2/SUR1 and Kir6.2/SUR2A-YS. (A–F) Data are the same as in Fig. 5 (A–F), but currents in the presence of gliclazide (I) are expressed as a fraction of that in drug- and nucleotide-free solution (Ic). (A and B) Concentration-response relationships for gliclazide inhibition of Kir6.2/SUR1 (A) and Kir6.2/SUR2A-YS (B) channels in the presence and absence of 100 µM MgADP. The lines are the best fit of Eq. 1 to the mean data: IC50 = 72 nM, h = 1.1, a = 0.42 (A, open circles; n = 6); IC50 = 187 nM, h = 1.1, a = 0.18, L = 2.5 (A, closed circles; n = 6); IC50 = 1.3 µM, h = 1.1, a = 0.65 (B, open circles; n = 5); IC50 = 1.6 µM, h = 1.2, a = 1.61, L = 1.7 (B, closed circles; n = 5). (C and D) Concentration-response relationships for glibenclamide inhibition of Kir6.2/SUR1 (C) and Kir6.2/SUR2A (D) channels in the presence and absence of 100 µM MgADP. The lines are the best fit of Eq. 1 to the mean data: IC50 = 2.8 nM, h = 0.93, a = 0.32 (C, open circles; n = 6); IC50 = 3.7 nM, h = 1.2, a = 0.12, L = 2.8 (C, closed circles; n = 6); IC50 = 13 nM, h = 0.94, a = 0.32 (D, open circles; n = 5); IC50 = 30 nM, h = 0.75, a = 1.02, L = 1.5 (D, closed circles; n = 5). (E and F) Concentration-response relationships for gliclazide inhibition of Kir6.2-G334D/SUR1 (E) and Kir6.2-G334D/SUR2A-YS (F) channels in the presence and absence of 1 mM MgATP. The lines are the best fit of Eq. 1 to the mean data: IC50 = 70 nM, h = 1.0, a = 0.39 (E, open squares; n = 6); IC50 = 210 nM, h = 1.0, a = 0.42, L = 2.0 (E, closed squares; n = 6); IC50 = 1.3 µM, h = 1.2, a = 0.64 (F, open squares; n = 5); IC50 = 2.5 µM, h = 1.0, a = 1.44, L = 1.8 (F, closed squares; n = 5). Mean ± SEM.
The relationship between channel activation and nucleotide concentration in Fig. 7 was fit with
| (2) |
where EC50 is the nucleotide concentration at which activation is half maximal, [X] is the test nucleotide concentration in the presence of gliclazide, h is the slope factor (Hill coefficient), and a is the ratio of the maximal stimulatory effect of the nucleotide in the presence and absence of gliclazide. In the absence of gliclazide, a = 1.
Figure 7.

Gliclazide reduces MgATP and MgADP activation of Kir6.2-G334D/SUR1. (A and C) Representative records showing activation of Kir6.2-G334D/SUR1 currents by 1 mM MgADP (A) or 10 mM MgATP (C) in the presence and absence of 30 µM gliclazide (as indicated). The dotted lines indicate the zero current level. (B and D) Concentration-activation relationships for MgADP (B) or MgATP (D) for Kir6.2-G334D/SUR1 channels in the absence (open circles; n = 6) or presence (closed circles; n = 6) of 30 µM gliclazide. (B) The lines are the best fit of Eq. 2 to the mean data. Open circles: EC50 = 9 µM, h = 1.3; a was fixed at 1. Closed circles: EC50 = 560 µM, h = 1.5, a = 0.3. (D) The lines are the best fit to the mean data of Eq. 2 with a set to 1 (open circles: EC50 = 124 µM, h = 1.3) or Eq. 2 with a set to 0.3 (closed circles: EC50 = 8.1 mM, h = 1.3). Mean ± SEM.
In Fig. 8, the relationship was fit with
| (3) |
where IN and I0 are steady-state current values in the presence and absence of the nucleotide, respectively, expressed as a fraction of the maximum current value reached after excision, a is the maximal stimulatory effect of the drug expressed as a fraction of the maximum current value reached after excision, and the other parameters are defined as in Eq. 2.
Figure 8.

Effects of mutations in the WA motif of SUR1 on Mg-nucleotide activation. (A and B) Concentration-activation relationships for MgADP (A) or MgATP (B) for channels composed of Kir6.2-G334D and SUR1 (open circles; n = 6), SUR1-KAKA (closed circles; n = 5), SUR1-K1A (closed squares; n = 5), or SUR1-K2A (open squares; n = 5) channels. The solid lines are the best fit of Eq. 3 to the mean data (A, open circles: a = 0.84, EC50 = 9 µM, h = 1.3; B, open circles: a = 0.88, EC50 = 124 µM, h = 1.3; closed squares: a = 0.52, EC50 = 150 µM, h = 1.3; open squares: a = 0.40, EC50 = 200 µM, h = 1.1; closed circles: a = 0.05, EC50 = 150 µM, h = 1.3) except for data for channels containing mutant SUR1 subunits in A, where the lines are drawn by hand. The dotted lines indicate the relationship obtained for Kir6.2-G334D/SUR1 in the presence of 30 µM gliclazide (shown in Fig. 7). Mean ± SEM.
The relationship between MgADP concentration and wild-type KATP currents (see Fig. 4, B, D, and F) was fit with
| (4) |
where IX is the steady-state KATP current in the presence of the test ADP concentration [X], IC is the current in nucleotide-free solution obtained by averaging the current before and after nucleotide application, IC50 and EC50 are nucleotide concentrations at which the inhibition and activation are half maximal, respectively, h1 and h2 are the Hill coefficients for inhibition and activation, respectively, and a is the maximal increase in KATP current caused by nucleotide activation.
The catalytic cycle of SUR2A-NBS2 was adapted from Bienengraeber et al. (2004):
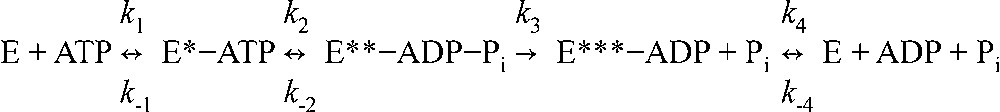 |
(Scheme 1) |
where E is NBS2 and asterisks indicate distinct conformations that occur during Mg2+-dependent ATP hydrolysis. Transitions are bidirectional, with the exception of the Pi liberation step (k3), which is irreversible (Bienengraeber et al., 2004).
Scheme 1 gives the following expressions for the fractional occupancy of NBS2 by MgADP in the presence of ADP (Eq. 5) and ATP (Eq. 6; after Bienengraeber et al. [2004]):
| (5) |
| (6) |
where [ADP] and [ATP] are the bulk ADP and ATP concentrations, respectively, EADP is the number of NBS2s occupied by ADP and ET is the total number of NBS2s.
For noise analysis, the macroscopic mean current (I) and variance (σ) were determined from data segments of 1-s duration. Control data were recorded immediately before gliclazide application, and test data were recorded once a steady-state condition was reached. PO and N were calculated as described previously (Proks et al., 2010).
Statistics
All values are given as mean ± SEM. Statistical significance was determined using Student’s t test.
Online supplemental material
Fig. S1 shows concentration-inhibition relationships for ATP inhibition of Kir6.2/SUR2A-YS channels in the absence and presence of Mg2+. Fig. S2 shows concentration-inhibition relationships for ATP and ADP inhibition of Kir6.2-G334D/SUR1 in the absence of Mg2+. Fig. S3 compares concentration-response relationships for gliclazide inhibition of SUR1 and SUR2A-YS channels with either Kir6.2 or Kir6.2-G334D as the pore-forming subunit. Fig. S4 simulates the concentration-inhibition relationships for sulfonylurea inhibition of SUR2A-containing channels using a Monod–Wyman–Changeux (MWC) model. Fig. S5 simulates the reduction in the maximal extent of MgATP activation of Kir6.2-G334D/SUR1 channels induced by gliclazide (assumed to be a transduction defect) using an MWC model. The supplemental text entitled “Simulations of the effect of PO on the EC50 for Mg-nucleotide activation” and Fig. S6 assess in detail the possible effects of gliclazide on Mg-nucleotide activation of Kir6.2-G334D/SUR1 channels that result from changes in channel gating. Table S1 gives mean values of PO in nucleotide-free solutions for various SUR1-containing channels. Online supplemental material is available at http://www.jgp.org/cgi/content/full/jgp.201411222/DC1.
RESULTS
The Y1206S mutation confers gliclazide sensitivity on SUR2A
Fig. 3 (A and B) compares high-affinity glibenclamide inhibition of recombinant β-cell (Kir6.2/SUR1) and cardiac (Kir6.2/SUR2A) types of KATP channel in inside-out patches excised from Xenopus oocytes. As previously reported (Gribble et al., 1998b), glibenclamide inhibited both types of channel with similar affinity, but inhibition of Kir6.2/SUR1 was not reversible on the time scale of our experiments, whereas glibenclamide inhibition of Kir6.2/SUR2A was partially reversible.
In contrast to glibenclamide, gliclazide produced reversible, high-affinity inhibition of Kir6.2/SUR1 but not Kir6.2/SUR2A channels (Gribble and Ashcroft, 1999). We therefore sought to confer sensitivity to gliclazide on Kir6.2/SUR2A. A previous study has shown that mutation of serine 1237 in SUR1 to tyrosine abolishes high-affinity tolbutamide inhibition and reduces glibenclamide binding (Ashfield et al., 1999). The reverse mutation in SUR2A (Y1206S) increases 3[H]glibenclamide binding to levels similar to those found for SUR1 (Hambrock et al., 2002; Winkler et al., 2007). We therefore hypothesized that the Y1206S mutation might also confer sensitivity to tolbutamide and gliclazide on SUR2A. Fig. 3 (C and D) shows that this is indeed the case: like Kir6.2/SUR1, Kir6.2/SUR2A-YS channels were reversibly inhibited by gliclazide. The Y1206S (YS) mutation did not alter the intrinsic (i.e., unliganded) open probability, which was 0.71 ± 0.04 (n = 5) for Kir6.2/SUR2A-YS and 0.71 ± 0.04 (n = 6) for Kir6.2/SUR2A. It also did not alter the IC50 for ATP inhibition (Fig. S1) or ADP inhibition, the EC50 for MgADP activation of Kir6.2/SUR2A (Table 1), or (as shown later) the interaction between sulfonylurea and MgADP activation. Consequently, it appears to be a useful tool for assessing interactions between sulfonylureas and nucleotides on Kir6.2/SUR2A.
Table 1.
Mean ± SEM for parameters used to fit ADP concentration-response relationships for the indicated KATP channels in the presence or absence of sulfonylureas
| Channel | Parameter | Mg2+ | No sulfonylurea | +Sulfonylurea |
| 30 µM gliclazide | ||||
| Kir6.2/SUR1 | IC50 (µM) | − | 62 ± 3 (n = 6) | 59 ± 7 (n = 6) |
| Kir6.2/SUR1 | IC50 (µM) | + | 258 ± 57 (n = 12) | 64 ± 3a (n = 6) |
| Kir6.2/SUR1 | EC50 (µM) | + | 13 ± 2 (n = 12) | n.a. |
| Kir6.2/SUR1 | a | + | 3.2 ± 0.7 (n = 12) | n.a. |
| Kir.2/SUR2A-YS | IC50 (µM) | − | 95 ± 5 (n = 6) | 88 ± 11 (n = 6) |
| Kir.2/SUR2A-YS | IC50 (µM) | + | 310 ± 56 (n = 6) | 286 ± 39 (n = 6) |
| Kir.2/SUR2A-YS | EC50 (µM) | + | 16 ± 5 (n = 6) | 19 ± 3 (n = 6) |
| Kir.2/SUR2A-YS | a | + | 1.2 ± 0.3 (n = 6) | 1.1 ± 0.1 (n = 6) |
| 1 µM glibenclamide | ||||
| Kir6.2/SUR2A | IC50 (µM) | − | 96 ± 19 (n = 6) | 80 ± 8 (n = 6) |
| Kir6.2/SUR2A | IC50 (µM) | + | 280 ± 70 (n = 7) | 315 ± 49 (n = 7) |
| Kir6.2/SUR2A | EC50 (µM) | + | 22 ± 8 (n = 7) | 17 ± 2 (n = 7) |
| Kir6.2/SUR2A | a | + | 1.1 ± 0.3 (n = 7) | 1.1 ± 0.2 (n = 7) |
Parameters were obtained from the data shown in Fig. 4, but all values are the mean of the fits to the individual dose–response curves. a is the maximal increase in KATP current caused by MgADP activation (Eq. 4; note the maximal amplitude of activation is given by 1 + a). n.a., not applicable.
P < 0.01 against control (Student’s t test).
Fig. 3 (C and D) also illustrates high-affinity gliclazide inhibition of Kir6.2/SUR1 and Kir6.2/SUR2A-YS channels. There was an ∼15-fold difference in the drug concentration causing half-maximal inhibition, which was 77 ± 11 nM (n = 6) for Kir6.2/SUR1 and 1.2 ± 0.3 µM (n = 5) for Kir6.2/SUR2A-YS. Similarly, the maximal extent of inhibition was greater (∼60%) for Kir6.2/SUR1 than Kir6.2/SUR2A-YS (35%). One possible explanation for these differences is that Kir6.2/SUR2A-YS binds gliclazide less tightly and transduces inhibition somewhat less efficiently than Kir6.2/SUR1. In addition, it is possible that the higher intrinsic (i.e., unliganded) open probability (PO) of Kir6.2/SUR2A-YS channels, which was 0.71 ± 0.4 (n = 5) compared with 0.43 ± 0.3 (n = 6) for Kir6.2/SUR, may contribute. However, this seems less likely as there was no substantial difference in the IC50 or maximal extent of glibenclamide inhibition for channels containing SUR1 and SUR2A.
The IC50 for gliclazide inhibition of Kir6.2/SUR2A-YS channels is ∼800-fold less than that reported for recombinant Kir6.2/SUR2A channels (800 µM; Gribble and Ashcroft, 1999). This is consistent with the idea that gliclazide inhibition of Kir6.2/SUR2A-YS is mediated via binding to SUR2A and that of Kir6.2/SUR2A by binding to Kir6.2. Interestingly, Lawrence et al. (2001) reported an IC50 of 19 µM for gliclazide inhibition of pinacidil-activated KATP channels in rat ventricular myocytes. This is much higher than expected for SUR2A and suggests that KATP channels in these cells may be composed of a mixture of SUR1 and SUR2A subunits, as recently suggested for cardiac KATP channels (see review by Zhang et al. [2010]).
Sulfonylurea modulation of nucleotide response
We next compared the effect of gliclazide on nucleotide modulation of Kir6.2/SUR1 and Kir6.2/SUR2A-YS. We used 30 µM gliclazide, a concentration at which high-affinity inhibition is near maximal (indicating most high-affinity binding sites on SUR are occupied) for both types of channel.
In Mg-free solution, where nucleotide activation is absent, gliclazide was without obvious effect on ADP inhibition of either Kir6.2/SUR1 or Kir6.2-SUR2A-YS (Fig. 4, A and C). In the presence of Mg2+, the ADP concentration-response relation followed a bell-shaped curve (Fig. 4, B and D, open symbols). This is the result of the simultaneous presence of activation (at SUR) and inhibition (at Kir6.2; Hopkins et al., 1992). Therefore, we fit the data with a function that is the product of activation and inhibition (Materials and methods, Eq. 4). For both types of channel, the calculated IC50 for ADP inhibition was greater in the presence of Mg2+ than in its absence, increasing from 62 to 258 µM for Kir6.2/SUR1 and from 95 to 310 µM for Kir6.2/SUR2A-YS (Table 1). This suggests that ADP inhibition is reduced in the presence of Mg2+, as previously suggested (Nichols et al., 1996; Shyng et al., 1997; Ribalet et al., 2000; Abraham et al., 2002; Proks et al., 2010). The calculated EC50 for MgADP activation of Kir6.2/SUR2A-YS (16 µM) was similar to that for Kir6.2/SUR1 (13 µM; Table 1); however, the maximal extent of MgADP activation was less (Fig. 4).
30 µM gliclazide produced strikingly different effects on MgADP activation of Kir6.2/SUR1 and Kir6.2/SUR2A-YS, completely eliminating the stimulatory effect of MgADP on Kir6.2/SUR1 but having no effect on Kir6.2/SUR2A-YS (Fig. 4, B and D, closed symbols). Gliclazide also abolished the decrease in ADP inhibition of Kir6.2-SUR1 produced by Mg2+: in the presence of the drug, the IC50 for MgADP inhibition was 64 µM, similar to that found in Mg2+- and drug-free solution (62 µM; Table 1). In contrast, gliclazide did not alter the calculated IC50 for MgADP inhibition of Kir6.2/SUR2A-YS.
We also examined the effect of glibenclamide on MgADP modulation of wild-type Kir6.2/SUR2A to test whether the SUR2A-YS mutation might have altered the interaction between nucleotides and sulfonylureas. We used 1 µM glibenclamide, a concentration at which high-affinity inhibition is near maximal (Ashfield et al., 1999). Glibenclamide was without obvious effect on either ADP inhibition or MgADP activation of Kir6.2/SUR2A (Fig. 4, E and F; and Table 1), as observed for gliclazide and Kir6.2/SUR2A-YS. Collectively, therefore, the data indicate that, in contrast to Kir6.2/SUR1, sulfonylureas do not influence MgADP activation of Kir6.2/SUR2A or Kir6.2/SUR2A-YS.
Nucleotide modulation of sulfonylurea inhibition
Fig. 5 (A–D) compares the effect of nucleotides on gliclazide and glibenclamide inhibition of β-cell and cardiac KATP channels: current is plotted as a fraction of that in the absence of the drug. IC50 values obtained from these experiments are given in Tables 2 and 3. At gliclazide concentrations <200 nM, there was little difference in inhibition of Kir6.2/SUR1 channels in the presence or absence of 100 µM MgADP (Fig. 5 A). However, the maximal extent of high-affinity gliclazide inhibition was substantially enhanced by MgADP. This is because the drug completely abolishes the activatory effect of MgADP (Fig. 4 B) and thereby reveals the inhibitory effect of the nucleotide (Fig. 1; Gribble et al., 1997a). This was not the case for Kir6.2/SUR2A-YS. As Fig. 5 B shows, the fraction of gliclazide-resistant current increased, rather than decreased, when MgADP was present. Similarly, glibenclamide inhibition of Kir6.2/SUR1 was increased, and that of Kir6.2/SUR2A was reduced, by MgADP (Fig. 5, C and D). These data are similar to those previously described for sulfonylurea inhibition for native and heterologously expressed wild-type KATP channels in the presence of MgADP (Venkatesh et al., 1991; Gribble et al., 1997a, 1998b; Proks et al., 2013).
Table 2.
IC50 values for gliclazide concentration-inhibition relationships of the indicated KATP channels
| Channel | Nucleotide | No nucleotide | +Nucleotide |
| Kir6.2/SUR1 | 100 µM MgADP | 77 ± 11 nM (n = 6) | 245 ± 29 nMa (n = 6) |
| Kir.2/SUR2A-YS | 100 µM MgADP | 1.2 ± 0.2 µM (n = 5) | 2.3 ± 0.6 µM (n = 5) |
| Kir6.2-G334D/SUR1 | 1 mM MgATP | 72 ± 12 nM (n = 6) | 240 ± 42 nMa (n = 6) |
| Kir.2-G334D/SUR2A-YS | 1 mM MgATP | 1.6 ± 0.5 µM (n = 5) | 2.9 ± 1.1 µM (n = 5) |
All values are the mean of the fits to the individual dose–response curves.
P < 0.01 against 0 mM nucleotide (by Student’s t test).
Table 3.
IC50 values for glibenclamide concentration-inhibition relationships of the indicated KATP channels
| Channel | Control | +100 µM MgADP |
| Kir6.2/SUR1 | 4.2 ± 1.1 nM (n = 6) | 3.8 ± 0.7 nM (n = 6) |
| Kir.2/SUR2A | 15.8 ± 3.0 nM (n = 6) | 29.2 ± 7.1 nM (n = 5) |
All values are the mean of the fits to the individual dose–response curves.
We next investigated whether MgATP has a similar effect on gliclazide inhibition to MgADP. For this purpose, it was essential to remove ATP inhibition at Kir6.2. We therefore exploited the fact that the Kir6.2-G334D mutation largely abolishes ATP and ADP inhibition (at Kir6.2) without affecting the single-channel kinetics in the absence of nucleotide (Fig. S2; Drain et al., 1998; Masia et al., 2007; Proks et al., 2010). The Kir6.2-G334D mutation had no effect on gliclazide inhibition of SUR1- or SUR2A-YS–containing channels in the absence of nucleotide (Fig. S3). Fig. 5 (E and F) shows that MgATP increased the maximal extent of high-affinity gliclazide inhibition of Kir6.2-G334D/SUR1 but reduced that of Kir6.2-G334D/SUR2A-YS. As found for MgADP, MgATP also affected the IC50 of gliclazide inhibition: the IC50 increased from 72 to 240 nM for Kir6.2-G334D/SUR1 and from 1.6 to 2.9 µM for Kir6.2-G334D/SUR2A-YS channels (Table 2). Thus, the interaction between MgATP and gliclazide qualitatively resembles that found for MgADP and gliclazide, for channels containing SUR1 and SUR2A-YS.
An increase in the IC50 for gliclazide block of Kir6.2-G334D/SUR1 channels by Mg-nucleotides has been previously reported (Proks et al., 2013) and was attributed to antagonism between sulfonylurea binding to SUR1 and MgADP binding to NBS2. A similar effect was observed for gliclazide block of Kir6.2/SUR1 channels in the presence of MgADP (Fig. 5 A; Proks et al., 2002).
We also plotted current in the presence of sulfonylurea as a fraction of that in the absence of both drug and nucleotide. Fig. 6 shows that although 100 µM MgADP stimulated both types of channel (Gribble et al., 1997a), the current amplitude in the presence of both MgADP and a maximally effective gliclazide concentration was approximately eightfold larger for Kir6.2/SUR2A-YS than for Kir6.2/SUR1 (compare A and B, closed circles). Similarly, in the case of glibenclamide and MgADP, inhibition was approximately eightfold larger for Kir6.2/SUR2A than for Kir6.2/SUR1 (Fig. 6, compare C and D, closed circles). This is expected to have dramatically different effects on the membrane potential of β-cells and cardiac cells.
When current in the presence of gliclazide was plotted as a fraction of that in the absence of both drug and nucleotide (Fig. 6 E), it became obvious that gliclazide abolished MgATP-induced activation of Kir6.2-G334D/SUR1 channels in a concentration-dependent manner but that the maximal extent of inhibition was unchanged (because these channels are not inhibited by MgATP; Proks et al., 2013). However, the current amplitude in the presence of both MgATP and a maximally effective gliclazide concentration was still approximately fourfold greater for Kir6.2-G334D/SUR2A-YS than Kir6.2-G334D/SUR1 (Fig. 6, compare E and F, closed squares).
Gliclazide suppresses Mg-nucleotide activation
Our data indicate that sulfonylureas impair Mg-nucleotide activation of β-cell but not cardiac KATP channels. In the rest of this paper, we therefore focused on the molecular mechanism by which gliclazide suppresses activation of the Kir6.2/SUR1 channel by Mg-nucleotides. We used the Kir6.2-G334D mutation (which abolishes nucleotide inhibition at Kir6.2) to isolate the effects of Mg-nucleotides on SUR1.
Gliclazide dramatically reduced MgADP activation of Kir6.2-G334D/SUR1 channels (Fig. 7, A and B; and Table 1). The concentration at which activation was half maximal (EC50) increased from 9 µM in the absence of the drug to 560 µM in its presence, and maximal activation was reduced to 30% of that in the absence of gliclazide. The smaller extent of activation suggests that gliclazide impairs the mechanism by which MgADP binding is communicated to the channel gate on Kir6.2. The mean time constant for MgADP deactivation (at 3 mM MgADP) decreased from 1.1 ± 0.1 s in the absence of gliclazide to 0.4 ± 0.1 s in its presence (n = 8; P < 0.01; not depicted).
Noise analysis of Kir6.2-G334D/SUR1 currents indicated that the drug reduced the ability of 1 mM MgADP (which produces near-maximal activation) to increase both the number of functional channels and the mean PO of active channels. In the presence of gliclazide and MgADP, the increase in the number of functional channels was only 42% of that in the presence of MgADP alone. The PO was 0.81 ± 0.02 in the absence and 0.59 ± 0.04 in the presence of 30 µM gliclazide when measured in the same patch (n = 6; 1 mM MgADP was present throughout).
Gliclazide also dramatically reduced MgATP activation (Fig. 7, C and D). At MgATP concentrations >30 mM, a residual inhibition prevented accurate estimation of the maximal stimulatory effect of MgATP in the presence of gliclazide. To fit the data we therefore assumed that in the presence of gliclazide MgATP produces the same maximal activation as MgADP (30%; Fig. 7 B). This does not seem unreasonable, as maximal activation is the same for MgADP and MgATP in the absence of gliclazide (Proks et al., 2010). When maximal activation was fixed at 30%, the EC50 was 8 mM in the presence of gliclazide compared with 124 µM in its absence. The relative increase in EC50 induced by gliclazide (∼80-fold) was similar to that observed for MgADP (∼70-fold). The time constant for 3 mM MgATP deactivation on washout of nucleotide was lower than that for MgADP and unaffected by gliclazide, being 1.9 ± 0.3 s in its absence and 1.7 ± 0.3 s (n = 8) in its presence (not depicted). This suggests the rate of MgATP deactivation is dominated by processes other than MgADP unbinding/deactivation.
It is noteworthy that at MgATP concentrations found in β-cells (1–10 mM) gliclazide did not completely abolish activation of Kir6.2-G334D/SUR1 currents. Thus, in the native environment, a small degree of nucleotide activation (<25% of maximal) may persist in the presence of the sulfonylurea.
The effects of mutations in the NBSs on nucleotide activation
It is well established that mutation of the WA lysine in either NBS1 or NBS2 of SUR1 markedly reduces the ability of MgADP to stimulate wild-type channels or to reverse the inhibition produced by MgATP (Gribble et al., 1997b). We therefore explored whether the effect of these mutations resembled that produced by gliclazide. We mutated the WA lysine to alanine in NBS1 (K1A) or NBS2 (K2A) or both NBS1 and NBS2 simultaneously (KAKA). To quantify the effect of these mutations on nucleotide activation in the absence of inhibition at Kir6.2, we coexpressed SUR1-KAKA, SUR1-K1A, and SUR1-K2A with Kir6.2-G334D. We refer to these channels as KAKA, K1A, and K2A channels.
The KAKA mutation abolished channel activation by MgADP (Fig. 8 A). Both the K1A and K2A mutations also markedly impaired channel activation. Although it was not possible to measure the concentration-activation curve fully, because of inhibition at MgADP concentrations >1 mM, it is clear that the EC50 is strikingly increased. It is also noteworthy that the effect of the K1A and K2A mutations was very similar to that of gliclazide (Fig. 8 A, dotted line).
The KAKA mutation also largely abolished channel activation by MgATP (Fig. 8 B). In striking contrast to their effects on MgADP activation, however, the K1A and K2A mutations had only a minor effect on the EC50 for MgATP activation (Fig. 8 B), although they reduced maximal activation by 55% and 68%, respectively. Thus, gliclazide produces a far greater reduction in MgATP activation than either the K1A or K2A mutations.
DISCUSSION
Our results provide a quantitative description of the complex interaction between the sulfonylureas gliclazide and glibenclamide and the Mg-nucleotides MgADP and MgATP on the activity of recombinant β-cell (Kir6.2/SUR1) and cardiac (Kir6.2/SUR2A) KATP channels.
We show that MgATP as well as MgADP produces an apparent increase in both gliclazide and glibenclamide inhibition of Kir6.2/SUR1 channels but reduces inhibition of Kir6.2/SUR2A and Kir6.2/SUR2A-YS channels. Our data provide strong evidence in support of the idea that sulfonylureas suppress Mg-nucleotide activation of Kir6.2/SUR1 channels and so lead to an “apparent” increase in drug inhibition when MgATP or MgADP is present. They also provide insight into nucleotide handling by the NBSs of SUR1 and how this is affected by mutation of the WA lysines.
Nucleotide regulation of sulfonylurea inhibition
MgADP decreased the effect of gliclazide and glibenclamide on SUR2A-containing channels at all drug concentrations. This can be explained by the effect of the nucleotide on the single-channel kinetics. As discussed above, it is well established that mutations, or agents, that stabilize the open state of the channel reduce the maximal extent of high-affinity sulfonylurea inhibition (Trapp et al., 1998; Koster et al., 1999; Krauter et al., 2001). Because MgADP at concentrations <100 µM increases the single-channel open probability (PO), it is predicted to impair sulfonylurea inhibition. We simulated the effect of PO using a MWC model (Proks et al., 2013). We found that the reduction in gliclazide inhibition of Kir6.2/SUR2A-YS produced by MgADP predicted an increase in PO that was very close to that observed experimentally (Fig. S4; calculated PO = 0.79 and measured PO = 0.79). Similar results were found for the effect of MgADP on glibenclamide inhibition of Kir6.2/SUR2A and for gliclazide inhibition of Kir6.2-G334D/SUR2A-YS. We point out that the Kir6.2-G334D mutation renders the channel insensitive to ATP inhibition, so that MgATP increases Kir6.2-G334D/SUR2A-YS channel activity; in native channels, of course, ATP binding to Kir6.2 would produce a profound inhibition.
The effect of Mg-nucleotides on gliclazide inhibition of Kir6.2-G334D/SUR1 was concentration dependent. At drug concentrations from ∼10 to 400 nM, MgATP reduced gliclazide inhibition. It is possible that this results from the enhanced PO produced by the nucleotide, as explained above, and/or from displacement of gliclazide binding by MgADP (Hambrock et al., 2002). At higher drug concentrations, however, MgATP strongly enhanced gliclazide inhibition of Kir6.2G334D/SUR1. We attribute this to the fact that gliclazide largely abolished MgATP activation of channel activity, thereby revealing the full extent of the inhibitory effect of ATP at Kir6.2.
In the case of MgADP, no significant reduction in gliclazide or glibenclamide inhibition of Kir6.2/SUR1 was observed at low drug concentrations. At high concentrations, MgADP enhanced current inhibition, which is attributable to the fact that both sulfonylureas abolished MgADP activation of Kir6.2/SUR1.
Why sulfonylureas prevent MgADP activation of Kir6.2/SUR1 but not Kir6.2/SUR2A-YS channels remains unclear. One possibility is that this reflects differences in the sulfonylurea-binding site and/or how this site couples to the NBS2 of SUR. Another is that it reflects differences in how nucleotide binding at NBS2 of SUR couples to Kir6.2.
Effects of gliclazide on Mg-nucleotide interactions with SUR1
Several mechanisms for the ability of gliclazide to impair MgADP activation of Kir6.2/SUR1 channels can be postulated: (a) the drug might displace MgADP from one or both NBSs of SUR1, (b) it might impair the transduction of MgADP binding into channel activation, or (c) its effect might be mediated indirectly, via a change in the equilibrium between gating states. Our data favor the idea that both nucleotide binding and transduction are impaired, but changes in gating equilibrium play little, if any, role.
The dramatic decrease in the maximal extent of MgADP activation produced by gliclazide is consistent with the idea that transduction of MgADP binding into channel activation is impaired by the drug. This is because all NBSs will be occupied by MgADP at concentrations that induce maximal nucleotide activation (note this will occur at much higher MgADP concentrations in the presence of the drug). We consider it less likely that a transduction defect can also account for the ∼60-fold increase in the EC50 for MgADP activation produced by gliclazide. This is because current evidence supports a concerted model of KATP channel gating and simple versions of such a model predict that a transduction defect would cause only minor changes in EC50 (see Fig. S5 for details). However, the possibility that impaired transduction contributes to the shift in EC50 cannot be completely excluded as KATP channel gating may be much more complex than in our model and the transduction mechanism that couples ligand binding to channel gating may be different for different ligands.
We also consider the changes in gating produced by sulfonylurea inhibition are unlikely to produce substantial shifts in the EC50 for Mg-nucleotide activation. This is primarily because the EC50 (in the absence of gliclazide) showed no obvious dependence on the PO measured before nucleotide application (Fig. S6 A): the PO ranged from 0.2 to 0.6, similar to the range of PO (0.1–0.4) observed in the presence of 30 µM gliclazide (see supplemental text for further discussion).
The marked reduction in the EC50 for MgADP activation produced by gliclazide is most simply explained by assuming that the drug decreases MgADP binding to SUR1. There is evidence that MgATP and MgADP impair glibenclamide binding to SUR1 (Schwanstecher et al., 1991, 1992; Ashcroft and Ashcroft, 1992; Russ et al., 2001; Hambrock et al., 2002; Ortiz et al., 2012), and from the principle of microscopic reversibility, it is therefore expected that sulfonylureas will impair Mg-nucleotide binding. Indeed, glibenclamide enhances dissociation of prebound 8-azido-ATP from the NBS1 of SUR1 when Mg-nucleotides are present (Ueda et al., 1999). It was previously proposed that this is because the presence of MgADP at NBS2 of SUR1 stabilizes high-affinity 8-azido-ATP binding to NBS1 and that this effect is suppressed by glibenclamide. Our data suggest that sulfonylureas might reduce this stabilizing effect by impairing MgADP binding to NBS2. Studies of equilibrium binding of MgADP and MgATP to the octameric KATP channel complex are now required to test this idea directly.
In contrast to gliclazide, the KA mutations had no obvious effect on PO in the absence of nucleotides (Table S1). These mutations also had little effect on the EC50 for MgATP. However, they dramatically affected the EC50 for MgADP, which strongly suggests they may influence MgADP binding. Thus, it is unlikely that the KA mutations affect the EC50 for Mg-nucleotide activation via changes in gating. In summary, our data favor the idea that the dramatic changes in EC50 we observe for both gliclazide and the KA mutations are primarily caused by reduced Mg-nucleotide binding and/or transduction.
Effects of WA mutations on Mg-nucleotide activation
Upon nucleotide binding, the nucleotide-binding domains (NBDs) of ABC proteins dimerize in a head-to-tail arrangement to form two NBSs at the dimer interface (Schneider and Hunke, 1998). Structural studies reveal that in the “closed” dimer the conserved WA lysine forms extensive hydrogen bonds with the terminal phosphate of ATP. Nucleotide hydrolysis breaks this dimer interaction, resulting in an “open” NBD. In other ATPases, the WA lysine is crucial not only for ATP hydrolysis, but also for nucleotide binding (reviewed by Hanson and Whiteheart [2005]).
We found that mutation of the WA lysine in either NBS1 or NBS2 of SUR1 produced a marked increase in the EC50 for MgADP activation. This might result from a reduction in MgADP binding, and/or a change in the mechanism by which binding is translated into channel activation. We favor the former idea, as previous studies have shown that mutation of lysine in NBS2 to methionine decreased MgADP binding to NBS2 of SUR1 (Ueda et al., 1997). It is not possible to say whether transduction is also affected because the maximal extent of Mg-nucleotide activation could not be determined.
In contrast to what was found for MgADP, the EC50 for MgATP activation was unaltered by mutation of the WA lysine in either NBS1 or NBS2 of SUR1. This suggests the mutations may not affect MgATP binding. A previous study has demonstrated that MgATP hydrolysis is reduced by the KA mutations (de Wet et al., 2007), which may underlie the reduction in the maximal extent of activation we observed. The dramatic decrease in the maximal extent of MgATP activation produced by the KAKA mutation suggests that mutation of both NBSs simultaneously may further impair the rate of MgATP hydrolysis and/or the transduction of MgATP binding into channel activation. Why the same mutation has such different effects on MgATP and MgADP handling is still unresolved and will probably require a high-resolution x-ray structure to determine.
Modeling the NBS catalytic cycle
A deeper understanding of the effects of gliclazide and the WA mutations on nucleotide activation requires detailed analysis of the NBS catalytic cycle. As a first approximation, we computed the MgADP occupancy of NBS2 of SUR1 (Fig. 9, A and B), using rate constants obtained for the catalytic cycle of SUR2A-NBD2 by Bienengraeber et al. (2004) and Scheme 1 described in Materials and methods.
Figure 9.

Simulations of the fractional occupancy of NBS2 by MgADP. (A and B) Simulations of the fractional occupancy of NBS2 by MgADP in the presence of either MgADP (A; Eq. 5) or MgATP (B; Eq. 6). The model and the values of the rate constants were taken from Bienengraeber et al. (2004). Calculated control curves for MgADP and MgATP predicted EC50 values for MgADP occupancy of NBS2 that were very similar to those measured experimentally for channel activation. (A) To simulate the effect of gliclazide or the K1A and K2A mutations (KA mutations) on MgADP activation, we assumed a threefold increase in the off rate for MgADP binding in the presence of gliclazide (as measured experimentally) or when K1A or K2A was mutated. A 30-fold decrease in the on rate of MgADP binding in the presence of gliclazide (or the K1A or K2A mutation) predicted the measured EC50 for MgADP activation in the presence of gliclazide. (B) We used the same values for the rate constants for MgADP binding as in A and assumed the binding affinity of MgATP was reduced by gliclazide to the same extent as MgADP binding. This resulted in a predicted EC50 for the fractional occupancy of NBS2 by MgADP in the presence of gliclazide of 8 mM, which was the same as that measured experimentally for MgATP activation. To model the effect of the K1A and K2A mutations, we assumed the mutations had no effect on MgATP binding (as observed experimentally) and that they reduced the rate of ATP hydrolysis 100-fold. This reduced the maximal fractional occupancy of NBS2 by MgADP, but had no effect on the EC50 (as observed experimentally).
We found a striking agreement between the predicted half-maximal occupancy of NBS2 by MgADP (8.6 µM) and the measured EC50 for MgADP activation of Kir6.2-G334D/SUR1 channels (9 µM). Likewise, half-maximal activation of Kir6.2-G334D/SUR1 by MgATP (124 µM) was similar to the calculated half-maximal occupancy of NBS2 by MgADP (110 µM) in MgATP solution. The latter is also close to the Km for MgATP hydrolysis by rat SUR1 (100 ± 30 µM; de Wet et al., 2007) and not substantially different from that for ATP binding to NBD2 of hamster SUR1 (60 ± 36 µM; Matsuo et al., 2000). This provides support for the idea that the EC50 for nucleotide activation of Kir6.2-G334D/SUR1 channels is primarily determined by the properties of the catalytic cycle at NBD2 and little affected by gating.
A shift in the half-maximal occupancy of NBS2 by MgADP, equivalent to that seen for channel activation in the presence of gliclazide, can be simulated by assuming that the sulfonylurea impairs MgADP and MgATP binding with equal efficacy (compare Fig. 7 with Fig. 9). To account for the reduction in maximal activation by MgADP it is necessary to postulate that the translation of receptor occupancy into pore opening is also impaired by gliclazide (by ∼70%).
We were able to reproduce the effects of the K1A and K2A mutations in our simulations if we assumed that they (a) impair MgADP binding at NBS2 to the same extent as gliclazide and (b) the mutations reduce MgATP hydrolysis but not MgATP binding (Fig. 9). The latter assumption is supported by ATP hydrolysis measurements on purified SUR1 (de Wet et al., 2007). To simulate the extent of maximal channel activation by MgATP, it was necessary to assume that the KA mutations (like gliclazide) have a much greater effect on the on rate of MgADP than that of MgADP dissociation. These simulations provide further support for the idea that the KA mutations impair MgADP, but not MgATP, binding to SUR1.
Therapy
The nucleotide regulation of sulfonylurea inhibition has important therapeutic implications. Therapeutic concentrations of gliclazide in the plasma of patients with type 2 diabetes are around 10 µM (Abdelmoneim et al., 2012), and they will be even higher in patients with neonatal diabetes. Free concentrations of the drug are expected to be substantially lower, as >95% of drug is bound to plasma proteins (Balant, 1981). Nevertheless, even at a concentration of 500 nM (5% free drug), nucleotides influence gliclazide inhibition of Kir6.2/SUR1 and Kir6.2/SUR2A-YS.
Cardiac KATP channels are thought to be largely closed under normoxic, normoglycemic conditions but open in response to ischemia and the transient hypoxia that occurs during cardiac contraction (as the coronary supply is reduced by compression of the coronary artery). Although gliclazide does not inhibit wild-type cardiac KATP channels, other sulfonylureas (e.g., glibenclamide) that do so are also modulated by Mg-nucleotides. Intracellular ATP normally lies in the millimolar range in cardiac myocytes, and during ischemia MgADP can rise to over 100 µM (Jennings and Reimer, 1991). This may explain why cardiac side effects of sulfonylureas are few: under resting conditions, the channels are shut and when they open during ischemia, intracellular MgADP renders the sulfonylurea substantially less effective.
In pancreatic β-cells, the combined concentrations of intracellular MgATP and MgADP will always be >100 µM, and thus sufficient to enhance gliclazide inhibition. It has been proposed that MgADP regulation of sulfonylurea inhibition may result in drug action varying with the metabolism of the cell, as MgADP levels rise when metabolism is low (Ghosh et al., 1991; Detimary et al., 1998; Ronner et al., 2001). However, our results suggest that even if this does occur, it is unlikely to be of functional importance because MgATP is also effective at enhancing gliclazide inhibition.
Supplementary Material
Acknowledgments
We thank Drs. C. Miller, M. Puljung, and G. Sachse for critical comments on the manuscript.
We thank the Wellcome Trust (grant no. 089795/Z/09/Z), the Royal Society, and the European Union (LSHM-CT-2006-518153 and 332620) for support. H. de Wet was a European Foundation for the Study of Diabetes/Lilly Research Fellow. F.M. Ashcroft holds a Royal Society-Wolfson Research Merit Award.
The authors declare no competing financial interests.
Lawrence G. Palmer served as editor.
Footnotes
Abbreviations used in this paper:
- MWC
- Monod–Wyman–Changeux
- NBD
- nucleotide-binding domain
- NBS
- nucleotide-binding site
- TM
- transmembrane domain
- WA
- Walker A
References
- Abdelmoneim, A.S., Hasenbank S.E., Seubert J.M., Brocks D.R., Light P.E., and Simpson S.H.. 2012. Variations in tissue selectivity amongst insulin secretagogues: a systematic review. Diabetes Obes. Metab. 14:130–138. 10.1111/j.1463-1326.2011.01496.x [DOI] [PubMed] [Google Scholar]
- Abraham, M.R., Selivanov V.A., Hodgson D.M., Pucar D., Zingman L.V., Wieringa B., Dzeja P.P., Alekseev A.E., and Terzic A.. 2002. Coupling of cell energetics with membrane metabolic sensing. Integrative signaling through creatine kinase phosphotransfer disrupted by M-CK gene knock-out. J. Biol. Chem. 277:24427–24434. 10.1074/jbc.M201777200 [DOI] [PubMed] [Google Scholar]
- Aguilar-Bryan, L., Nichols C.G., Wechsler S.W., Clement J.P. IV, Boyd A.E. III, González G., Herrera-Sosa H., Nguy K., Bryan J., and Nelson D.A.. 1995. Cloning of the beta cell high-affinity sulfonylurea receptor: a regulator of insulin secretion. Science. 268:423–426. 10.1126/science.7716547 [DOI] [PubMed] [Google Scholar]
- Alekseev, A.E., Brady P.A., and Terzic A.. 1998. Ligand-insensitive state of cardiac ATP-sensitive K+ channels. Basis for channel opening. J. Gen. Physiol. 111:381–394. 10.1085/jgp.111.2.381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashcroft, F.M., and Rorsman P.. 2013. KATP channels and islet hormone secretion: new insights and controversies. Nat. Rev. Endocrinol. 9:660–669. 10.1038/nrendo.2013.166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashcroft, F.M., Harrison D.E., and Ashcroft S.J.. 1984. Glucose induces closure of single potassium channels in isolated rat pancreatic β-cells. Nature. 312:446–448. 10.1038/312446a0 [DOI] [PubMed] [Google Scholar]
- Ashcroft, S.J., and Ashcroft F.M.. 1992. The sulfonylurea receptor. Biochim. Biophys. Acta. 1175:45–59. 10.1016/0167-4889(92)90008-Y [DOI] [PubMed] [Google Scholar]
- Ashfield, R., Gribble F.M., Ashcroft S.J., and Ashcroft F.M.. 1999. Identification of the high-affinity tolbutamide site on the SUR1 subunit of the KATP channel. Diabetes. 48:1341–1347. 10.2337/diabetes.48.6.1341 [DOI] [PubMed] [Google Scholar]
- Balant, L.1981. Clinical pharmacokinetics of sulphonylurea hypoglycaemic drugs. Clin. Pharmacokinet. 6:215–241. 10.2165/00003088-198106030-00003 [DOI] [PubMed] [Google Scholar]
- Barrett-Jolley, R., and Davies N.W.. 1997. Kinetic analysis of the inhibitory effect of glibenclamide on KATP channels of mammalian skeletal muscle. J. Membr. Biol. 155:257–262. 10.1007/s002329900178 [DOI] [PubMed] [Google Scholar]
- Bienengraeber, M., Olson T.M., Selivanov V.A., Kathmann E.C., O’Cochlain F., Gao F., Karger A.B., Ballew J.D., Hodgson D.M., Zingman L.V., et al. 2004. ABCC9 mutations identified in human dilated cardiomyopathy disrupt catalytic KATP channel gating. Nat. Genet. 36:382–387. 10.1038/ng1329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokvist, K., Ammälä C., Ashcroft F.M., Berggren P.O., Larsson O., and Rorsman P.. 1991. Separate processes mediate nucleotide-induced inhibition and stimulation of the ATP-regulated K+-channels in mouse pancreatic β-cells. Proc. Biol. Sci. 243:139–144. 10.1098/rspb.1991.0022 [DOI] [PubMed] [Google Scholar]
- Dabrowski, M., Wahl P., Holmes W.E., and Ashcroft F.M.. 2001. Effect of repaglinide on cloned beta cell, cardiac and smooth muscle types of ATP-sensitive potassium channels. Diabetologia. 44:747–756. 10.1007/s001250051684 [DOI] [PubMed] [Google Scholar]
- de Wet, H., Mikhailov M.V., Fotinou C., Dreger M., Craig T.J., Vénien-Bryan C., and Ashcroft F.M.. 2007. Studies of the ATPase activity of the ABC protein SUR1. FEBS J. 274:3532–3544. 10.1111/j.1742-4658.2007.05879.x [DOI] [PubMed] [Google Scholar]
- Detimary, P., Dejonghe S., Ling Z., Pipeleers D., Schuit F., and Henquin J.C.. 1998. The changes in adenine nucleotides measured in glucose-stimulated rodent islets occur in β cells but not in α cells and are also observed in human islets. J. Biol. Chem. 273:33905–33908. 10.1074/jbc.273.51.33905 [DOI] [PubMed] [Google Scholar]
- Drain, P., Li L., and Wang J.. 1998. KATP channel inhibition by ATP requires distinct functional domains of the cytoplasmic C terminus of the pore-forming subunit. Proc. Natl. Acad. Sci. USA. 95:13953–13958. 10.1073/pnas.95.23.13953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh, A., Ronner P., Cheong E., Khalid P., and Matschinsky F.M.. 1991. The role of ATP and free ADP in metabolic coupling during fuel-stimulated insulin release from islet beta-cells in the isolated perfused rat pancreas. J. Biol. Chem. 266:22887–22892 [PubMed] [Google Scholar]
- Gribble, F.M., and Ashcroft F.M.. 1999. Differential sensitivity of beta-cell and extrapancreatic KATP channels to gliclazide. Diabetologia. 42:845–848. 10.1007/s001250051236 [DOI] [PubMed] [Google Scholar]
- Gribble, F.M., and Reimann F.. 2003. Sulphonylurea action revisited: the post-cloning era. Diabetologia. 46:875–891. 10.1007/s00125-003-1143-3 [DOI] [PubMed] [Google Scholar]
- Gribble, F.M., Tucker S.J., and Ashcroft F.M.. 1997a. The interaction of nucleotides with the tolbutamide block of cloned ATP-sensitive K+ channel currents expressed in Xenopus oocytes: a reinterpretation. J. Physiol. 504:35–45. 10.1111/j.1469-7793.1997.00035.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gribble, F.M., Tucker S.J., and Ashcroft F.M.. 1997b. The essential role of the Walker A motifs of SUR1 in K-ATP channel activation by Mg-ADP and diazoxide. EMBO J. 16:1145–1152. 10.1093/emboj/16.6.1145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gribble, F.M., Tucker S.J., Haug T., and Ashcroft F.M.. 1998a. MgATP activates the β cell KATP channel by interaction with its SUR1 subunit. Proc. Natl. Acad. Sci. USA. 95:7185–7190. 10.1073/pnas.95.12.7185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gribble, F.M., Tucker S.J., Seino S., and Ashcroft F.M.. 1998b. Tissue specificity of sulfonylureas: studies on cloned cardiac and beta-cell KATP channels. Diabetes. 47:1412–1418. 10.2337/diabetes.47.9.1412 [DOI] [PubMed] [Google Scholar]
- Hambrock, A., Löffler-Walz C., and Quast U.. 2002. Glibenclamide binding to sulphonylurea receptor subtypes: dependence on adenine nucleotides. Br. J. Pharmacol. 136:995–1004. 10.1038/sj.bjp.0704801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen, A.M.K., Christensen I.T., Hansen J.B., Carr R.D., Ashcroft F.M., and Wahl P.. 2002. Differential interactions of nateglinide and repaglinide on the human β-cell sulphonylurea receptor 1. Diabetes. 51:2789–2795. 10.2337/diabetes.51.9.2789 [DOI] [PubMed] [Google Scholar]
- Hansen, A.M.K., Hansen J.B., Carr R.D., Ashcroft F.M., and Wahl P.. 2005. Kir6.2-dependent high-affinity repaglinide binding to β-cell KATP channels. Br. J. Pharmacol. 144:551–557. 10.1038/sj.bjp.0706082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson, P.I., and Whiteheart S.W.. 2005. AAA+ proteins: have engine, will work. Nat. Rev. Mol. Cell Biol. 6:519–529. 10.1038/nrm1684 [DOI] [PubMed] [Google Scholar]
- Hopkins, W.F., Fatherazi S., Peter-Riesch B., Corkey B.E., and Cook D.L.. 1992. Two sites for adenine-nucleotide regulation of ATP-sensitive potassium channels in mouse pancreatic β-cells and HIT cells. J. Membr. Biol. 129:287–295. 10.1007/BF00232910 [DOI] [PubMed] [Google Scholar]
- Inagaki, N., Gonoi T., Clement J.P., Wang C.Z., Aguilar-Bryan L., Bryan J., and Seino S.. 1996. A family of sulfonylurea receptors determines the pharmacological properties of ATP-sensitive K+ channels. Neuron. 16:1011–1017. 10.1016/S0896-6273(00)80124-5 [DOI] [PubMed] [Google Scholar]
- Jennings, R.B., and Reimer K.A.. 1991. The cell biology of acute myocardial ischemia. Annu. Rev. Med. 42:225–246. 10.1146/annurev.me.42.020191.001301 [DOI] [PubMed] [Google Scholar]
- Koster, J.C., Sha Q., and Nichols C.G.. 1999. Sulfonylurea and K+-channel opener sensitivity of KATP channels. Functional coupling of Kir6.2 and SUR1 subunits. J. Gen. Physiol. 114:203–213. 10.1085/jgp.114.2.203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krauter, T., Ruppersberg J.P., and Baukrowitz T.. 2001. Phospholipids as modulators of KATP channels: distinct mechanisms for control of sensitivity to sulphonylureas, K+ channel openers, and ATP. Mol. Pharmacol. 59:1086–1093 [PubMed] [Google Scholar]
- Kühner, P., Prager R., Stephan D., Russ U., Winkler M., Ortiz D., Bryan J., and Quast U.. 2012. Importance of the Kir6.2 N-terminus for the interaction of glibenclamide and repaglinide with the pancreatic KATP channel. Naunyn Schmiedebergs Arch. Pharmacol. 385:299–311. 10.1007/s00210-011-0709-8 [DOI] [PubMed] [Google Scholar]
- Lawrence, C.L., Proks P., Rodrigo G.C., Jones P., Hayabuchi Y., Standen N.B., and Ashcroft F.M.. 2001. Gliclazide produces high-affinity block of KATP channels in mouse isolated pancreatic beta cells but not rat heart or arterial smooth muscle cells. Diabetologia. 44:1019–1025. 10.1007/s001250100595 [DOI] [PubMed] [Google Scholar]
- Li, L., Geng X., and Drain P.. 2002. Open state destabilization by ATP occupancy is mechanism speeding burst exit underlying KATP channel inhibition by ATP. J. Gen. Physiol. 119:105–116. 10.1085/jgp.119.1.105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Löffler-Walz, C., Hambrock A., and Quast U.. 2002. Interaction of KATP channel modulators with sulfonylurea receptor SUR2B: implication for tetramer formation and allosteric coupling of subunits. Mol. Pharmacol. 61:407–414. 10.1124/mol.61.2.407 [DOI] [PubMed] [Google Scholar]
- Masia, R., Koster J.C., Tumini S., Chiarelli F., Colombo C., Nichols C.G., and Barbetti F.. 2007. An ATP-binding mutation (G334D) in KCNJ11 is associated with a sulfonylurea-insensitive form of developmental delay, epilepsy, and neonatal diabetes. Diabetes. 56:328–336. 10.2337/db06-1275 [DOI] [PubMed] [Google Scholar]
- Matsuo, M., Tanabe K., Kioka N., Amachi T., and Ueda K.. 2000. Different binding properties and affinities for ATP and ADP among sulfonylurea receptor subtypes, SUR1, SUR2A, and SUR2B. J. Biol. Chem. 275:28757–28763. 10.1074/jbc.M004818200 [DOI] [PubMed] [Google Scholar]
- Nichols, C.G., Shyng S.L., Nestorowicz A., Glaser B., Clement J.P. IV, Gonzalez G., Aguilar-Bryan L., Permutt M.A., and Bryan J.. 1996. Adenosine diphosphate as an intracellular regulator of insulin secretion. Science. 272:1785–1787. 10.1126/science.272.5269.1785 [DOI] [PubMed] [Google Scholar]
- Ortiz, D., Voyvodic P., Gossack L., Quast U., and Bryan J.. 2012. Two neonatal diabetes mutations on transmembrane helix 15 of SUR1 increase affinity for ATP and ADP at nucleotide binding domain 2. J. Biol. Chem. 287:17985–17995. 10.1074/jbc.M112.349019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson, E.R., Flechtner I., Njølstad P.R., Malecki M.T., Flanagan S.E., Larkin B., Ashcroft F.M., Klimes I., Codner E., Iotova V., et al. . Neonatal Diabetes International Collaborative Group. 2006. Switching from insulin to oral sulfonylureas in patients with diabetes due to Kir6.2 mutations. N. Engl. J. Med. 355:467–477. 10.1056/NEJMoa061759 [DOI] [PubMed] [Google Scholar]
- Proks, P., Reimann F., Green N., Gribble F., and Ashcroft F.. 2002. Sulfonylurea stimulation of insulin secretion. Diabetes. 51:S368–S376. 10.2337/diabetes.51.2007.S368 [DOI] [PubMed] [Google Scholar]
- Proks, P., Girard C., Haider S., Gloyn A.L., Hattersley A.T., Sansom M.S., and Ashcroft F.M.. 2005. A gating mutation at the internal mouth of the Kir6.2 pore is associated with DEND syndrome. EMBO Rep. 6:470–475. 10.1038/sj.embor.7400393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proks, P., de Wet H., and Ashcroft F.M.. 2010. Activation of the KATP channel by Mg-nucleotide interaction with SUR1. J. Gen. Physiol. 136:389–405. 10.1085/jgp.201010475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proks, P., de Wet H., and Ashcroft F.M.. 2013. Molecular mechanism of sulphonylurea block of KATP channels carrying mutations that impair ATP inhibition and cause neonatal diabetes. Diabetes. 62:3909–3919. 10.2337/db13-0531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimann, F., Dabrowski M., Jones P., Gribble F.M., and Ashcroft F.M.. 2003. Analysis of the differential modulation of sulphonylurea block of β-cell and cardiac ATP-sensitive K+ (KATP) channels by Mg-nucleotides. J. Physiol. 547:159–168. 10.1113/jphysiol.2002.031625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribalet, B., John S.A., and Weiss J.N.. 2000. Regulation of cloned ATP-sensitive K channels by phosphorylation, MgADP, and phosphatidylinositol bisphosphate (PIP2): a study of channel rundown and reactivation. J. Gen. Physiol. 116:391–410. 10.1085/jgp.116.3.391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronner, P., Naumann C.M., and Friel E.. 2001. Effects of glucose and amino acids on free ADP in βHC9 insulin-secreting cells. Diabetes. 50:291–300. 10.2337/diabetes.50.2.291 [DOI] [PubMed] [Google Scholar]
- Russ, U., Lange U., Löffler-Walz C., Hambrock A., and Quast U.. 2001. Interaction of the sulfonylthiourea HMR 1833 with sulfonylurea receptors and recombinant ATP-sensitive K+ channels: comparison with glibenclamide. J. Pharmacol. Exp. Ther. 299:1049–1055 [PubMed] [Google Scholar]
- Schneider, E., and Hunke S.. 1998. ATP-binding-cassette (ABC) transport systems: functional and structural aspects of the ATP-hydrolyzing subunits/domains. FEMS Microbiol. Rev. 22:1–20. 10.1111/j.1574-6976.1998.tb00358.x [DOI] [PubMed] [Google Scholar]
- Schwanstecher, M., Löser S., Rietze I., and Panten U.. 1991. Phosphate and thiophosphate group donating adenine and guanine nucleotides inhibit glibenclamide binding to membranes from pancreatic islets. Naunyn Schmiedebergs Arch. Pharmacol. 343:83–89. 10.1007/BF00180681 [DOI] [PubMed] [Google Scholar]
- Schwanstecher, M., Löser S., Brandt C., Scheffer K., Rosenberger F., and Panten U.. 1992. Adenine nucleotide-induced inhibition of binding of sulphonylureas to their receptor in pancreatic islets. Br. J. Pharmacol. 105:531–534. 10.1111/j.1476-5381.1992.tb09014.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyng, S., and Nichols C.G.. 1997. Octameric stoichiometry of the KATP channel complex. J. Gen. Physiol. 110:655–664. 10.1085/jgp.110.6.655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyng, S., Ferrigni T., and Nichols C.G.. 1997. Regulation of KATP channel activity by diazoxide and MgADP. Distinct functions of the two nucleotide binding folds of the sulfonylurea receptor. J. Gen. Physiol. 110:643–654. 10.1085/jgp.110.6.643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapp, S., Proks P., Tucker S.J., and Ashcroft F.M.. 1998. Molecular analysis of ATP-sensitive K channel gating and implications for channel inhibition by ATP. J. Gen. Physiol. 112:333–349. 10.1085/jgp.112.3.333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker, S.J., Gribble F.M., Zhao C., Trapp S., and Ashcroft F.M.. 1997. Truncation of Kir6.2 produces ATP-sensitive K+ channels in the absence of the sulphonylurea receptor. Nature. 387:179–183. 10.1038/387179a0 [DOI] [PubMed] [Google Scholar]
- Ueda, K., Inagaki N., and Seino S.. 1997. MgADP antagonism to Mg2+-independent ATP binding of the sulfonylurea receptor SUR1. J. Biol. Chem. 272:22983–22986. 10.1074/jbc.272.37.22983 [DOI] [PubMed] [Google Scholar]
- Ueda, K., Komine J., Matsuo M., Seino S., and Amachi T.. 1999. Cooperative binding of ATP and MgADP in the sulfonylurea receptor is modulated by glibenclamide. Proc. Natl. Acad. Sci. USA. 96:1268–1272. 10.1073/pnas.96.4.1268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesh, N., Lamp S.T., and Weiss J.N.. 1991. Sulfonylureas, ATP-sensitive K+ channels, and cellular K+ loss during hypoxia, ischemia, and metabolic inhibition in mammalian ventricle. Circ. Res. 69:623–637. 10.1161/01.RES.69.3.623 [DOI] [PubMed] [Google Scholar]
- Vila-Carriles, W.H., Zhao G., and Bryan J.. 2007. Defining a binding pocket for sulfonylureas in ATP-sensitive potassium channels. FASEB J. 21:18–25. 10.1096/fj.06-6730hyp [DOI] [PubMed] [Google Scholar]
- Winkler, M., Stephan D., Bieger S., Kühner P., Wolff F., and Quast U.. 2007. Testing the bipartite model of the sulfonylurea receptor binding site: binding of A-, B-, and A + B-site ligands. J. Pharmacol. Exp. Ther. 322:701–708. 10.1124/jpet.107.123224 [DOI] [PubMed] [Google Scholar]
- Zhang, H., Flagg T.P., and Nichols C.G.. 2010. Cardiac sarcolemmal KATP channels: Latest twists in a questing tale! J. Mol. Cell. Cardiol. 48:71–75. 10.1016/j.yjmcc.2009.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zingman, L.V., Alekseev A.E., Bienengraeber M., Hodgson D., Karger A.B., Dzeja P.P., and Terzic A.. 2001. Signaling in channel/enzyme multimers: ATPase transitions in SUR module gate ATP-sensitive K+ conductance. Neuron. 31:233–245. 10.1016/S0896-6273(01)00356-7 [DOI] [PubMed] [Google Scholar]
- Zünkler, B.J., Lenzen S., Männer K., Panten U., and Trube G.. 1988. Concentration-dependent effects of tolbutamide, meglitinide, glipizide, glibenclamide and diazoxide on ATP-regulated K+ currents in pancreatic B-cells. Naunyn Schmiedebergs Arch. Pharmacol. 337:225–230. 10.1007/BF00169252 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.