

A mild strain induced by matrix remodeling mechanically primes latent TGF-β1 for its subsequent activation and release in response to contractile forces.
Abstract
Integrin-mediated force application induces a conformational change in latent TGF-β1 that leads to the release of the active form of the growth factor from the extracellular matrix (ECM). Mechanical activation of TGF-β1 is currently understood as an acute process that depends on the contractile force of cells. However, we show that ECM remodeling, preceding the activation step, mechanically primes latent TGF-β1 akin to loading a mechanical spring. Cell-based assays and unique strain devices were used to produce a cell-derived ECM of controlled organization and prestrain. Mechanically conditioned ECM served as a substrate to measure the efficacy of TGF-β1 activation after cell contraction or direct force application using magnetic microbeads. The release of active TGF-β1 was always higher from prestrained ECM as compared with unorganized and/or relaxed ECM. The finding that ECM prestrain regulates the bioavailability of TGF-β1 is important to understand the context of diseases that involve excessive ECM remodeling, such as fibrosis or cancer.
Introduction
Myofibroblasts contribute to normal tissue repair by replacing and contracting the provisional ECM that fills tissue defects after injury (Hinz et al., 2012). When ECM remodeling activities of myofibroblasts are deregulated, repair proceeds into adverse and pathological fibrosis affecting all organs, including skin, heart, lung, liver, and kidney (Hinz et al., 2012; Wynn and Ramalingam, 2012). TGF-β1 is the most potent profibrotic cytokine known and the main growth factor inducing myofibroblast differentiation from a variety of different precursor cells (Hinz et al., 2007). Fibroblasts secrete TGF-β1 noncovalently associated with its latency-associated propeptide (LAP). This small latent complex covalently binds to the LTBP-1, an integral component of the ECM that stores and presents latent TGF-β1 for subsequent activation (Jenkins, 2008; Worthington et al., 2011; Zilberberg et al., 2012; Robertson and Rifkin, 2013). Binding of LAP to the ECM through the LTBP-1 is the structural precondition for mechanical activation by integrins (Annes et al., 2004; Wipff et al., 2007; Shi et al., 2011). The LTBP-1 binding site of LAP is directly opposite to the RGD site in LAP for integrin attachment; integrin-mediated force transmission induces a conformational change in LAP that liberates active TGF-β1 (Buscemi et al., 2011; Shi et al., 2011). All αv integrins bind to RGD in LAP (Jenkins, 2008; Wipff and Hinz, 2008; Nishimura, 2009; Henderson and Sheppard, 2013; Hinz, 2013). Integrins αvβ3, αvβ5, αvβ6, and possibly αvβ1 activate latent TGF-β1 by transmitting cell contractile forces (Wipff et al., 2007; Giacomini et al., 2012; Henderson et al., 2013).
We have previously demonstrated that the acute contractile state, i.e., the force exerted by fibroblastic cells, determines the quantity of TGF-β1 that is activated from the ECM (Wipff et al., 2007; Buscemi et al., 2011). Here, we propose that the changes in ECM organization produced by fibroblastic cells over days, weeks, and months in fibrotic lesions will augment the bioavailability of TGF-β1. We show that myofibroblasts mechanically prime TGF-β1 for activation by actively organizing the latent complex in the ECM during and after secretion, analogous to the loading of a mechanical spring. High levels of experimentally controlled ECM organization and mechanical load always resulted in high levels of TGF-β1 activated by acutely contracting myofibroblasts. Our results suggest that the excessive remodeling activity of fibroblastic cells in the early stages of tissue repair will set the stage for the development of fibrosis by adjusting the mechanical trigger point for latent TGF-β1 activation.
Results
Myofibroblast differentiation leads to increased ECM organization and TGF-β1 activation
To test whether de novo formation of myofibroblasts and increased tissue stress in vivo are associated with higher fibrillar organization of ECM in general and LTBP-1 in particular, we used a rat model of mechanically enhanced wound healing (Hinz et al., 2001b). The dermis of normal rat skin exhibited negligible levels of the fibronectin (FN) splice variant ED-A FN, and LTBP-1 and no α-smooth muscle actin (α-SMA)–positive myofibroblasts (Fig. 1 A). After dermal wounding, neoexpression of ED-A FN (day 3–4) preceded the first appearance of LTBP-1 and myofibroblasts (day 6–7) in the granulation tissue, with all proteins reaching peak expression at day 9 (Fig. 1 A). The alignment of ECM fibrils in parallel to the skin surface moderately increased over time of normal healing (Fig. 1 A). In contrast, mechanically restraining the wound edges with splints accelerated ED-A FN, LTBP-1, and α-SMA expression by ∼3 d and led to substantially higher fibril organization at any given time compared with normal wounds. Differences between normal and splinted wounds were most pronounced 9 d after wounding, as shown by quantifying LTBP-1 fibril density by image analysis (Fig. 1 A). Enhanced LTBP-1 organization correlated with the enhanced TGF-β1 downstream signaling (pSmad2/3 phosphorylation) and α-SMA expression reported in our previous studies using the same rat model (Hinz et al., 2001b; Wipff et al., 2007).
Figure 1.

Changes in ECM composition and structure during myofibroblast differentiation. (A) Granulation tissue sections of splinted and nonsplinted wounds from female Wistar rats were stained for α-SMA (blue), ED-A FN (red), and LTBP-1 (green). Normal rat skin is compared with 3-d-old nonsplinted wounds. Additionally, the arrowhead indicates the region of granulation tissue that is compared from splinted and nonsplinted wounds at day 3, 6, and 9. Insets show LTBP-1 only. Bars, 10 µm. (B) hDfs (control) were differentiated into hDMfs using 2 ng/ml TGF-β1 for 5 d in passage 1 (+TGF-β1) and used at passage 4 after 6 d of growth. Cells were labeled for α-SMA and F-actin and for LTBP-1, ED-A FN, and nuclei (blue). Bars, 50 µm. (C) Expression of ED-A FN, LTBP-1, vinculin, and α-SMA was determined by Western blotting of cells grown without (control [con]) and with TGF-β1 and quantified as a percentage of control normalized to vimentin as a loading control. (D) The mean density of fibril events per image field was quantified from 14 LTBP-1–stained images per three independent experiments. (E and F) Active TGF-β1 was measured by directly co-culturing fibroblastic cells with TMLC reporter cells either after inducing cell contraction with thrombin (E) or without inducing cell contraction (F, baseline). (G) Levels of active TGF-β1 are presented as a percentage of total TGF-β1 determined from heat-activated ECMs and cells. Graphs show mean values and SDs from at least three independent experiments (*, P ≤ 0.05; two-tailed paired t test). a.u., arbitrary unit.
To discriminate the role of the ECM strain in guiding fibrillar organization of LTBP-1 and availability of associated latent TGF-β1, we established controlled experimental conditions. High-contractile human dermal myofibroblasts (hDMfs) were generated by treating primary human dermal fibroblasts (hDfs) with active TGF-β1 once for 5 d in passage 1. TGF-β1 used to differentiate hDMfs was not detectable in passage 4 when hDMfs were assessed experimentally (Fig. S1, A and B). In contrast to the small population of cells (7 ± 5%) expressing the myofibroblast marker α-SMA in contractile stress fibers in standard culture conditions, the hDMf fraction represented 40 ± 15% of the cell population, even three passages after TGF-β1 stimulation (Fig. 1 B and Fig. S1, A and B). Higher protein expression levels of α-SMA in hDMfs correlated with higher expression of LTBP-1 and ED-A FN (Fig. 1, B and C). Organization of LTBP-1 and colocalization with ED-A FN in the myofibroblast ECM was threefold higher than in the ECM produced by low contractile hDfs, as quantified by LTBP-1 density in fibrils (Fig. 1, B and D). To compare the potential of hDfs and hDMfs to activate TGF-β from their differently organized ECM, we induced cell contraction with thrombin. Active TGF-β was measured using directly co-cultured transformed mink lung epithelial cells (TMLCs) and normalized to total levels of TGF-β measured in heat-activated cell/ECM lysates. TMLC reporting was abolished by adding TGF-β1–blocking antibodies to contraction-stimulated hDMf/TMLC co-cultures (Fig. S1 C), identifying TGF-β1 as the main isoform in hDMf cultures. Low concentrations of thrombin (0.5 U/ml) exerted no proteolytic action on either ECM or latent TGF-β1 (Fig. S1, D and E), confirming previous controls with other fibroblast sources (Wipff et al., 2007; Sarrazy et al., 2014). Thrombin-induced cell contraction and ECM deformation resulted in mechanical TGF-β1 activation similar to inducing cell contraction with TRAP-6 (thrombin receptor activating peptide 6), bearing no catalytic activity (Fig. S1, F and G). Contraction-induced active TGF-β1 levels were threefold higher in hDMf (36% of total TGF-β) than in hDf (11%) cultures (Fig. 1 E). Baseline levels of active TGF-β without addition of thrombin were low in both cell types (≤2%; Fig. 1 F). Total levels of TGF-β were only moderately higher in hDMf than in 6-d-old hDf cultures (Fig. 1 G).
Mechanical activation of TGF-β1 increases with increasing maturation of the ECM
Next, we tested whether the higher levels of active TGF-β1 in hDMf cultures are related to ECM properties rather than to the TGF-β1–activating fibroblast phenotype. hDfs and hDMfs were cultured to produce latent TGF-β1–containing ECM for 2, 3, and 6 d. LTBP-1 expression in hDMf cultures was detectable by 3 d and increased by 6 d; FN (total and ED-A FN) was detected already after 2 d and increased only moderately over culture time (Fig. 2 A). LTBP-1 and FN expression levels in hDf cultures followed the same time course but were lower than in hDMf cultures (not depicted and Fig. 1). The differently produced and matured ECMs were then decellularized using desoxycholate (DOC), and total TGF-β1 levels were measured. In the 3-d- and 6-d-old decellularized ECM, total TGF-β1 levels were similar and only moderately higher (1.25-fold) in hDMf than in hDf cultures (Fig. 2 B). However, when hDMfs were reseeded onto all decellularized ECMs and induced to contract, an approximately twofold increase in active TGF-β1 (percentage of total) was observed on the 6-d mature versus the 3-d immature ECM (Fig. 2 C). Moreover, approximately twofold more TGF-β1 was released by reseeded hDMfs from hDMf-remodeled ECM than from hDf-remodeled ECM of the same “age” (Fig. 2 C). To relate TGF-β1 activation with LTBP-1 organization in the ECM, we transfected EGFP-tagged LTBP-1 into hDMfs. LTBP-1–EGFP colocalized with endogenous LTBP-1 in the ECM (Fig. 2 D). In 2-d-old ECM, LTBP-1 was almost exclusively localized in patches at the substrate surface that became increasingly organized into ED-A FN-containing fibrils after 3- and 6-d culture (Fig. 2, E and F). These results indicate that the efficacy of mechanical activation of TGF-β1 correlates with the level of FN fibril formation and organization of the ECM in addition to the acute contractile state of the fibroblastic cells.
Figure 2.

ECM maturation and preremodeling affects TGF-β1 activation by myofibroblasts. (A) hDMfs were grown for 2, 3, and 6 d, and lysates were used for Western blotting with vimentin as a loading control. (B) hDf- or hDMf-derived ECMs were decellularized using DOC and heat activated before total amounts of TGF-β1 were determined by luciferase reporter assay. (C) hDMfs were seeded onto different preproduced ECMs and stimulated to contract with thrombin, and active TGF-β1 was measured as a percentage of total TGF-β1. (D) hDMfs were transfected with LTBP-1–EGFP and stained for LTBP-1 and EGFP after 12 d. (E) EGFP, ED-A FN, and nuclei (blue) were stained in hDMf cultures after 2, 3, and 6 d. Bottom rows show magnified ECM regions. (F) The mean density of LTBP-1–EGFP fibrils was quantified from stained images. Bars: (D) 150 µm; (E, top) 25 µm; (E, bottom) 5 µm. Graphs show mean values and SDs from at least three independent experiments (*, P ≤ 0.05; and ***, P ≤ 0.005 using ANOVA followed by a post-hoc Tukey’s multiple comparison test). a.u., arbitrary unit.
Myofibroblasts incorporate LTBP-1 into strained ECM fibrils in an integrin-mediated and actin–myosin-dependent process
Thus far, our results suggest that a higher degree of ECM organization contributes to the increased ability of hDMfs to generate active TGF-β1 compared with hDfs. To determine the remodeling capacities of hDMfs and hDfs independently of LTBP-1/ECM secretion, we preproduced LTBP-1–containing ECM in three different ways (Fig. 3). First, HEK293 (human embryonic kidney-293) cells were stably transfected with LTBP-1–EGFP and used to generate an ECM enriched in EGFP-tagged LTPB-1. The HEK293 cells were not able to organize FN or the secreted LTBP-1–EGFP into fibrils (Fig. 3 A). Second, at variance with the first setup, the ECM-producing HEK293 cells were removed using DOC, leaving behind LTBP-1–EGFP patches on the substrate surface (Fig. 3 B). Third, LTBP-1–EGFP was purified from HEK293 cell supernatants and used to coat culture substrates (Fig. 3 C and Fig. S2). Scanning electron microscopy demonstrated the ultrastructurally different ECM (Fig. 3). hDMfs and hDfs were then added to process the prefabricated ECM in FN-depleted medium for 2 d (Videos 1 and 2 and Fig. S3 A). During this time, the endogenous production of LTBP-1 was negligible (Fig. 2). Organization of the supplied LTBP-1–EGFP into ECM fibrils was significantly higher in hDMf than in hDf cultures in all three experimental models (Fig. 3).
Figure 3.

Myofibroblasts are more efficient in LTBP-1 ECM organization than fibroblasts. (A–C) LTBP-1 substrates were produced by using LTBP-1–EGFP-transfected HEK293 cells that were grown for 7 d (A), ECM was obtained after DOC treatment of LTBP-1–EGFP-transfected HEK293 cells that were grown for 7 d (B), and substrates coated with LTBP-1–EGFP were purified from supernatants of LTBP-1–EGFP-transfected HEK293 cells (C). The resulting ECM was used for scanning electron microscopy (SEM) and as substrates for hDfs and hDMfs. Fibroblastic cells were stained after 2-d growth for EGFP (green) and ED-A FN (red). Bars: (scanning electron microscopy images) 5 µm; (insets) 500 nm; (immunofluorescence images) 20 µm. LTBP-1–EGFP fibrils were quantified by image analysis from at least five images per three independent experiments to calculate mean values and SDs (***, P ≤ 0.005 using ANOVA followed by a post-hoc Tukey’s multiple comparison test). Insets show higher magnification views of cell-free ECM areas. a.u., arbitrary unit.
LTBP-1 incorporation into the ECM was previously shown to depend on FN secretion (Taipale et al., 1996; Dallas et al., 2000; Massam-Wu et al., 2010; Todorovic and Rifkin, 2012; Zilberberg et al., 2012), and binding of LTBP-1 to FN is essential for integrin-mediated activation of TGF-β1 (Annes et al., 2004). To test whether fibroblastic cells organize LTBP-1 directly or indirectly by hitchhiking on FN fibril formation, we knocked down FN in hDMfs using specific siRNA (Fig. S3 B). FN-deficient hDMfs were able to attach and spread onto LTBP-1–coated substrates but failed to form LTBP-1 fibrils (Fig. 4 A). Similar results were obtained using FN−/− mouse embryonic fibroblasts (MEFs; Fig. 4 B), confirming that the presence of FN is indeed crucial for LTBP-1 fibril formation. hDMfs produced higher amounts of FN than hDfs during LTBP-1 fibril formation (Fig. 1 and Fig. 3). To evaluate whether higher FN quantities accounted for the higher efficiency of hDMfs to produce LTBP-1–containing fibrils, we restricted the culture time of hDfs (Fig. 4 C) and hDMfs (Fig. 4 D) to 4 h on LTBP-1–EGFP-coated substrates in FN-free culture medium. Quantification of LTBP-1–EGFP immunostaining and video microscopy analysis (Video 3) demonstrated that hDMfs assembled LTBP-1–EGFP almost instantaneously into fibrils, which were always positive for cell-derived ED-A FN. In accordance with previous observations, the LTBP-1 fibril count was higher for hDMfs than for hDfs (Fig. 4, C and D; and Fig. S3 C). Importantly, hDMfs also incorporated approximately twofold more LTBP-1–EGFP into fibrils than hDfs when cultures were supplemented with an excess of plasma FN (100 µg/cm2), either added to the medium (not depicted) or adsorbed onto the culture surface together with LTBP-1–EGFP (Fig. 4, C and D; and Fig. S3 C).
Figure 4.
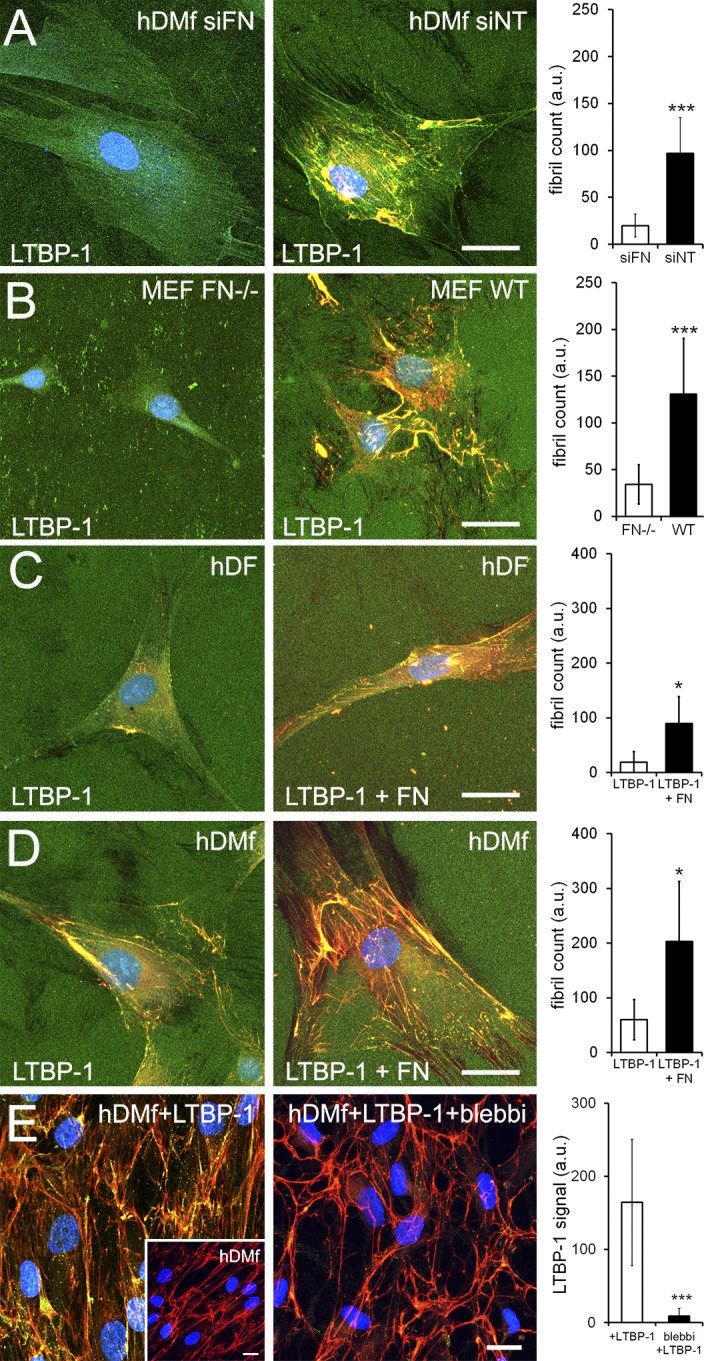
FN mediates LTBP-1 fibril formation and incorporation. (A and B) Substrates were coated with purified LTBP-1–EGFP and used to culture hDMfs transfected with siRNA against FN (siFN) or nonrelated control RNA (siNT; A) and FN−/− MEFs or wild-type (WT) MEFs (B). (C) hDfs and hDMfs were seeded onto substrates coated with LTBP-1–EGFP alone or LTBP-1–EGFP mixed with an excess of FN (100 µg/cm2) and grown for 4 h. (D) hDMfs were pregrown for 7 d before purified LTBP-1–EGFP was added to the culture medium for an additional 24 h in the presence or absence of 20 µM blebbistatin (blebbi). Experiments were performed in FN-depleted culture medium. All samples were stained for EGFP (green), FN (red), and nuclei (blue). (A–E) LTBP-1–EGFP fibrils (A–D) and staining intensity (E) were quantified by image analysis from at least five images per three independent experiments to calculate mean values and SDs (*, P ≤ 0.05; and ***, P ≤ 0.005 using ANOVA followed by a post-hoc Tukey’s multiple comparison test). Inset in E shows FN organization after 7 d of hDMfs without addition of LTBP-1–EGFP. Bars, 25 µm. a.u., arbitrary unit.
A previous study has demonstrated that LTBP-1 is increasingly transferred from FN to fibrillin-1 during fibroblast culture ECM maturation (Zilberberg et al., 2012). In our fibroblast cultures, FN and fibrillin-1 strongly colocalized ≤6 d of cell culture (Fig. S4 A). Moreover, mouse dermal myofibroblasts, being defective for fibrillin-1 incorporation into the ECM and deficient in LTBP-1 binding to fibrillin-1 (Judge et al., 2004) formed LTBP-1/FN fibrillar structures, indicating that FN was sufficient for initial LTBP-1 incorporation into ECM fibrils (Fig. S4, B and C). To test whether fibrillin-1 is important in mechanical priming of the ECM for TGF-β1 activation, we produced ECM using mouse dermal fibroblasts bearing a fibrillin-1 mutation in LTBP-1 binding. Fibrillin-1 mutant murine fibroblasts produced and organized LTBP-1 (Fig. S4 C); however, the amount of total TGF-β1 in the ECM was approximately fourfold lower than in wild-type cultures. Consequently, preorganization did not change the amount of TGF-β1 activated by hDMfs from fibrillin-1 mutant ECM, whereas preorganization improved TGF-β1 activation by 1.5-fold from wild-type mouse fibroblast ECM (Fig. S4 D).
We next investigated the incorporation of LTBP-1 into an established ECM network by adding purified, soluble LTBP-1–EGFP to hDMfs that were precultured for 7 d to produce their own ECM (Fig. 4 E). The supplemented LTBP-1–EGFP preferentially colocalized with preexisting ED-A FN fibrils (Fig. 4 E, inset), and the incorporation process was abolished in the presence of 20 µM blebbistatin (Fig. 4 E). Collectively, these results show that (a) the higher efficacy of hDMfs to form LTBP-1–containing fibrils is not caused by higher production of FN, (b) hDMfs incorporate exogenous LTBP-1 into the ECM during FN fibrillogenesis, and (c) this process is cell mediated and dependent on actin–myosin activity.
Because human LTBP-1 contains an RGD consensus site, it is conceivable that αv integrins participate in LTBP-1 incorporation into ECM fibrils and cell attachment to LTBP-1–coated substrates. To identify the integrin putatively mediating adhesion to LTBP-1, we generated microcontact printed islet arrays of purified LTBP-1–EGFP with typical focal adhesion features (10 × 4 × 1.5 µm). hDMfs adhered specifically to the printed LTBP-1 at sites of vinculin-positive focal adhesions when seeded for 4 h in serum-free medium (Fig. 5 A). Focal adhesions forming on LTBP-1 islets contained β1 integrin, to a lesser extent β3 integrin, and no integrin β5. Control prints using FN as a ligand demonstrated that all tested integrins were expressed in hDMfs and localized to focal adhesions (Fig. 5 B). Next, we seeded hDMfs onto fully LTBP-1–coated substrates for 4 h in the presence of specific integrin-blocking antibodies and different RGD peptides (Fig. 5 C). Focal adhesion formation and cell spreading were reduced by all integrin blocking antibodies in order from strongest to weakest: β1, β3, and β5 integrin. All competitive integrin-blocking peptides, but not controls, inhibited focal adhesion formation and cell spreading on LTBP-1 (Fig. 5 C). Strongest blocking was achieved with RGD as confirmed by quantifying the number of adherent hDMfs on LTBP-1–coated substrates (Fig. 5 D). These results indicate that hDMfs bind directly to the RGD sequence in LTBP-1 via integrins that possibly aid the active organization of LTBP-1 into ECM fibrils in a cell tension–dependent manner. Of the RGD-recognizing integrins expressed in hDMfs, β5 integrin seem to bind weakest by far.
Figure 5.

Integrins mediate binding of myofibroblasts to LTBP-1. (A and B) Arrays of an islet with typical focal adhesion features (10 × 4 × 1.5 µm) were microcontact printed using purified LTBP-1–EGFP (A) or plasma FN as a control (B). After attachment for 4 h to microcontact printed arrays in the absence of serum, hDMfs were immunostained for the printed protein (red, false colored for LTBP-1–EGFP), vinculin, and integrin subunits β1, β3, and β5 (green). Insets show higher magnification views of select focal adhesion. (C) hDMfs were seeded onto LTBP-1–EGFP-coated substrates in the presence of blocking antibodies or cyclic peptides against integrin subunits β1, β3, and β5 or the RGD site. Focal adhesion formation was visualized with vinculin (green), and nuclei were stained with DAPI (blue). (D) The number of adherent hDMfs on LTBP-1–EGFP-coated substrates was quantified by image analysis. Graph shows mean values and SDs from at least three independent experiments (***, P ≤ 0.005; two-tailed paired t test). con, control. Bars: (main images) 20 µm; (insets) 5 µm.
Incorporation of LTBP-1 into disorganized ECM impairs mechanical activation of TGF-β1
To further investigate how the higher preorganization of the hDMf ECM affects the efficacy of TGF-β1 activation, we generated ECM with MEFs exhibiting cell–ECM protein interaction defects. MEFs derived from mice deficient for FAK (FAK−/−; Rajshankar et al., 2012) and β1 integrin (β1−/−; Fässler and Meyer, 1995) and MEFs stably knocked down for filamin A (Kim et al., 2008) all produced disorganized ECM compared with wild-type MEF after 6 d of culture (Fig. S5 and Fig. 6 A). We continued with FAK−/− MEFs that produced a disorganized ECM with low FN fibril density (Fig. 6 A) but expressed LTBP-1 and FN at levels similar to those of wild-type MEFs (Fig. 6 B). This phenotype confirmed a previous study relating poor FN fibrillogenesis to the central role of FAK in fibrillar adhesion formation (Ilić et al., 2004). In addition to these studies, we herein show that FAK−/− MEFs exhibit significantly lower expression levels of α-SMA (Fig. 6 B) and exert lower contraction forces to deformable culture substrates (Fig. 6 C) as compared with wild-type MEFs. Despite the organizational differences, the amounts of total TGF-β1 in the ECM of DOC-treated FAK−/− and wild-type MEFs were similar (Fig. 6 D). In contrast, active TGF-β1 levels differed significantly when hDMfs were seeded onto the decellularized murine ECM and induced to contract. hDMf contraction activated 50% of the total TGF-β1 stored in the ECM from wild-type MEFs but only 20% from the FAK−/− ECM (Fig. 6 E). Baseline TGF-β1 activation in the absence of contraction was low (∼15%) and comparable on both ECM types (Fig. 6 E). Addition of the contraction agonist thrombin to decellularized MEF ECM in the absence of hDMfs did not release any TGF-β1, confirming the complete removal of MEFs and the specific action of thrombin on hDMf contraction (unpublished data). Control experiments performed with active TGF-β1–stimulated TMLCs verified that the reporter cell activity did not depend on the organization level of their ECM substrate (Fig. 6 F).
Figure 6.

ECM disorganization suppressed TGF-β1 activation FAK−/− MEFs, and wild-type MEFs were cultured for 6 d. (A) ECM production was assessed by immunofluorescence staining for FN (green) and nuclei (blue), and the mean density of FN fibrils was calculated from six images per three independent experiments. Bar, 100 µm. (B) Culture lysates were immunoblotted using vimentin and GAPDH as loading controls. (C) Cells were seeded for 1 d onto silicone substrates, and wrinkle formation as a result of substrate deformation was quantified from phase-contrast images. Bar, 25 µm. (D) After 6-d culture, MEFs were removed using DOC, and total TGF-β1 was measured after heating the remaining ECM. (E) hDMfs were seeded onto the decellularized MEF ECMs; active TGF-β1 levels were measured as a percentage of total TGF-β1 without (baseline) and with thrombin-induced contraction. (F) TMLCs alone were seeded onto decellularized ECM produced by MEF wild type (WT) and FAK−/− and stimulated for 1 h with 0, 0.03, 0.06, 0.12, 0.25, 0.50, and 1.0 ng/ml active TGF-β1. Cells were further processed and assessed for luciferase production (luminescence). The data shown are from a single representative experiment, and the experiment was completed once with three independent measurements. (G) Decellularized MEF ECM was incubated for 1 h with magnetic beads coated with BSA or anti-LAP (aLAP) antibody before magnetic force was applied, and active TGF-β1 was measured in the supernatant. Graphs show mean values and SDs from at least three independent experiments (***, P ≤ 0.005 using ANOVA followed by a post-hoc Tukey’s multiple comparison test). a.u., arbitrary unit.
Although attachment and spreading of hDMfs was similar on the different MEF-derived ECMs, ECM-induced phenotypic changes in hDMfs may have affected TGF-β1 activation. To exclude this possibility, we mechanically released active TGF-β1 using a cell-free assay (Fig. 6 F; Buscemi et al., 2011). Decellularized MEF-derived ECM was incubated with anti-LAP–coated ferromagnetic beads for 1 h before force was applied through a magnetic field and active TGF-β1 was measured (Fig. 6 G). Force-induced release of active TGF-β1 from FAK−/− MEF ECM was low (10% of total TGF-β1) and not different from no-force and BSA-coated bead controls (Fig. 6 G). Conversely, pulling magnetic beads coated with the anti-LAP antibody resulted in an approximately threefold higher release of active TGF-β1 from wild-type MEF ECM as compared with FAK−/− MEF ECM (Fig. 6 G). Collectively, these data demonstrated that mechanical activation of TGF-β1 from preorganized and mature ECM is more efficient than from disordered ECM.
Mechanical loading of LTBP-1 fibrils enhances TGF-β1 release
Fibroblast-mediated ECM maturation involves straining of secreted fibrils, bundling, and proteolytic remodeling. To test whether prestraining the ECM alone would facilitate TGF-β1 activation, we used a mechanical strain device and highly expandable silicone culture membranes allowing an up to eightfold strain (Majd et al., 2011; Rosenzweig et al., 2012). The device was used to simulate and accelerate strain in the ECM that is induced by cells after several days or weeks in contrast to conventional strain devices and membranes that allow a maximum linear expansion of ≤1.3-fold, corresponding to length change associated with single cell contractions (Wipff et al., 2009). hDfs were grown on nonstrained membranes (onefold) to produce ECM and removed after 6 d using DOC. The remaining LTBP-1/TGF-β1–rich ECM was then strained in the absence of any cells. Immunofluorescence video microscopy of live-strained LTBP-1 revealed that, starting after a prestrain of 1.4-fold, LTBP-1–containing fibrils straightened linearly with membrane strain to approximately twofold of their initial length at a membrane strain of 2.8-fold (Fig. 7, A and B; and Video 3). At strains larger than threefold, the ECM was detaching from the membrane and thus not investigated (unpublished data). hDMfs were then seeded on the differently strained ECM and induced to contract with thrombin and TRAP-6 in select conditions (Fig. 7 C). hDMf contraction activated 7% of total TGF-β1 at a prestrain of 1.7-fold, 13% from 1.8-fold prestrained ECM, and 61% from 1.9-fold prestrained ECM (Fig. 7 C). Western blotting for phospho-Smad2 in subsequently lysed hDMfs confirmed that increasing prestrain in the ECM resulted in increasing levels of active TGF-β1 and downstream signaling (Fig. 7 C).
Figure 7.
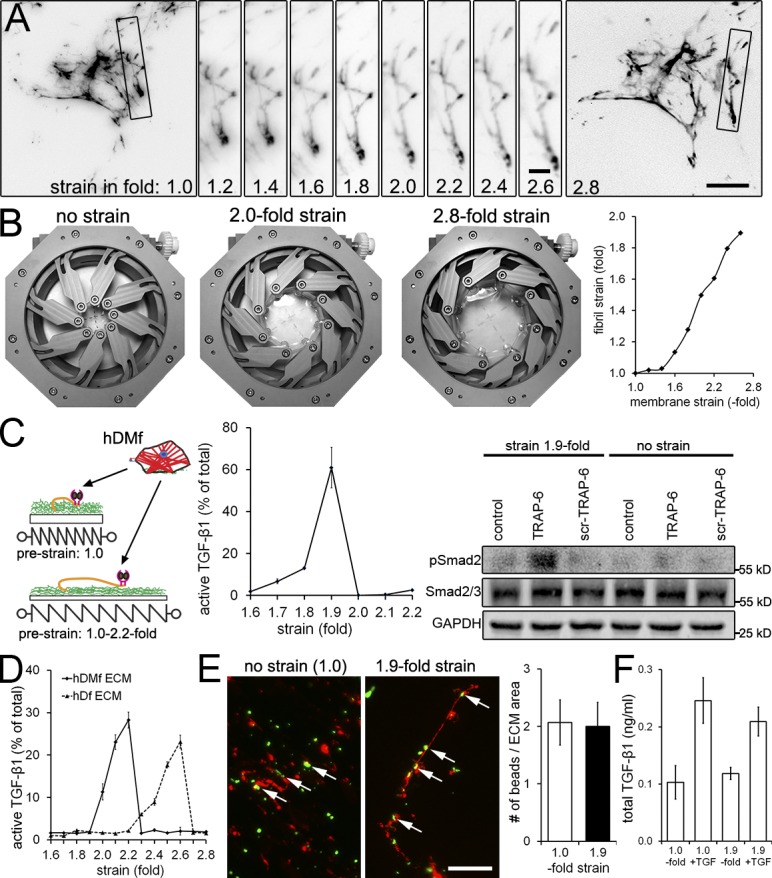
Prestraining LTBP-1–containing ECM enhances subsequent TGF-β1 release. hDMfs grown on relaxed highly expandable silicone membranes were removed after 6 d using DOC. (A) The cell-free DOC-insoluble ECM was stained without fixation for LTBP-1 and visualized during membrane expansion from relaxed (onefold) to 2.8-fold strains. Selected fibrils (boxes) were magnified and followed in incremental steps of a 0.2-fold membrane strain. (B) Illustration of the strain device opening and membrane expansion. Strain of LTBP-1–containing fibrils was measured as fold length change compared with initial length and plotted against membrane surface expansion. The data shown are from a single representative experiment out of three repeats. (C) hDMfs were seeded onto nonstrained (onefold) and prestrained decellularized ECM (≤2.2-fold). Cell contraction was induced in every condition using thrombin and TRAP-6 in select conditions, and release of active TGF-β1 was quantified as the percentage of total TGF-β1. Western blotting was performed for phospho- and total Smad2 in subsequently lysed myofibroblasts. (D) The decellularized ECM produced by either hDMfs or hDfs was strained in the absence of cells by expanding the membrane ≤2.8-fold, and active TGF-β1 released into the supernatant was measured at every 0.1-fold increment. (E) Decellularized ECM labeled for LTBP-1 (red) and green fluorescent microspheres at strains of 1.0- and 2.8-fold. Arrows indicate microspheres bound to LTBP-1 fibrils. The number of microspheres in the image field was quantified and normalized to the area covered by LTBP-1 fibrils. (F) 1 ng/ml active TGF-β1 was added for 1 h to cell-free hDMf-derived ECM that was either nonstrained (onefold) or strained 1.9-fold. Samples were rigorously washed three times, ECM was subsequently lysed, and levels of ECM-contained TGF-β1 were measured using TMLCs. The graph shows mean values and SDs from at least three independent experiments. Bars: (main images) 20 µm; (magnified images) 4 µm.
Because no TGF-β1 was activated by contraction of reseeded hDMf from hDf ECM that has been prestrained by more than twofold, we hypothesized that larger strains can lead to release of TGF-β1 from the latent complex even in the absence of counteracting cell integrins. We tested this possibility by producing LTBP-1 ECM using hDfs and hDMfs and removing the cells after 6 d with DOC. The cell-free ECM was then strained from 1.0–2.8-fold. Active TGF-β1 levels in the supernatant were measured after each incremental strain of 0.1-fold (Fig. 7 D). After strains of 2.0, 2.1, and 2.2, respectively, 11, 23, and 28% of the total TGF-β1 was activated and released from hDMf ECM. In contrast, release of active TGF-β1 from hDf ECM did not occur at strains <2.4-fold. Collectively, these results indicate that (a) prestraining ECM alone is sufficient to increase the release of active TGF-β1 by subsequent hDMf contraction, (b) high strain is able to mechanically open the latent complex for active TGF-β1 release even in the absence of cells, and (c) hDMf ECM reaches the TGF-β1 activation threshold at lower strains than hDf ECM.
Alternatively, it is possible that strained ECM fibrils reveal a higher number of latent TGF-β1 complexes that are available for integrin binding. To test this possibility, we produced onefold (no strain) and 1.9-fold prestrained hDf ECM as described in this paper that was incubated with anti-LAP antibody–coated fluorescent microspheres for 5 min. After rigorous washing, the number of beads remaining adherent per LTBP-1 fibril area in the image field was similar in both conditions, demonstrating that strain did not reveal “cryptic” LAP (Fig. 7 E). We further excluded that active TGF-β1 absorbs differently to ECM under different strain conditions (Fig. 7 F). Therefore, we conclude that the strain introduced into LTBP-1 fibrils primes the latent complex for TGF-β1 by myofibroblasts contraction.
Discussion
In a previous publication, we showed that fibroblasts directly activate TGF-β1 by exerting forces to the latent complex and that cultured rat lung myofibroblasts release more active TGF-β1 from their ECM than from fibroblasts (Wipff et al., 2007). These findings were explained with the higher contractile activity of myofibroblasts over fibroblasts (Hinz et al., 2001a). However, recent structural and single molecule force spectroscopy studies have indicated that forces as low as 40 pN, corresponding to the action of few myosin II motors, are sufficient to induce conformational changes in LAP that will lead to the release of TGF-β1 (Buscemi et al., 2011; Shi et al., 2011). Consistently, low contractile epithelial cells are also able to activate TGF-β1 through integrin-mediated force transduction (Giacomini et al., 2012). One possible explanation for this apparent contradiction is that the efficacy of force-mediated TGF-β1 activation depends on the mechanical properties of the myofibroblast ECM that are established before the actual activation step occurs. Here, we demonstrate in vitro and in vivo that myofibroblasts deposit similar levels of latent TGF-β1 into the ECM but organize the LTBP-1 into straighter and denser fibrils as compared with fibroblasts. By offering myofibroblasts latent TGF-β1–containing ECM of identical composition but different prestrain, we revealed a clear dependence of TGF-β1 activation on the level of ECM organization at the time of force exertion to the latent complex. We propose a model in which cell-mediated remodeling leads to gradual straining of LTBP-1/TGF-β1–containing ECM fibrils, analogous to loading a mechanical spring (Fig. 8).
Figure 8.

ECM prestrain generated by myofibroblast contraction affects TGF-β1 activation. Fibroblasts secrete ECM that is rich in FN (green) and the large latent complex of TGF-β1 (LTBP-1, LAP, and TGF-β1). Fibroblast-to-myofibroblast differentiation occurs during physiological (normal wound healing) and pathological (fibrosis) tissue remodeling. Myofibroblasts are characterized by α-SMA–positive stress fibers (red) enabling these cells to exert high contractile activity and forces transmitted to the ECM at sites of integrins. The gradual straitening and straining of ECM fibrils, containing FN and LTBP-1, prime the latent TGF-β1 complex for subsequent activation. At sufficient prestrain, minimal additional length changes in the ECM (i.e., small contractions) will be sufficient to release active TGF-β1 by inducing a conformational change in LAP. Hence, the mechanical preloading of the ECM determines the trigger point for TGF-β1 activation driving the vicious loop of myofibroblast self-activation.
Mechanical preloading of the ECM will have important implications for the availability of profibrotic TGF-β1 during normal tissue repair and development of fibrosis. Organ fibrosis comprises the excessive secretion and contraction of ECM by fibroblastic cells and augments tissue stiffness (Wynn, 2008; Hinz, 2012; Henderson and Sheppard, 2013; Klingberg et al., 2013). We and others have previously shown that growth on culture substrates with low elastic modulus reduces TGF-β1 activation by cell contraction (Wipff et al., 2007; Giacomini et al., 2012). These findings led to the conclusion that the ECM has to be sufficiently stiff to resist the cell forces needed to release TGF-β1 from its LAP straitjacket. Consistently, activation of TGF-β1 from the ECM depends on binding of LAP to LTBP-1, which in turn has to be linked to the ECM (Taipale et al., 1994; Annes et al., 2004; Fontana et al., 2005). However, the view of stiffness-controlled TGF-β1 activation may be oversimplified in light of our results showing that prestraining latent TGF-β1–containing ECM on silicone membranes with unchanged elastic modulus was sufficient to enhance TGF-β1 activation by subsequent cell contraction. Nevertheless, the bulk stiffness of the material may have a secondary effect by determining the organization state of the cell-derived ECM by regulating the remodeling activity of the adhering cells before TGF-β1 activation (Godbout et al., 2013).
In contrast to elastic cell culture polymers, the ECM of normal and fibrotic connective tissues is subject to strain stiffening (Storm et al., 2005; Discher et al., 2009). Our results suggest that a cell remodeling strain stiffens LTBP-1–containing ECM and thereby primes latent TGF-β1 for subsequent activation. The pathological remodeling and stiffening of scar tissue can be understood as the overall outcome of subcellular contractions with small length changes (∼400 nm) but repetitive occurrence (∼40 contractions/h) as measured in cultured fibroblasts (Follonier Castella et al., 2010a). Because such subcellular contractions theoretically accumulate to ∼2.5 mm per week in vitro, it may take weeks, months, or years for fibrotic scars to mature (Tomasek et al., 2002; Follonier Castella et al., 2010b). ECM fibril straightening is greatly accelerated in cultures of myofibroblasts that are contracting with higher frequency and amplitude than fibroblasts (Follonier Castella et al., 2010a). In our experiments, mechanical release of TGF-β1 was dependent on the extent of ECM preremodeling and thus more efficient from a myofibroblast-produced ECM than from fibroblast-produced ECM. The fact that induced acute myofibroblast contraction results in larger absolute length changes than fibroblast contraction is likely responsible for their higher capacity to activate TGF-β1 from the same preformed ECM.
Prestraining ECM on highly expandable culture membranes demonstrated that even the quasi–two-dimensional ECM of cultured fibroblasts provides sufficient buffer (slack) to allow ∼1.4-fold length changes before fibrils are visibly engaged, and it requires an ∼2.8-fold strain to liberate TGF-β1 in the absence of cells. In contrast, myofibroblast ECM is ∼25% more prestrained than the ECM of fibroblasts in our culture conditions as extrapolated from the release of TGF-β1 at a lower strain (twofold). It remains elusive whether TGF-β1 release in the absence of any cells was caused by ECM damage by “overstrain” or whether a physiological mechanism is conceivable that allows stress liberation of TGF-β1 without integrin pulling on the RGD site of LAP. A similar, cell cytoskeleton-independent effect was reported for the strained tendon, which is characterized by highly organized and strained collagen fibrils (Maeda et al., 2011). It has been proposed that a normal ECM strain in the tendon releases physiological levels of TGF-β1 that regulate tenocyte function. Tendon injury and loss of the mechanoprotective collagen structure were shown to result in dramatically increased active TGF-β1 levels (Maeda et al., 2011).
Linking the activation of latent TGF-β1 to the organization state of the ECM will provide a mechanical threshold to generate and/or sustain myofibroblasts, which develop their contractile activity by expressing α-SMA in response to TGF-β1 (Hinz et al., 2001a). In the poorly organized but latent TGF-β1–rich provisional ECM established after acute injury (Brunner and Blakytny, 2004), TGF-β1 activation by cell traction will be inefficient, and myofibroblasts will not develop. In a sufficiently prestrained ECM, even the low contractile forces exerted by migrating fibroblastic cells will promote latent TGF-β1 activation. This model can answer the classical hen-and-egg question of whether ECM stiffening must occur first to induce myofibroblast differentiation or whether myofibroblast contraction must occur first to stiffen the ECM. Importantly, secretion of latent TGF-β1 by myofibroblasts into an already straightened fibrotic ECM will provide a much faster trigger for TGF-β1 activation than required during fibrosis development. This property may at least partly explain the fact that decellularized ECM derived from fibrotic but not normal lung tissue instructs de novo seeded cells to become fibrotic even in the absence of exogenous TGF-β1 (Booth et al., 2012). It becomes indeed increasingly accepted that the biochemical and biophysical properties of fibrotic ECM provide sufficient cues to drive resident and recruited cells into a disease state (Berry et al., 2006; Shimbori et al., 2013).
The question remains open whether enhanced activation of TGF-β1 requires specific prestrain in LTBP-1 itself or whether LTBP-1 passively piggybacks on overall ECM remodeling. To date, there is no evidence for formation of LTBP-1–exclusive fibrils. Our results show that LTBP-1 is secreted independently of FN, but its fibrillar organization appears to depend on the presence of FN in the ECM. This interdependence was particularly evident in fibroblasts that overexpress LTBP-1 under a constitutively active promoter; in these cells, the physiological sequence of FN preceding LTBP-1 secretion (Dallas et al., 2005; Koli et al., 2005) is uncoupled. Our observations are consistent with the idea of FN acting as a master template in immature ECM for the subsequent recruitment and organization of other ECM proteins, including latent TGF-β1 binding proteins (LTBPs; Dallas et al., 2005; Koli et al., 2005), fibrillin-1 (Isogai et al., 2003; Chaudhry et al., 2007; Ono et al., 2009; Sabatier et al., 2009, 2013), fibulin-1 (Godyna et al., 1995), and collagen (Velling et al., 2002; Isogai et al., 2003) as well as the indirect role of heparan sulfate proteoglycans in LTBP-1 to FN binding (Chen et al., 2007). It is still unclear whether fibrillin-1 is another essential component to mechanically prime the ECM for subsequent TGF-β1 activation. The ECM produced by fibrillin-1 mutant murine fibroblasts in our experiments was too low in total TGF-β1 to exhibit clear differences in TGF-β1 activation before and after prestrain. Furthermore, because fibrillin-1 and FN largely colocalize in 6-d fibroblast cultures, it is reasonable to assume that the mechanism of ECM strain controlling TGF-β1 activation is principally independent from the molecular nature of the LTBP-1 binding protein provided that cell force can be transmitted.
Our new finding that fibroblastic cells incorporate nonendogenous purified LTBP-1 into preexisting or developing FN ECM shows that LTBP-1 fibril assembly can occur after secretion. Importantly, we show that blocking fibroblastic cell contraction inhibited the incorporation of endogenous purified LTBP-1 into a preexisting FN network, indicating a cell-driven and active process. It remains to be shown whether fibroblasts use integrins to promote LTBP-1 fibril assembly or whether a cell-derived strain generates assembly sites in FN for LTBP-1 binding, similar for what has been described for FN autofibrillogenesis (Zhong et al., 1998; Smith et al., 2007; Singh et al., 2010). Experiments performed with prestrained cell-free ECM render it unlikely that strain-induced FN autofibrillogenesis allows increased LTBP-1 binding and thereby contributes to increased TGF-β1 activation, at least in our culture experiments. In vivo, however, this possibility has yet to be tested. The fact that myofibroblasts formed focal adhesions with microcontact-printed LTBP-1 and that blocking of RGD binding integrins reduced adhesion of myofibroblasts to LTBP-1–coated substrates suggests that fibroblastic cells directly bind LTBP-1. The RGD sequence described in human LTBP-1 serves as a possible recognition site (Hyytiäinen et al., 2004). However, rodent LTBP-1 and widely expressed LTBP-3 (Saharinen et al., 1999) do not contain RGD, which may thus not be the only cell binding site in LTBPs. Although direct integrin binding may contribute to LTBP-1 recruitment to existing FN fibrils, it seems insufficient for LTBP-1 fibrillogenesis in the absence of FN.
It will be a future challenge to develop specific strategies interfering with the mechanical loading of the fibroblast/myofibroblast ECM with respect to TGF-β1 activation. It is amply clear that defects in essential components of the FN and microfibril ECM lead to a variety of TGF-β1–related diseases (Ramirez and Rifkin, 2009; Doyle et al., 2012; Baldwin et al., 2013). The sole inhibition of LTBPs binding to the ECM is unlikely to be efficient as an antifibrosis therapy because fibrillin-1 mutants defective for LTBP-1 binding exhibit TGF-β1 hyperactive phenotypes. Alternatively, integrins represent possible antifibrotic targets that provide relative cell and tissue specificity in the context of latent TGF-β1 activation (Gerber et al., 2013; Henderson et al., 2013; Hinz, 2013).
Materials and methods
Ethics statement
Human material was provided by B.A. Alman, and the use was approved by the Institutional Review Board of the Hospital for Sick Children. Written informed consent was obtained from patients for the use of the material.
Animal experiments
On a total of 20 female Wistar rats (200–220 g), full-thickness 20 × 20–mm wounds were produced in the middle of the dorsum. Wounds were allowed to heal either spontaneously (control) or were subject to mechanical stress by fixing the edges of the wound tissue on a plastic frame that prevents wound closure and retains the size of the open wound over time (stressed). Authorization of the local animal ethic committees was obtained as previously described (Hinz et al., 2001b). Rats were sacrificed by CO2 anesthesia, and wound granulation tissue was harvested after 3, 6, and 9 d after wounding. For immunofluorescence staining of rat tissues, 5-µm sections were fixed with 100% acetone for 15 min at −20°C and then dried for 30 min at RT. After adjusting to RT, sections were rinsed in PBS, blocked with 1% BSA for 30 min, and stained with primary antibodies ED-A FN (1:200; sc-59826; Santa Cruz Biotechnology, Inc.) and LTBP-1 (1:100 [MAB388; R&D Systems] or 1:250 [Ab39; a gift from C.-H. Heldin, Uppsala University, Uppsala, Sweden]) followed by 30-min secondary fluorescently labeled antibodies.
Cell culture and reagents
Fibroblasts were explanted from human dermal tissue–derived human tissue sections. In brief, tissues were cut into 1-mm3 cubes, attached to tissue culture plates, and immersed in standard DMEM (Life Technologies), supplemented with 10% fetal bovine serum (Sigma-Aldrich), and penicillin/streptomycin (Life Technologies). Cells were allowed to migrate out of tissues for 10 d before the first passage. To promote hDf-to-hDMf differentiation, 2 ng/ml TGF-β1 (100-B-001; R&D Systems) was added for 6 d. The Flp-In 293 cell line (gift from P. Jurdic, Université de Lyon, Lyon, France) was maintained in standard medium supplemented with Zeocin (1:1,000; R250-05; Life Technologies). Flp-In 293 cells from a single-cell clone were transfected with pSecTag–LTBP-1–EGFP, and a stable HEK293–LTBP-1–EGFP (HEK293–LTBP-1) cell line was derived by fluorescence-activated cell sorting and limited dilution. HEK293–LTBP-1 cells were maintained under hygromycin B (1:50; 10687-010; Life Technologies) selection. hDfs and myofibroblasts, wild-type MEFs (CRL-2645T; ATCC), FAK−/− (CRL-2644; ATCC), filamin A knockdown, created by stable transfection of NIH3T3 cells with shRNA against filamin A were a gift (Kiema et al., 2006; Shifrin et al., 2009), and integrin β1−/− knockout cells (GD25; Fässler et al., 1995) were gifts from C. McCulloch (University of Toronto, Toronto, Ontario, Canada). For integrin β1−/− knockout cells, the second exon of the β1integrin gene in embryonic stem (ES) cells was disrupted with a gene trap vector with a β-galactosidase–neomycin fusion DNA. ES cells were immortalized with recombinant retroviruses that transduced the SV-40 large T. A single clone was established that was mutated in both alleles. The homozygous mutant clone did not produce integrin β1 mRNA or protein (Fässler et al., 1995). FN−/− MEFs were developed together with R. Fässler (Max Planck Institute of Biochemistry, Munich, Germany). In brief, ES cells were isolated form embryonic day 14.5 FN floxed embryos, immortalized via retroviral transduction of SV-40 large T antigen, cloned, and treated with adenoviral Cre to delete the floxed FN alleles. The FN targeting vector spanned the region from the promoter to the second intron. For the floxed FN allele, loxP sites were confirmed within the 5′-untranslated region and within the first intron. Cre-mediated recombination at these two loxP sites removed the start codon, signal sequence, and the exon/intron border of exon 1 to generate the null allele (Sakai et al., 2001). Wild-type and fibrillin-1 C1039G/+ mutant mouse dermal myofibroblasts (gift from H. Dietz, Johns Hopkins University School of Medicine, Baltimore, MD; Judge et al., 2004) were maintained in standard culture media.
Plasmids and purification of LTBP-1–EGFP
The pSecTag–LTBP-1–EGFP plasmid was created by subcloning the previously published LTBP-1–EGFP sequence in frame of pSecTag/FRT/V5-His-TOPO vector (Life Technologies; Buscemi et al., 2011). Correct integration was confirmed by sequencing the insert both ways at 5′-TCAG-3′ (SickKids Hospital, Toronto, Canada). LTBP-1 was purified from serum-free conditioned media of HEK293–LTBP-1. In brief, conditioned medium was collected and dialyzed against Dulbecco’s PBS (Life Technologies) before it was run over an ion metal affinity chromatography column with HIS-Select Nickel Affinity Gel (Sigma-Aldrich). Columns were washed with wash buffer containing 0, 10, or 15 mM imidazole. Fractions containing LTBP-1 were eluted with 250 mM imidazole (Fig. S2).
Immunofluorescence
Before immunofluorescence staining of cultured cells, samples were simultaneously fixed, and permeabilized in 3% paraformaldehyde. In vitro samples were fixed with 3% paraformaldehyde for 10 min, washed with PBS, and then permeabilized with 0.2% Triton X-100 for 5 min. Primary antibodies used in this study were α-SMA (mouse IgG2a; clone SM1; a gift of G. Gabbiani, University of Geneva, Geneva, Switzerland), vinculin (rabbit; 1:400; ab11194; Abcam), FN (rabbit; 1:400; F3648; Sigma-Aldrich), ED-A FN (mouse IgG1;1:200; sc-59826; Santa Cruz Biotechnology, Inc.), LTBP-1 (mouse IgG1 [1:100; MAB388; R&D Systems] or rabbit [1:250; Ab39]), fibrillin-1 (rabbit; 1:500; raised against the recombinant, soluble N-terminal half of fibrillin-1 fragment; gift from D.P. Reinhardt, McGill University, Montreal, Quebec, Canada; Sabatier et al., 2009), integrin β1 (mouse IgG1; 1:50; ab30394; Abcam), integrin β3 (mouse IgG1; 1:50; MAB1974; EMD Millipore), and integrin β5 (rabbit; 1:50; ab15459; Abcam). Recombinant LTBP-1 was stained with anti-GFP antibodies (rabbit [1:200; ab290; Abcam] and mouse IgG1 [1:200; ab291; Abcam]) to achieve higher detection sensitivity than anti–LTBP-1 antibodies. Secondary antibodies used were goat anti–mouse IgG Alexa Fluor 568 (1:100; A-11004; Life Technologies), goat anti–mouse IgG1 FITC (1:100; 1070-02; SouthernBiotech), goat anti–mouse IgG2a TRITC (1:100; 1080-03; SouthernBiotech), and goat anti–rabbit-TRITC and -FITC (1:100; F9887; Sigma-Aldrich). To stain F-actin and nuclear DNA, phalloidin Alexa Fluor 488 and 568 (1:100; Life Technologies) and DAPI (1:50; Sigma-Aldrich) were used, respectively.
Image acquisition, processing, and quantitative analysis
Fluorescence microscopy images were acquired with an upright microscope (Axio Imager; Carl Zeiss) equipped with a camera (AxioCam HRm; Carl Zeiss), ApoTome.2 structured illumination, and ZEN software (Zeiss-1; Carl Zeiss). Fig. 1 A images were taken with Plan Apochromat objectives (40×, NA 1.2 [Fig. 1 A, top two rows] and 63×, NA 1.4, oil differential interference contrast at RT; Carl Zeiss) on Zeiss-1 using ApoTome.2 structured illumination to calculate confocal optical sections. Fig. 1 B (top) images were taken with a Plan Fluar objective (20×, NA 0.75 at RT, air; Carl Zeiss) on Zeiss-1. Fig. 1 B (bottom) images were taken with a Plan Fluar objective (40× oil immersion, NA 1.30 at RT; Nikon) at the Center for Microfluidics in Chemistry and Biology at the University of Toronto with a confocal microscope (A1; Nikon) equipped with two camera systems, a Retiga 2000R Fast 1394 camera (QImaging) and a confocal imaging system running NIS-Elements software (Confocal-1; Nikon). All Fig. 2 images were acquired on Confocal-1 with a Plan Fluar objective (40×, NA 1.30, oil immersion at RT; Nikon). Fig. 3 scanning electron microscope images were acquired on an environmental scanning electron microscope (XL-30; Phillips) at the Department of Pathology and Laboratory Medicine (Mount Sinai Hospital). Samples were prepared according to imaging facility requirements. In brief, samples were fixed with glutaraldehyde and dehydrated stepwise to 100% ethanol. After critical point drying, samples were gold coated with a low-vacuum sputter. Fig. 3 (A and B) confocal images were taken on a confocal microscope system (DMIRE2; Leica), from here on referred as Confocal-2, with an HCX Plan Apochromat CS (40×, NA 1.25, oil immersion at RT; Leica) objective and processed with LAS Software (Leica). Fig. 3 C and all confocal images from Fig. 4 were taken on Confocal-1 with a Plan Fluar objective (40×, NA 1.30, oil immersion at RT; Nikon). Fig. 5 images of microcontact printing were taken on Zeiss-1 with a Plan Apochromat objective (63×, NA 1.4, oil differential interference contrast at RT; Carl Zeiss). Images in Fig. 6 A were taken on Zeiss-1 with a Plan Fluar objective (20×, NA 0.75 at RT; Carl Zeiss), and Fig. 6 C images were taken live with Zeiss-1 and a water immersion Apochromat objective (40×, NA 1.0 at RT; Carl Zeiss). Fig. 7 images are obtained from Video 1 processed with MetaMorph software (Molecular Devices). Video 1 was taken on a microscope (Axiovert 135M; Carl Zeiss) with a camera (C10600 ORCA-R2; Hamamatsu Photonics) and a Plan Fluar objective (10×, NA 0.5 at RT; Carl Zeiss). Fig. S4 B images were taken on Confocal-1 with a Plan Fluar objective (40×, NA 1.30, oil immersion at RT; Nikon). Fig. S5 images were taken on Zeiss-1 with a Fluar objective (20×, NA 0.75 at RT; Carl Zeiss). All Carl Zeiss microscopes were operated with ZEN software.
Quantitative image analysis was performed using ImageJ (National Institutes of Health) and customized macros (supplemental material). Figures were assembled with Photoshop CS5 (Adobe). ED-A FN and LTBP-1 fibril count and density were determined from images taken from tissue slices and cell cultures. In brief, single-channel images were converted to 8-bit grayscale, and thresholding was applied to remove background depending on experimental conditions. Particle events were counted, and density was analyzed for events with a size larger than 2 × 2 pixels and 0–1 circularity.
Immunoprecipitation and Western blotting
For Western blots, vinculin, α-SMA, and vimentin were separated on 10%, and FN, ED-A FN, FAK, GPF (reduced conditions), and LTBP-1 (nonreduced conditions) were separated on 8% SDS-PAGE gels. Gels were transferred to nitrocellulose membranes using semidry transfer technique at 18 mAmps/gel and 20 V for 16 h or overnight. Protein membranes were blocked with 5% skim milk, and primary antibodies were detected with fluorescently labeled anti–mouse 680-nm and anti–rabbit 800-nm IRDye secondary antibodies (1:10,000; LIC-926-68020 and LIC-926-32211; LI-COR Biosciences). Signals were detected with an Fx imaging system (Odyssey; LI-COR Biosciences).
TGF-β1 bioassay
Active TGF-β1 was quantified using TMLCs, which produce luciferase under control of the PAI-1 promoter in response to TGF-β1 (provided by D.B. Rifkin, New York University, New York, NY; Abe et al., 1994). Co-culture or decellularized ECM experiments were performed by seeding TMLCs directly onto the first cell layer or DOC-insoluble ECM. Fibroblast and myofibroblast contraction was then induced by 0.5 U/ml thrombin for 1 h with subsequent media change for 16 h. TMLCs were lysed with cell culture lysis reagent (1:5; Promega) and assessed with a luciferase assay kit (Promega) using a luminometer (Centro LB; Berthold Technologies). To asses TGF-β levels from culture supernatants, TMLCs (60,000 cells/cm2) were allowed to adhere for 4 h before being subjected to conditioned media, which was native (active TGF-β) or heat activated (total TGF-β) for 10 min at 80°C for 1 h. TMLCs were then grown in medium containing 1% FBS for 16 h. 5 ng/ml TGF-β1–specific blocking antibody (AF-101-NA; R&D Systems) was used to confirm that TGF-β1 is the main TGF-β isoform activated in our cultures. If not stated otherwise, all results were corrected for TMLC baseline luciferase production in the absence of TGF-β1 and thrombin activation. In all conditions, active TGF-β1 was normalized to total TGF-β1.
ECM labeling with fluorescent microspheres
Red fluorescence microspheres with 1-µm diameter (Life Technologies) were coated with the anti–LAP-1 antibody. ECM on highly elastic silicone rubber membranes was decellularized and stained for LTBP-1 and anti–mouse IgG1-FITC as described in this paper. ECM was decellularized with DOC buffer (150 mM NaCl, 50 mM Tris-HCl, 1% NP-40, and 0.5% sodium DOC) treatment. In brief, cells were washed three times with PBS, ice-cold DOC buffer was added for 5–10 min at 4°C, and cell debris was aspirated. Subsequent washes with PBS cleaned ECM of remaining debris. Nonstrained and 1.9-fold strained ECMs were incubated with an excess of microspheres for 5 min followed by extensive washing to remove unbound spheres. Five random images of LTBP-1 fibril area and spheres were taken in three independent experiments and analyzed for the amount of LTBP-1 area and bound spheres.
Microcontact printing and cell adhesion assay
To visualize direct binding of cells to purified LTBP-1, we used microcontact printing (Goffin et al., 2006). In brief, polydimethylsiloxane stamps exhibiting islet topographies with dimensions of 10-µm length, 1.5-µm width, and 4-µm spacing were coated with 0.2 µg/ml LTBP-1 or FN (FC010; EMD Millipore) for 1 h. Protein prints were created on plastic coverslips, and protein-free areas were passivated with poly-l-lysine–polyethylene glycol for 10 min. Myofibroblasts were seeded on prints in serum-free media for 4 h before being processed for immunostaining. For cell adhesion quantification, tissue culture plastic wells were coated with 20 µg/ml LTBP-1, and hDMfs were seeded for 4 h in the presence of cyclic peptides antagonizing 10 µM RGD (12135-010; Gibco), 10 µM control RGE (12139-010; Gibco), integrins αvβ3/αvβ5 (EMD121974; Cilengitide), αvβ3 integrin (EMD66203), and scrambled control (EMD135981; Merck). Blocking antibodies were used directed against 10 µg/ml integrins β1 (MAB1965; EMD Millipore), β3 (MAB1976; EMD Millipore), and β5 (MAB1961; EMD Millipore). Additional controls were 10 µM RGE and human IgG (I9135; Sigma-Aldrich). Samples were rigorously washed three times before cells were fixed for quantification and staining.
ECM prestrain assay
To strain decellularized ECM, we continuously expanded silicone culture substrates with a mechanical strain device (Cellerator; Cytomec GmbH) and highly expandable silicone rubber culture membranes, chemically functionalized for cell adhesion and coated with covalently bound rat tail collagen type I at 10 µg/ml (354236; BD) as previously described (Wipff et al., 2009). Fibroblastic cells were grown for 7 d and removed with DOC to preserve ECM only (Wipff et al., 2007). Membranes and attached ECM were then uniaxially strained from 1.0 (nonstrained)- to 2.8-fold in 0.1% increments before hDMfs were seeded and grown for 4 h on the DOC-insoluble ECM. Finally, cell contraction was induced, and TGF-β1 activation was measured. In one series of experiments, TGF-β1 activation was measured during ECM prestrain in the absence of any cells.
Cell contraction assay
Cell contractility was assessed using deformable silicone substrates as previously described (Godbout et al., 2013). In brief, polydimethylsiloxane substrates with a Young’s modulus of 2 kPa were coated with 10 ng/ml FN. Cells were seeded at a concentration of 2,500 cells/cm2, and wrinkle formation on substrates was observed after 24 h in culture. Live-phase contrast images were acquired with an inverted microscope (Axiovert 135M; 40× objective) and analyzed using ImageJ software (National Institutes of Health) by thresholding for phase-bright wrinkles and analyzing the surface area covered by identified particles in the resulting binary images. Relative contraction was expressed as the image area covered by wrinkles as previously described (Balestrini et al., 2012).
Statistical analysis
When applicable, data are presented as means ± SD. We assessed differences between groups with an analysis of variance (ANOVA) followed by a post-hoc Tukey’s multiple comparison test, and we set the significance level at P = 0.05. For experiments comparing fibroblasts versus myofibroblasts, we performed a two-tailed paired t test. When applicable, differences were considered to be statistically significant and indicated with *, P ≤ 0.05; **, P ≤ 0.01; and ***, P ≤ 0.005. Error bars represent SD.
Online supplemental material
Fig. S1 provides control experiments demonstrating that the TGF-β1 used to produce hDMfs in early passages does not interfere with active TGF-β1 measurements in later passages. Fig. S2 characterizes purified LTBP-1. Fig. S3 shows dynamic organization of HEK293 cell-derived LTBP-1 ECM by myofibroblasts. Fig. S4 elucidates the role of fibrillin-1 in LTBP-1 organization using MEFs that produce a LTBP-1 binding-deficient mutant of fibrillin-1. Fig. S5 summarizes the different mouse fibroblast models that were evaluated to produce an ECM with identical composition but defective organization compared with wild-type cells. Video 1 corresponds to the image sequence shown in Fig. 7 and demonstrates dynamic strain of LTBP-1 ECM. Video 2 illustrates dynamic organization of LTBP-1 by hDMfs grown on top of LTBP-1–producing HEK293 cells. Video 3 shows LTBP-1 organization by myofibroblasts in co-culture with HEK293 cells. A ZIP file is also provided that contains a supplementary macro to quantify cell contractile activity from wrinkling substrates. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.201402006/DC1.
Supplementary Material
Acknowledgments
We thank Dr. Pierre Jurdic for providing Flp-In 293 cells, Dr. Reinhard Fässler for FN−/− MEFs, and Dr. Hal Dietz for fibrillin-1 C1039G/+ mutant mouse dermal myofibroblasts. Thanks go to Drs. Christine Chaponnier and Giulio Gabbiani for kindly providing antibodies against α-SMA, Dr. Carl-Hendrik Heldin for anti–LTBP-1 (Ab39), Dr. Dieter P. Reinhardt for anti–fibrillin-1 antibodies, Dr. Daniel B. Rifkin for TGF-β1 reporter cells, and Dr. Benny Geiger for pLifeAct-RFP. Dr. Christopher A.G. McCulloch and Dr. Craig Simmons (University of Toronto, Toronto, Ontario, Canada) are acknowledged for technical support and advice, and Dr. Cay Kielty (University of Manchester, Manchester, England, UK) is acknowledged for critically reading of our first manuscript draft.
This research was supported by the Canadian Institutes of Health Research (CIHR; grants 210820 and 286920), the Collaborative Health Research Program (CIHR/Natural Sciences and Engineering Research Council grants 1004005 and 413783), the Canada Foundation for Innovation and Ontario Research Fund (grant 26653), and the Heart and Stroke Foundation Ontario (grant NA7086), all to B. Hinz. Data presented herein were further funded from the European Union’s Seventh Framework Program (FP7/2007–2013; under grant agreement 237946) and the CIHR Cell Signals Training program.
The authors declare no competing financial interests.
Footnotes
Abbreviations used in this paper:
- α-SMA
- α-smooth muscle actin
- ANOVA
- analysis of variance
- DOC
- desoxycholate
- ES
- embryonic stem
- FN
- fibronectin
- hDf
- human dermal fibroblast
- hDMf
- human dermal myofibroblast
- LAP
- latency-associated propeptide
- LTBP
- latent TGF-β1 binding protein
- MEF
- mouse embryonic fibroblast
- TMLC
- transformed mink lung epithelial cell
References
- Abe, M., Harpel J.G., Metz C.N., Nunes I., Loskutoff D.J., and Rifkin D.B.. 1994. An assay for transforming growth factor-β using cells transfected with a plasminogen activator inhibitor-1 promoter-luciferase construct. Anal. Biochem. 216:276–284. 10.1006/abio.1994.1042 [DOI] [PubMed] [Google Scholar]
- Annes, J.P., Chen Y., Munger J.S., and Rifkin D.B.. 2004. Integrin αVβ6-mediated activation of latent TGF-β requires the latent TGF-β binding protein-1. J. Cell Biol. 165:723–734. 10.1083/jcb.200312172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin, A.K., Simpson A., Steer R., Cain S.A., and Kielty C.M.. 2013. Elastic fibres in health and disease. Expert Rev. Mol. Med. 15:e8. 10.1017/erm.2013.9 [DOI] [PubMed] [Google Scholar]
- Balestrini, J.L., Chaudhry S., Sarrazy V., Koehler A., and Hinz B.. 2012. The mechanical memory of lung myofibroblasts. Integr. Biol. (Camb.). 4:410–421. 10.1039/c2ib00149g [DOI] [PubMed] [Google Scholar]
- Berry, M.F., Engler A.J., Woo Y.J., Pirolli T.J., Bish L.T., Jayasankar V., Morine K.J., Gardner T.J., Discher D.E., and Sweeney H.L.. 2006. Mesenchymal stem cell injection after myocardial infarction improves myocardial compliance. Am. J. Physiol. Heart Circ. Physiol. 290:H2196–H2203. 10.1152/ajpheart.01017.2005 [DOI] [PubMed] [Google Scholar]
- Booth, A.J., Hadley R., Cornett A.M., Dreffs A.A., Matthes S.A., Tsui J.L., Weiss K., Horowitz J.C., Fiore V.F., Barker T.H., et al. 2012. Acellular normal and fibrotic human lung matrices as a culture system for in vitro investigation. Am. J. Respir. Crit. Care Med. 186:866–876. 10.1164/rccm.201204-0754OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunner, G., and Blakytny R.. 2004. Extracellular regulation of TGF-β activity in wound repair: growth factor latency as a sensor mechanism for injury. Thromb. Haemost. 92:253–261 [DOI] [PubMed] [Google Scholar]
- Buscemi, L., Ramonet D., Klingberg F., Formey A., Smith-Clerc J., Meister J.J., and Hinz B.. 2011. The single-molecule mechanics of the latent TGF-β1 complex. Curr. Biol. 21:2046–2054. 10.1016/j.cub.2011.11.037 [DOI] [PubMed] [Google Scholar]
- Chaudhry, S.S., Cain S.A., Morgan A., Dallas S.L., Shuttleworth C.A., and Kielty C.M.. 2007. Fibrillin-1 regulates the bioavailability of TGFβ1. J. Cell Biol. 176:355–367. 10.1083/jcb.200608167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Q., Sivakumar P., Barley C., Peters D.M., Gomes R.R., Farach-Carson M.C., and Dallas S.L.. 2007. Potential role for heparan sulfate proteoglycans in regulation of transforming growth factor-beta (TGF-β) by modulating assembly of latent TGF-β-binding protein-1. J. Biol. Chem. 282:26418–26430. 10.1074/jbc.M703341200 [DOI] [PubMed] [Google Scholar]
- Dallas, S.L., Keene D.R., Bruder S.P., Saharinen J., Sakai L.Y., Mundy G.R., and Bonewald L.F.. 2000. Role of the latent transforming growth factor β binding protein 1 in fibrillin-containing microfibrils in bone cells in vitro and in vivo. J. Bone Miner. Res. 15:68–81. 10.1359/jbmr.2000.15.1.68 [DOI] [PubMed] [Google Scholar]
- Dallas, S.L., Sivakumar P., Jones C.J., Chen Q., Peters D.M., Mosher D.F., Humphries M.J., and Kielty C.M.. 2005. Fibronectin regulates latent transforming growth factor-beta (TGF β) by controlling matrix assembly of latent TGF β-binding protein-1. J. Biol. Chem. 280:18871–18880. 10.1074/jbc.M410762200 [DOI] [PubMed] [Google Scholar]
- Discher, D., Dong C., Fredberg J.J., Guilak F., Ingber D., Janmey P., Kamm R.D., Schmid-Schönbein G.W., and Weinbaum S.. 2009. Biomechanics: cell research and applications for the next decade. Ann. Biomed. Eng. 37:847–859. 10.1007/s10439-009-9661-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle, J.J., Gerber E.E., and Dietz H.C.. 2012. Matrix-dependent perturbation of TGFβ signaling and disease. FEBS Lett. 586:2003–2015. 10.1016/j.febslet.2012.05.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fässler, R., and Meyer M.. 1995. Consequences of lack of beta 1 integrin gene expression in mice. Genes Dev. 9:1896–1908. 10.1101/gad.9.15.1896 [DOI] [PubMed] [Google Scholar]
- Fässler, R., Pfaff M., Murphy J., Noegel A.A., Johansson S., Timpl R., and Albrecht R.. 1995. Lack of β1 integrin gene in embryonic stem cells affects morphology, adhesion, and migration but not integration into the inner cell mass of blastocysts. J. Cell Biol. 128:979–988. 10.1083/jcb.128.5.979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Follonier Castella, L.F., Buscemi L., Godbout C., Meister J.J., and Hinz B.. 2010a. A new lock-step mechanism of matrix remodelling based on subcellular contractile events. J. Cell Sci. 123:1751–1760. 10.1242/jcs.066795 [DOI] [PubMed] [Google Scholar]
- Follonier Castella, L., Gabbiani G., McCulloch C.A., and Hinz B.. 2010b. Regulation of myofibroblast activities: calcium pulls some strings behind the scene. Exp. Cell Res. 316:2390–2401. 10.1016/j.yexcr.2010.04.033 [DOI] [PubMed] [Google Scholar]
- Fontana, L., Chen Y., Prijatelj P., Sakai T., Fässler R., Sakai L.Y., and Rifkin D.B.. 2005. Fibronectin is required for integrin αvβ6-mediated activation of latent TGF-β complexes containing LTBP-1. FASEB J. 19:1798–1808. 10.1096/fj.05-4134com [DOI] [PubMed] [Google Scholar]
- Gerber, E.E., Gallo E.M., Fontana S.C., Davis E.C., Wigley F.M., Huso D.L., and Dietz H.C.. 2013. Integrin-modulating therapy prevents fibrosis and autoimmunity in mouse models of scleroderma. Nature. 503:126–130. 10.1038/nature12614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giacomini, M.M., Travis M.A., Kudo M., and Sheppard D.. 2012. Epithelial cells utilize cortical actin/myosin to activate latent TGF-β through integrin α(v)β(6)-dependent physical force. Exp. Cell Res. 318:716–722. 10.1016/j.yexcr.2012.01.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godbout, C., Follonier Castella L., Smith E.A., Talele N., Chow M.L., Garonna A., and Hinz B.. 2013. The mechanical environment modulates intracellular calcium oscillation activities of myofibroblasts. PLoS ONE. 8:e64560. 10.1371/journal.pone.0064560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godyna, S., Mann D.M., and Argraves W.S.. 1995. A quantitative analysis of the incorporation of fibulin-1 into extracellular matrix indicates that fibronectin assembly is required. Matrix Biol. 14:467–477. 10.1016/0945-053X(95)90004-7 [DOI] [PubMed] [Google Scholar]
- Goffin, J.M., Pittet P., Csucs G., Lussi J.W., Meister J.J., and Hinz B.. 2006. Focal adhesion size controls tension-dependent recruitment of α-smooth muscle actin to stress fibers. J. Cell Biol. 172:259–268. 10.1083/jcb.200506179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson, N.C., and Sheppard D.. 2013. Integrin-mediated regulation of TGFβ in fibrosis. Biochim. Biophys. Acta. 1832:891–896. 10.1016/j.bbadis.2012.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson, N.C., Arnold T.D., Katamura Y., Giacomini M.M., Rodriguez J.D., McCarty J.H., Pellicoro A., Raschperger E., Betsholtz C., Ruminski P.G., et al. 2013. Targeting of αv integrin identifies a core molecular pathway that regulates fibrosis in several organs. Nat. Med. 19:1617–1624. 10.1038/nm.3282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinz, B.2012. Mechanical aspects of lung fibrosis: a spotlight on the myofibroblast. Proc. Am. Thorac. Soc. 9:137–147. 10.1513/pats.201202-017AW [DOI] [PubMed] [Google Scholar]
- Hinz, B.2013. It has to be the αv: myofibroblast integrins activate latent TGF-β1. Nat. Med. 19:1567–1568. 10.1038/nm.3421 [DOI] [PubMed] [Google Scholar]
- Hinz, B., Celetta G., Tomasek J.J., Gabbiani G., and Chaponnier C.. 2001a. α-smooth muscle actin expression upregulates fibroblast contractile activity. Mol. Biol. Cell. 12:2730–2741. 10.1091/mbc.12.9.2730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinz, B., Mastrangelo D., Iselin C.E., Chaponnier C., and Gabbiani G.. 2001b. Mechanical tension controls granulation tissue contractile activity and myofibroblast differentiation. Am. J. Pathol. 159:1009–1020. 10.1016/S0002-9440(10)61776-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinz, B., Phan S.H., Thannickal V.J., Galli A., Bochaton-Piallat M.L., and Gabbiani G.. 2007. The myofibroblast: one function, multiple origins. Am. J. Pathol. 170:1807–1816. 10.2353/ajpath.2007.070112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinz, B., Phan S.H., Thannickal V.J., Prunotto M., Desmoulière A., Varga J., De Wever O., Mareel M., and Gabbiani G.. 2012. Recent developments in myofibroblast biology: paradigms for connective tissue remodeling. Am. J. Pathol. 180:1340–1355. 10.1016/j.ajpath.2012.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyytiäinen, M., Penttinen C., and Keski-Oja J.. 2004. Latent TGF-β binding proteins: extracellular matrix association and roles in TGF-β activation. Crit. Rev. Clin. Lab. Sci. 41:233–264. 10.1080/10408360490460933 [DOI] [PubMed] [Google Scholar]
- Ilić, D., Kovacic B., Johkura K., Schlaepfer D.D., Tomasević N., Han Q., Kim J.B., Howerton K., Baumbusch C., Ogiwara N., et al. 2004. FAK promotes organization of fibronectin matrix and fibrillar adhesions. J. Cell Sci. 117:177–187. 10.1242/jcs.00845 [DOI] [PubMed] [Google Scholar]
- Isogai, Z., Ono R.N., Ushiro S., Keene D.R., Chen Y., Mazzieri R., Charbonneau N.L., Reinhardt D.P., Rifkin D.B., and Sakai L.Y.. 2003. Latent transforming growth factor β-binding protein 1 interacts with fibrillin and is a microfibril-associated protein. J. Biol. Chem. 278:2750–2757. 10.1074/jbc.M209256200 [DOI] [PubMed] [Google Scholar]
- Jenkins, G.2008. The role of proteases in transforming growth factor-β activation. Int. J. Biochem. Cell Biol. 40:1068–1078. 10.1016/j.biocel.2007.11.026 [DOI] [PubMed] [Google Scholar]
- Judge, D.P., Biery N.J., Keene D.R., Geubtner J., Myers L., Huso D.L., Sakai L.Y., and Dietz H.C.. 2004. Evidence for a critical contribution of haploinsufficiency in the complex pathogenesis of Marfan syndrome. J. Clin. Invest. 114:172–181. 10.1172/JCI200420641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiema, T., Lad Y., Jiang P., Oxley C.L., Baldassarre M., Wegener K.L., Campbell I.D., Ylänne J., and Calderwood D.A.. 2006. The molecular basis of filamin binding to integrins and competition with talin. Mol. Cell. 21:337–347. 10.1016/j.molcel.2006.01.011 [DOI] [PubMed] [Google Scholar]
- Kim, H., Sengupta A., Glogauer M., and McCulloch C.A.. 2008. Filamin A regulates cell spreading and survival via β1 integrins. Exp. Cell Res. 314:834–846. 10.1016/j.yexcr.2007.11.022 [DOI] [PubMed] [Google Scholar]
- Klingberg, F., Hinz B., and White E.S.. 2013. The myofibroblast matrix: implications for tissue repair and fibrosis. J. Pathol. 229:298–309. 10.1002/path.4104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koli, K., Hyytiäinen M., Ryynänen M.J., and Keski-Oja J.. 2005. Sequential deposition of latent TGF-β binding proteins (LTBPs) during formation of the extracellular matrix in human lung fibroblasts. Exp. Cell Res. 310:370–382. 10.1016/j.yexcr.2005.08.008 [DOI] [PubMed] [Google Scholar]
- Maeda, T., Sakabe T., Sunaga A., Sakai K., Rivera A.L., Keene D.R., Sasaki T., Stavnezer E., Iannotti J., Schweitzer R., et al. 2011. Conversion of mechanical force into TGF-β-mediated biochemical signals. Curr. Biol. 21:933–941. 10.1016/j.cub.2011.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majd, H., Quinn T.M., Wipff P.J., and Hinz B.. 2011. Dynamic expansion culture for mesenchymal stem cells. Methods Mol. Biol. 698:175–188. 10.1007/978-1-60761-999-4_14 [DOI] [PubMed] [Google Scholar]
- Massam-Wu, T., Chiu M., Choudhury R., Chaudhry S.S., Baldwin A.K., McGovern A., Baldock C., Shuttleworth C.A., and Kielty C.M.. 2010. Assembly of fibrillin microfibrils governs extracellular deposition of latent TGF β. J. Cell Sci. 123:3006–3018. 10.1242/jcs.073437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura, S.L.2009. Integrin-mediated transforming growth factor-β activation, a potential therapeutic target in fibrogenic disorders. Am. J. Pathol. 175:1362–1370. 10.2353/ajpath.2009.090393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono, R.N., Sengle G., Charbonneau N.L., Carlberg V., Bächinger H.P., Sasaki T., Lee-Arteaga S., Zilberberg L., Rifkin D.B., Ramirez F., et al. 2009. Latent transforming growth factor β-binding proteins and fibulins compete for fibrillin-1 and exhibit exquisite specificities in binding sites. J. Biol. Chem. 284:16872–16881. 10.1074/jbc.M809348200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajshankar, D., Downey G.P., and McCulloch C.A.. 2012. IL-1β enhances cell adhesion to degraded fibronectin. FASEB J. 26:4429–4444. 10.1096/fj.12-207381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez, F., and Rifkin D.B.. 2009. Extracellular microfibrils: contextual platforms for TGFβ and BMP signaling. Curr. Opin. Cell Biol. 21:616–622. 10.1016/j.ceb.2009.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson, I.B., and Rifkin D.B.. 2013. Unchaining the beast; insights from structural and evolutionary studies on TGFβ secretion, sequestration, and activation. Cytokine Growth Factor Rev. 24:355–372. 10.1016/j.cytogfr.2013.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenzweig, D.H., Matmati M., Khayat G., Chaudhry S., Hinz B., and Quinn T.M.. 2012. Culture of primary bovine chondrocytes on a continuously expanding surface inhibits dedifferentiation. Tissue Eng. Part A. 18:2466–2476. 10.1089/ten.tea.2012.0215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatier, L., Chen D., Fagotto-Kaufmann C., Hubmacher D., McKee M.D., Annis D.S., Mosher D.F., and Reinhardt D.P.. 2009. Fibrillin assembly requires fibronectin. Mol. Biol. Cell. 20:846–858. 10.1091/mbc.E08-08-0830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatier, L., Djokic J., Fagotto-Kaufmann C., Chen M., Annis D.S., Mosher D.F., and Reinhardt D.P.. 2013. Complex contributions of fibronectin to initiation and maturation of microfibrils. Biochem. J. 456:283–295. 10.1042/BJ20130699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saharinen, J., Hyytiäinen M., Taipale J., and Keski-Oja J.. 1999. Latent transforming growth factor-β binding proteins (LTBPs)—structural extracellular matrix proteins for targeting TGF-β action. Cytokine Growth Factor Rev. 10:99–117. 10.1016/S1359-6101(99)00010-6 [DOI] [PubMed] [Google Scholar]
- Sakai, T., Johnson K.J., Murozono M., Sakai K., Magnuson M.A., Wieloch T., Cronberg T., Isshiki A., Erickson H.P., and Fässler R.. 2001. Plasma fibronectin supports neuronal survival and reduces brain injury following transient focal cerebral ischemia but is not essential for skin-wound healing and hemostasis. Nat. Med. 7:324–330. 10.1038/85471 [DOI] [PubMed] [Google Scholar]
- Sarrazy, V., Koehler A., Chow M.L., Zimina E., Li C.X., Kato H., Caldarone C.A., and Hinz B.. 2014. Integrins αvβ5 and αvβ3 promote latent TGF-β1 activation by human cardiac fibroblast contraction. Cardiovasc. Res. 102:407–417. 10.1093/cvr/cvu053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi, M., Zhu J., Wang R., Chen X., Mi L., Walz T., and Springer T.A.. 2011. Latent TGF-β structure and activation. Nature. 474:343–349. 10.1038/nature10152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shifrin, Y., Arora P.D., Ohta Y., Calderwood D.A., and McCulloch C.A.. 2009. The role of FilGAP-filamin A interactions in mechanoprotection. Mol. Biol. Cell. 20:1269–1279. 10.1091/mbc.E08-08-0872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimbori, C., Gauldie J., and Kolb M.. 2013. Extracellular matrix microenvironment contributes actively to pulmonary fibrosis. Curr. Opin. Pulm. Med. 19:446–452. 10.1097/MCP.0b013e328363f4de [DOI] [PubMed] [Google Scholar]
- Singh, P., Carraher C., and Schwarzbauer J.E.. 2010. Assembly of fibronectin extracellular matrix. Annu. Rev. Cell Dev. Biol. 26:397–419. 10.1146/annurev-cellbio-100109-104020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, M.L., Gourdon D., Little W.C., Kubow K.E., Eguiluz R.A., Luna-Morris S., and Vogel V.. 2007. Force-induced unfolding of fibronectin in the extracellular matrix of living cells. PLoS Biol. 5:e268. 10.1371/journal.pbio.0050268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storm, C., Pastore J.J., MacKintosh F.C., Lubensky T.C., and Janmey P.A.. 2005. Nonlinear elasticity in biological gels. Nature. 435:191–194. 10.1038/nature03521 [DOI] [PubMed] [Google Scholar]
- Taipale, J., Miyazono K., Heldin C.H., and Keski-Oja J.. 1994. Latent transforming growth factor-β1 associates to fibroblast extracellular matrix via latent TGF-β binding protein. J. Cell Biol. 124:171–181. 10.1083/jcb.124.1.171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taipale, J., Saharinen J., Hedman K., and Keski-Oja J.. 1996. Latent transforming growth factor-beta 1 and its binding protein are components of extracellular matrix microfibrils. J. Histochem. Cytochem. 44:875–889. 10.1177/44.8.8756760 [DOI] [PubMed] [Google Scholar]
- Todorovic, V., and Rifkin D.B.. 2012. LTBPs, more than just an escort service. J. Cell. Biochem. 113:410–418. 10.1002/jcb.23385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasek, J.J., Gabbiani G., Hinz B., Chaponnier C., and Brown R.A.. 2002. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat. Rev. Mol. Cell Biol. 3:349–363. 10.1038/nrm809 [DOI] [PubMed] [Google Scholar]
- Velling, T., Risteli J., Wennerberg K., Mosher D.F., and Johansson S.. 2002. Polymerization of type I and III collagens is dependent on fibronectin and enhanced by integrins α11β1 and α2β1. J. Biol. Chem. 277:37377–37381. 10.1074/jbc.M206286200 [DOI] [PubMed] [Google Scholar]
- Wipff, P.J., and Hinz B.. 2008. Integrins and the activation of latent transforming growth factor β1 - an intimate relationship. Eur. J. Cell Biol. 87:601–615. 10.1016/j.ejcb.2008.01.012 [DOI] [PubMed] [Google Scholar]
- Wipff, P.J., Rifkin D.B., Meister J.J., and Hinz B.. 2007. Myofibroblast contraction activates latent TGF-β1 from the extracellular matrix. J. Cell Biol. 179:1311–1323. 10.1083/jcb.200704042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wipff, P.J., Majd H., Acharya C., Buscemi L., Meister J.J., and Hinz B.. 2009. The covalent attachment of adhesion molecules to silicone membranes for cell stretching applications. Biomaterials. 30:1781–1789. 10.1016/j.biomaterials.2008.12.022 [DOI] [PubMed] [Google Scholar]
- Worthington, J.J., Klementowicz J.E., and Travis M.A.. 2011. TGFβ: a sleeping giant awoken by integrins. Trends Biochem. Sci. 36:47–54. 10.1016/j.tibs.2010.08.002 [DOI] [PubMed] [Google Scholar]
- Wynn, T.A.2008. Cellular and molecular mechanisms of fibrosis. J. Pathol. 214:199–210. 10.1002/path.2277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynn, T.A., and Ramalingam T.R.. 2012. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat. Med. 18:1028–1040. 10.1038/nm.2807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong, C., Chrzanowska-Wodnicka M., Brown J., Shaub A., Belkin A.M., and Burridge K.. 1998. Rho-mediated contractility exposes a cryptic site in fibronectin and induces fibronectin matrix assembly. J. Cell Biol. 141:539–551. 10.1083/jcb.141.2.539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilberberg, L., Todorovic V., Dabovic B., Horiguchi M., Couroussé T., Sakai L.Y., and Rifkin D.B.. 2012. Specificity of latent TGF-β binding protein (LTBP) incorporation into matrix: role of fibrillins and fibronectin. J. Cell. Physiol. 227:3828–3836. 10.1002/jcp.24094 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.