

In Drosophila, AKT1 and the Dcp-1 caspase calibrate basal autophagy levels and cellular metabolism by regulating Acn stability.
Abstract
How cellular stresses up-regulate autophagy is not fully understood. One potential regulator is the Drosophila melanogaster protein Acinus (Acn), which is necessary for autophagy induction and triggers excess autophagy when overexpressed. We show that cell type–specific regulation of Acn depends on proteolysis by the caspase Dcp-1. Basal Dcp-1 activity in developing photoreceptors is sufficient for this cleavage without a need for apoptosis to elevate caspase activity. On the other hand, Acn was stabilized by loss of Dcp-1 function or by the presence of a mutation in Acn that eliminates its conserved caspase cleavage site. Acn stability also was regulated by AKT1-mediated phosphorylation. Flies that expressed stabilized forms of Acn, either the phosphomimetic AcnS641,731D or the caspase-resistant AcnD527A, exhibited enhanced basal autophagy. Physiologically, these flies showed improvements in processes known to be autophagy dependent, including increased starvation resistance, reduced Huntingtin-induced neurodegeneration, and prolonged life span. These data indicate that AKT1 and caspase-dependent regulation of Acn stability adjusts basal autophagy levels.
Introduction
Two modes of macroautophagy, referred to as autophagy from here on, support cell survival by alleviating distinct problems. A first predominantly catabolic mode of autophagy enables acutely starving cells to nonspecifically degrade cellular content and reclaim necessary building blocks. The lysosome-localized mammalian target of rapamycin (TOR; mTOR) signaling complex plays a major role in the regulation of this mode of autophagy (Settembre et al., 2012; Yan and Lamb, 2012; Efeyan et al., 2013; Jewell et al., 2013). A second mode, sometimes called quality control autophagy, appears to operate at low levels in most cells to eliminate deleterious cellular content and debris, ranging in size from invading pathogens and dysfunctional mitochondria to misfolded proteins (Kroemer et al., 2010). Both of these modes use the core autophagy proteins to initiate the generation of isolation membranes (also known as phagophores), promote their growth to autophagosomes, and finally promote their fusion with lysosomes to initiate degradation of the captured content (Mizushima et al., 2011).
Studies of starvation-induced autophagy in yeast and in the context of tumorigenesis led to the discovery of most of the core autophagy proteins (Klionsky and Ohsumi, 1999; Liang et al., 1999). Since then, significant progress has been made in understanding the homeostatic regulation of autophagy in response to nutrient deprivation (Yang and Klionsky, 2010; White, 2012). Less well understood, however, is the regulation of basal autophagy levels. The importance of the quality control function of basal autophagy was highlighted by autophagy-deficient mice and flies, which undergo neurodegeneration (Hara et al., 2006; Komatsu et al., 2006; Juhász and Neufeld, 2008) and cardiac hypertrophy (Nakai et al., 2007). Clearance of aggregation-prone proteins is a key aspect of autophagy in preventing disease and extending life span (Meléndez et al., 2003; Vellai et al., 2009; Demontis and Perrimon, 2010; Kroemer et al., 2010).
Basal autophagy and life span can be enhanced by increased expression of autophagy genes (Simonsen et al., 2008). For their acute up-regulation, lysosome-bound TFEB (transcription factor EB) plays an important role in transcriptional responses to metabolic changes through its mTOR-regulated nuclear relocalization (Settembre et al., 2012). Long-term adjustment of basal autophagy relies on multiple factors regulating transcriptional and posttranscriptional processes (Füllgrabe et al., 2014).
One such regulator of autophagy is encoded by the acinus (acn) gene (Haberman et al., 2010). Acn proteins are highly conserved between flies and mammals, but their function is incompletely understood. Acn was originally discovered as a protein that induces DNA condensation or fragmentation after its activation by Caspase-3 in apoptotic cells (Sahara et al., 1999; Joselin et al., 2006). Acn is, however, not required for apoptosis, which progresses unimpeded in Drosophila melanogaster and mammalian Acn loss-of-function cells (Joselin et al., 2006; Haberman et al., 2010). Acn, RNPS1, and SAP18 form the nuclear apoptosis- and splicing-associated protein (ASAP) complex (Schwerk et al., 2003; Joselin et al., 2006; Murachelli et al., 2012), which modulates RNA metabolism through interactions with the exon junction complex, spliceosomes, and messenger ribonucleoprotein particles (Tange et al., 2005; Joselin et al., 2006; Singh et al., 2010; Hayashi et al., 2014; Malone et al., 2014).
Drosophila acn alleles were identified in a screen for mutants affecting endolysosomal trafficking (Haberman et al., 2010). In the same study, a role of Acn in regulating autophagy was revealed by loss- and gain-of-function experiments in Drosophila larval fat bodies, a tissue poised for robust up-regulation of autophagy in response to environmental or developmental cues (Rusten et al., 2004; Scott et al., 2004). In fat body cells lacking acn function, early autophagosomes stall and fail to fuse with lysosomes. Conversely, systemic Acn overexpression causes excess autophagy that is lethal to flies. Genetic epistasis experiments placed the requirement of Acn for regulating autophagy downstream or in a pathway parallel to the TOR complex: up-regulation of autophagy in response to dominant-negative TOR is suppressed in acn mutants, but enhanced autophagy as a result of Acn gain of function is not repressed by activated TOR (Haberman et al., 2010). A high-content RNAi screen for genes involved in viral autophagy in human cells revealed a conserved role for Acn in autophagy (Orvedahl et al., 2011).
Here, we explore the regulation of Acn function as part of a pathway regulating basal levels of autophagy. We find that Acn integrates multiple inputs as its activity is inhibited by Caspase-mediated cleavage and enhanced by AKT1-dependent phosphorylation. Genetically stabilized forms of Acn increase the level of basal autophagy sufficiently to prolong life span and partially suppress Huntingtin-induced neurodegeneration.
Results
Acn is dynamically regulated at the protein level
Acn levels in developing photoreceptor cells are dynamically regulated in a cell type–specific manner (Haberman et al., 2010). To investigate the mechanisms that regulate Acn in eye discs, we expressed two GFP transgenes under control of the endogenous 4-kb acn genomic region (Fig. 1 A), which is sufficient to rescue all acn phenotypes (Fig. S1; Haberman et al., 2010). A transgene in which GFP was fused to the N terminus of Acn mimicked the expression pattern of endogenous Acn (Fig. 1, B and C) and was sufficient to rescue acn lethality. In contrast, when GFP was expressed from the same control region but separated from Acn by a stop codon, cell type–specific regulation was lost (Fig. 1 D). This argues against a major contribution of transcriptional control to dynamic changes of Acn expression in developing photoreceptors. To directly test whether Acn, when separated from its genomic control regions and 5′ and 3′ UTRs, is regulated at the protein level, we used the eye-specific glass multimer reporter (GMR)–Gal4 driver to coexpress Myc-tagged Acn and an endoplasmic reticulum–targeted GFP-KDEL from upstream activating sequence (UAS) transgenes with identical UTRs (Fig. 1 E). As expected, GFP-KDEL was expressed in all cells posterior to the furrow, but Acn levels varied greatly: some cells displayed strong expression, whereas others were devoid of it (e.g., Fig. 1 E, arrowheads). These results indicate that Acn is dynamically regulated at the protein level.
Figure 1.
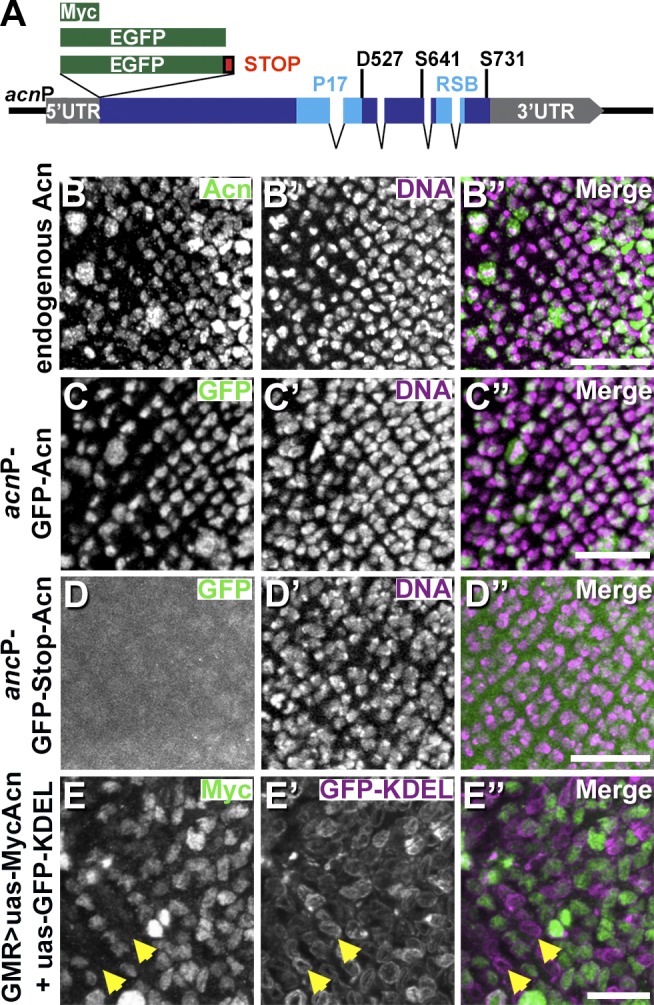
Acn is regulated on the protein level. (A) Schematic of acn locus and genomic transgenes all inserted at 96F3 for this study. RSB is the conserved RNPS1 and SAP18 binding site (Martelli et al., 2012). (B–E″) Projections of confocal sections of eye discs showing early stages of photoreceptor development. Regions of eye discs shown are just posterior to the furrow as in the white frame of Fig. 2 D. Images show wild-type discs stained for endogenous Acn (B) and DNA (B′) or discs with indicated transgenes stained for GFP (C and D) and DNA (C′ and D′) or for Myc (E) and GFP (E′). Arrows in E–E″ point to cells expressing GFP-KDEL but lacking Myc-Acn. Merged images are to the right. Bars, 20 µm. Detailed genotypes are given in Table S2.
Acn is cleaved at the conserved D527 site
Acn is highly conserved; Drosophila and human Acn share 53% identity in their P17 domains (Fig. 1 A). This includes D527 (Fig. 2 A), which aligns with the aspartate residue cleaved by Caspase-3 in apoptotic mammalian cells (Sahara et al., 1999). To test whether Drosophila Acn is proteolytically cleaved at this site in vivo, we generated flies in which endogenous Acn was replaced by Myc-tagged AcnD527A or Acnwt as a control. Expression was under control of the endogenous acn promoter and enhancers within a 4-kb acn genomic region (Fig. 1 A). All genomic acn transgenes were inserted at 96F3 using the site-specific unidirectional PhiC31 recombinase to avoid insertion site–specific differences in expression levels (Groth et al., 2004; Venken et al., 2006). When assessed by quantitative RT-PCR, the level of expression was indistinguishable for both transgenes, and both rescued lethality and endocytic trafficking defects of acn-null alleles (Fig. S1). We will refer to these rescued flies that, in an acn-null background, express exclusively transgenic forms of Acn as Acnwt or AcnD527A flies from here on. This system allows us to test in vivo the properties of various Acn mutants without interference from the endogenous Acn protein. In such rescued larvae, levels of AcnD527A were about three times higher when compared with Acnwt (Fig. 2, B and C), indicating that the larval Acn protein was stabilized by loss of the caspase cleavage site.
Figure 2.

Acn is cleaved and inactivated at aspartate D527. (A) Conserved aspartate D527 of Drosophila melanogaster (Dm) Acn is shown in alignment with Drosophila yakuba (Dy), Anopheles gambiae (Ag), Xenopus tropicalis (Xt), mouse (Mus musculus [Mm]), and human (Homo sapiens [Hs]) proteins. Color code for amino acid groups: brown for aromatic, white for hydrophobic, salmon for hydroxyls, red for acidic, blue for basic, cyan for amido, and yellow for sulfur. (B) Western blot of lysates from larvae probed for Acn and Actin. (C) Quantification of blots for Acn levels relative to Actin (n = 3). Error bars show means ± SD. *, P < 0.05; ***, P < 0.001. (D) Micrograph of eye disc stained for DNA (magenta) and Acn (green). The frame just posterior to the furrow (arrow) indicates the region shown in high magnification images (E and F). (D′) Micrograph of an eye disc expressing nuclear GFP. Dotted lines encircle R3/4 and R2/5/8 photoreceptors just posterior to the furrow. (E–F″) Dynamic changes in Acn expression in photoreceptors visualized in projections of confocal sections of eye discs from acnP-Myc-Acnwt (E) and acnP-Myc-AcnD527A (F) larvae double labeled for Myc and C-terminal Acn epitopes of Myc-Acn (merged images are shown in E″ and F″). Arrows point to R3/4 photoreceptor pairs just posterior to the furrow. Bars, 10 µm. (G and H) Quantification of Acn and Myc levels in early R3/4 and R2/5/8 cells measured from ≥180 ommatidia from four eye discs, each for Acnwt and AcnD527A. (G) Cumulative frequency histograms of normalized fluorescence intensities for Myc and Acn immunoreactivity. (H) Intensity blots of normalized integrated intensities of Acn and Myc immunoreactivity for individual groups of R3/4 and R2/5/8 cells in Myc-Acnwt and Myc-AcnD527A discs and calculated Pearson correlation coefficients r. fluor. int., fluorescence intensities. (I–N) SEM images of wild-type control (I) and eyes expressing under GMR-Gal4 control the indicated UAS transgenes: none (J), UAS-Acnwt (K), UAS-AcnD527A (L), UAS-Acn1–527 (M), and UAS-Acn528–739 (N). Bar, 50 µm. Detailed genotypes and roughness quantification are given in Tables S1 and S2. wt, wild type.
Acn stabilization was even more obvious in eye discs (Fig. 2, D–H) double stained with antibodies against the N-terminal Myc epitope and an Acn epitope close to the C terminus (Haberman et al., 2010). In eye discs expressing Myc-Acnwt, the ratio of Myc to Acn levels changed dynamically in photoreceptor nuclei. In emerging R3/R4 cells, identified by their shape and position in ommatidia close to the furrow (Fig. 2 D′; Wolff and Ready, 1993), the ratio of Myc to Acn was high. That ratio dropped in older R3/R4 cells more posterior to the furrow. Loss of the Myc-tagged N-terminal part of Acn relative to the C-terminal region is consistent with cleavage of Acn and preferential degradation of the N-terminal fragment. In AcnD527A eye discs, in contrast, Myc immunoreactivity was increased in R3/R4 as well as in R2/R5/R8 cells (Fig. 2, E–G). Stabilization of AcnD527 was also revealed by the increased correlation of Myc and Acn immunoreactivity in early photoreceptor cells when compared with Acnwt (Fig. 2 H). These data indicate that in developing photoreceptors Acn is cleaved at asparate527.
In apoptotic mammalian cells, a chromatin-modifying function of Acn is activated by caspase-mediated cleavage (Sahara et al., 1999; Hu et al., 2005; Joselin et al., 2006). To test whether Drosophila Acn was activated by its cleavage, we compared the effects of UAS-controlled transgenes expressing full-length Acn or truncated versions that would be generated by cleavage at aspartate527 (Fig. 2, I–N). These UAS transgenes were expressed under control of the eye-specific GMR-Gal4 driver. Compared with wild-type (Fig. 2 I) or GMR-Gal4 control eyes (Fig. 2 J), eyes expressing wild-type UAS-Acn were mildly rough (Fig. 2 K). Expression of stabilized AcnD527A caused enhanced roughness (Fig. 2 L). In contrast, eyes expressing the truncated UAS-Acn1–527 (Fig. 2 M) or UAS-Acn528–739 (Fig. 2 N) appeared unchanged from controls. Furthermore, Daughterless (Da)-Gal4–driven expression of truncated Acn proteins, unlike the corresponding UAS-Acnwt, could not rescue lethality of acn-null alleles. In summary, our data indicate that in developing photoreceptors, Acn is inactivated by cleavage at asparate527.
Acn cleavage regulates autophagy
We have previously shown that Acn function is required downstream of TOR for autophagy, whereas elevated levels of Acn induce autophagy independent of TOR activity (Haberman et al., 2010). We therefore tested whether larvae expressing only stabilized AcnD527A from the endogenous promoter exhibited altered levels of autophagy. In these AcnD527A eye discs, Atg8-positive autophagosomes formed at rates significantly higher than in Acnwt, indicating increased autophagy (Fig. 3, A–C).
Figure 3.

Stabilized AcnD527A enhances basal autophagy. (A and B) Micrographs of fed Acnwt or AcnD527A larval eye discs stained for Atg8a. (C) Quantification of Atg8a punctae in eye discs from five larvae. (D) Western blot of fat body lysates from 96-h fed larvae probed for Acn and Actin. (E) Quantification of blots for Acn levels relative to Actin (n = 3). (F–G′ and I–J′) Micrographs of Acnwt or AcnD527A fat bodies encompassing six to eight cells from 96-h fed or starved size-matched larvae. (F–G′) Fat bodies stained with antibodies against Atg8a. (H) Quantification of Atg8a punctae averaged from five larvae from one representative experiment out of three repeats. (I–J′) GFP-Atg8a fluorescence detected in live fat bodies. (K) Quantification of GFP-Atg8a punctae averaged from five larvae from one representative experiment out of three repeats. (L–M″) TEMs of fed Acnwt or AcnD527A fat bodies. Examples of dense lysosomes (arrowheads) and membrane-enriched autolysosomes (arrows) are shown at higher magnification. (N) Quantification of percentage of autolysosomal area averaged from 25 images per genotype. (O) Quantification of diameters of ≥100 lysosomes and autolysosomes per genotype. Detailed genotypes are given in Table S2. wt, wild type. Error bars show means ± SD. **, P < 0.01; ***, P < 0.001.
For a more detailed investigation, we turned to fat bodies, which constitute a well-established autophagy model in Drosophila (Rusten et al., 2004; Scott et al., 2004). Compared with Acnwt, levels of AcnD527A are elevated in fat bodies (Fig. 3, D and E). In fed larvae, few Atg8-positive punctae accumulate in Acnwt fat bodies, and upon amino acid starvation, they increase in number (Fig. 3, F, F′, and H). In AcnD527A larval fat bodies, however, numerous Atg8-positive structures were detected even without starvation (Fig. 3, G and H), and their number was further increased by starvation (Fig. 3, G′ and H). Elevated autophagy in fed AcnD527A larvae was also observed by a significantly increased Atg8a-I to Atg8a-II conversion (Fig. S2, L and M). Interestingly, starvation increased levels of Acn proteins only modestly by ∼20% (Fig. S1, I and J).
To distinguish between effects of Acn on autophagosomes and autolysosomes, we quantified GFP-Atg8a–positive punctae in live fat bodies. GFP-Atg8a preferentially detects autophagosomes, as GFP fluorescence is quenched by the acidic pH of autolysosomes. As expected, GFP-Atg8a punctae were rare in fed Acnwt larvae (Fig. 3, I and K), and their number increased upon starvation (Fig. 3, I′ and K). In contrast, in AcnD527A larvae, GFP-Atg8a punctae were detected without starvation (Fig. 3, J and K) and further increased upon starvation (Fig. 3, J′ and K). Furthermore, an increase in autolysosomes was detected using LysoTracker staining of fed and starved AcnD527A fat bodies (Fig. S2, A, B, and E). Finally, we analyzed these changes on the ultrastructural level. Transmission EM (TEM) of fed 96-h larvae revealed that lysosomes and autolysosomes in fed AcnD527A larval fat bodies were significantly larger and more abundant compared with Acnwt (Fig. 3, L and M). The area occupied by lysosomal and autolysosomal structures was increased almost 10-fold (Fig. 3 N), and their mean diameter was about fourfold larger in AcnD527A larvae (Fig. 3 O). Together, our data indicate an elevated level of starvation-independent basal autophagy at all stages when Acn cleavage at asparate527 is blocked.
Dcp-1 cleaves Acn
To test which protease is responsible for cleaving Drosophila Acn, we performed a targeted RNAi screen. We took advantage of the rough eye phenotype resulting from eye-specific GMR-Gal4–driven expression of UAS-Acnwt at elevated temperature (28°C; Fig. 4, A and B). This phenotype was further enhanced when stabilized UAS-AcnD527A was expressed instead (Fig. 4 C). We reasoned that reduced activity of the protease responsible for Acn cleavage should enhance the roughness induced by UAS-Acnwt to a similar level to that of cleavage-resistant UAS-AcnD527A.
Figure 4.

Acn levels and activity is regulated by Dcp-1. (A–G) Micrographs of wild type (A) and eyes in which GMR-Gal4 drives expression of Acnwt (B), AcnD527A (C), Acnwt + Dcp-1–RNAi (D), Dcp-1-RNAi (E), Acnwt + Dredd-RNAi (F), Acnwt + Dronc-RNAi (G). Bar, 50 µm. (H–K′) Micrographs of larval eye discs stained for DNA and Acn (green) from OreR (H and H′), dcp-1 (I and I′), GMR-Gal4, UAS–Dcp-1–RNAi (J and J′), and GMR-Gal4, UAS-Dredd-1–RNAi (K and K′). (L–M″′) Micrographs of eye (L–L″′) and antennal (M–M″′) discs with clones of cells expressing Dcp-1–RNAi marked by RFP and stained for Acn and DNA. Arrow indicates the morphogenetic furrow and the broken line indicates the clone boundary. (N) Western blots of wild type (+/+) or dcp-1 larvae (−/−) expressing Myc-Acnwt or Myc-AcnD527A as indicated. Blots were probed for Myc or Actin. (O) Quantification of blots (n = 3) as shown in N. Bars: (H–M) 10 µm. Detailed genotypes and roughness quantification are given in Tables S1 and S2. Error bars show means ± SD. **, P < 0.01; ***, P < 0.001.
Among the eight caspases tested (Table S1), only dcp-1 (death caspase-1) genetically interacted with Acn. Two different UAS–Dcp-1–RNAi transgenes, when coexpressed with UAS-Acnwt using the GMR-Gal4 driver enhanced roughness and reduced eye size similar to UAS-AcnD527A (Fig. 4 D and not depicted). Importantly, expression of Dcp-1 RNAi by itself did not cause eye roughness (Fig. 4 E). For all other caspases tested in the eye, RNAi coexpression with UAS-Acnwt did not obviously modify the eye roughness (Fig. 4, F and G, shows Dredd-RNAi and Dronc-RNAi as examples).
To investigate the role of Dcp-1 further, we analyzed its effect on Acn levels. Eyes mutant for dcp-1 (Fig. 4 I′; Laundrie et al., 2003) or expressing Dcp-1–RNAi (Fig. 4 J′), but not Dredd-RNAi (Fig. 4 K′), exhibited increased Acn levels in photoreceptor cells in larval eye discs compared with wild type (Fig. 4 H′). Moreover, flippase-induced Dcp-1 knockdown clones (marked by RFP) in eye-antennal discs demonstrated enhanced Acn levels compared with surrounding wild-type tissue (Fig. 4, L and M). Furthermore, we compared the level of Myc-tagged Acnwt and AcnD527A in both wild-type and dcp-1 larvae by Western blotting. In a wild-type background, the level of Myc-tagged AcnD527A is higher than Acnwt (Fig. 4, N and O). This difference is significantly reduced, and the level of Acnwt is significantly higher in the dcp-1 background (Fig. 4, N and O). These data identify the Caspase-3 homologue Dcp-1 as the main protease necessary for cleavage of Acn at asparate527 in nonapoptotic cells, such as developing photoreceptors.
Dcp-1−dependent cleavage of Acn regulates autophagy
Next, we tested whether loss of Dcp-1 function and the resulting Acn stabilization induces autophagy. We compared autophagosome levels in 96-h fat bodies using Atg8a staining. Compared with wild type, dcp-1 mutant or Dcp-1 knockdown fat bodies displayed robustly induced autophagy in fed larvae (Fig. 5, A–C and F). A similar enhancement was observed for the formation of autolysosomes detected by LysoTracker staining (Fig. S2, F–K). Enhanced autophagy was specific for Dcp-1 caspase, as knockdown of Dredd did not induce autophagy in fed larvae (Fig. 5, E and F; and Fig. S2, J and K). For mutant and RNAi-induced dcp-1 loss-of-function larvae, the number of Atg8a-positive punctae was further increased upon starvation (Fig. 5, B′ and C′). To test whether the increased basal autophagy in response to loss of Dcp-1 function is mediated through elevated levels of Acn, we generated animals in which Dcp-1 and Acn were knocked down together. In those larvae, fed or starved, autophagy was reduced back to wild-type levels (Fig. 5, D and F; and Fig. S2, I and K), indicating that the remaining protein after Acn knockdown (Fig. S1 H) is sufficient to support starvation-induced autophagy (Fig. 5, D′ and F; and Fig. S2, I′ and K) but not sufficient to drive elevated levels of basal autophagy in a fed dcp-1 background (Fig. 5, D and F; and Fig. S2, I and K). Together, these data are consistent with a requirement for Dcp-1 in nonapoptotic cells to control the level of Acn protein and thereby the basal starvation-independent level of autophagy.
Figure 5.
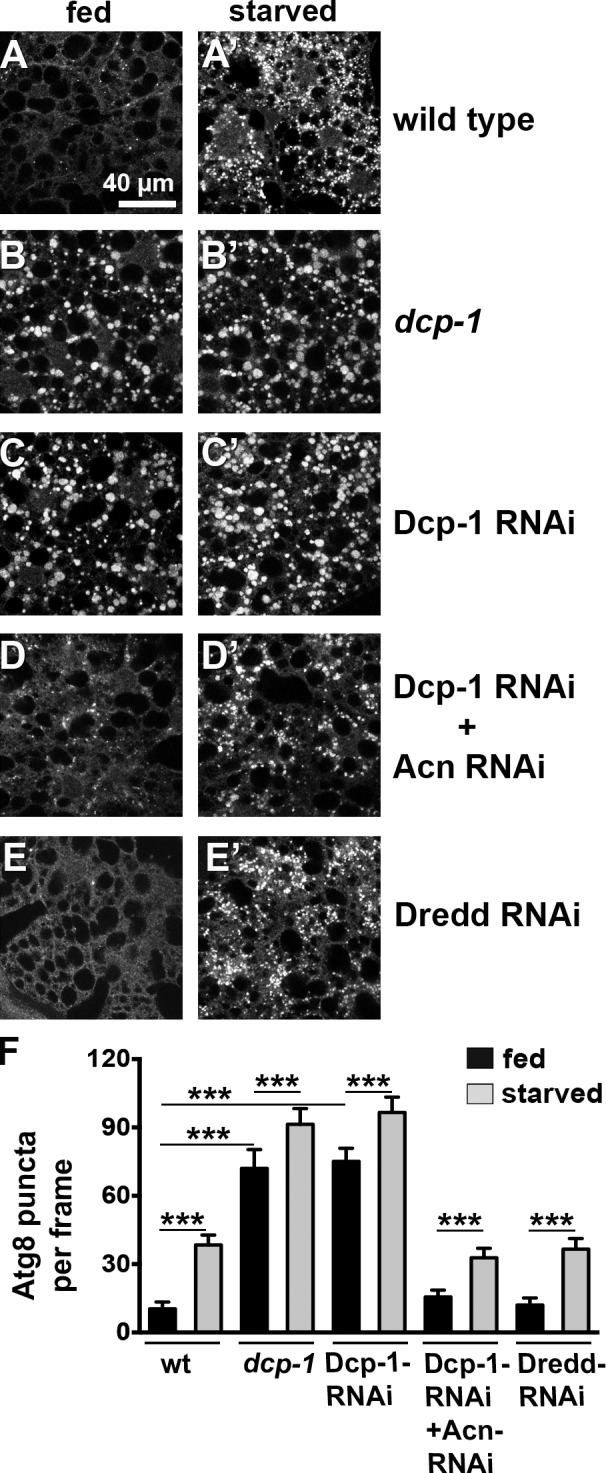
Dcp-1 regulates basal autophagy. (A–E′) Micrographs show fat bodies from 96-h fed and starved size-matched larvae stained for Atg8a. Genotypes are wild type (A), dcp-1 (B), Da-Gal4, UAS–Dcp-1–RNAi (C), Da-Gal4, UAS–Dcp-1–RNAi + UAS-Acn-RNAi (D), and Da-Gal4, UAS-Dredd-RNAi (E). Each image encompasses six to eight cells. (F) Quantification of Atg8a punctae averaged from five larvae from one representative experiment out of three repeats. wt, wild type. See also Fig. S1 H for efficiency of Acn knockdown. Detailed genotypes are given in Table S2. Error bars show means ± SD. ***, P < 0.001.
AKT1 regulates Acn stability
High-level overexpression of Acn causes autophagy-dependent lethality (Haberman et al., 2010). Therefore, we tested whether the Acn-induced rough eye (Figs. 2 K and 6 A; compare with wild-type eyes in Figs. 2 I and 4 A) is also a reflection of overactive autophagy. We found that knockdown of Atg1 or Atg7 suppressed the UAS-Acnwt–induced rough eye (Fig. 6, B and D; and Table S1), whereas expression of RNAi for Atg1 or Atg7 by itself resulted in an eye indistinguishable from wild type (Fig. 6, C and E). This indicates that the Acn-induced rough eye, at least in part, is caused by overactive autophagy.
Figure 6.

AKT1 phosphorylates Acn. (A–I) Micrographs of eyes expressing the indicated UAS transgenes under control of GMR-Gal4. Bar, 50 µm. (J) Western blots of anti-Myc immunoprecipitates of Myc-Acnwt and Myc-AcnS641,731A expressed in S2 cells and detected with antibodies against Acn or Acn-pS641. S2 cell lysates were probed for Actin. IP, immunoprecipitation; wt, wild type. (K) Western blots of lysates from S2 cells expressing Myc-Acnwt that were treated without (−) or with calf intestinal phosphatase (CIP) and probed for Acn, Acn-pS641, or Actin. (L–M″) Apical most regions of eye discs that include cone cell nuclei (marked CC) are depicted in z projections of confocal sections. Eye discs were from larvae expressing Myc-Acnwt (L–L″) or Myc-AcnS641,731A (M–M″) and were stained for Acn-pS641, Myc, and DNA. Arrows point to unspecific staining in mitotic cells. (N–N″) Projection of confocal sections of eye disc expressing AKT1-RNAi within RFP-marked clones (magenta) and stained for Acn-pS641 (N′) and DNA (N″). The broken line indicates the clone boundary. Detailed genotypes and roughness quantification are given in Tables S1 and S2.
Next, we used this sensitized system to test whether AKT1 functions to regulate Acn-mediated autophagy. Two findings suggested such a role of AKT1: (1) Acn contains two conserved AKT1 consensus targets sites (RxRxxS/T) at S641 and S731 (Fig. 1 A), and (2) AKT1 can phosphorylate mammalian Acn in vitro and reduce its proteolytic cleavage (Hu et al., 2005). Importantly, the UAS-Acnwt–induced rough eye (Fig. 6 A) was suppressed when UAS-AKT1-RNAi was coexpressed (Fig. 6 F). In contrast, by themselves, AKT1 knockdown (Fig. 6 G) or UAS-AKT1 overexpression in the eye (Fig. 6 H) did not result in visible phenotypes. GMR-Gal4–driven coexpression of UAS-Acnwt and UAS-AKT1, however, enhanced the rough eye (Fig. 6 I). These strong genetic interactions between Acn and AKT1 are consistent with AKT1 phosphorylating Acn and thereby enhancing its activity.
To further analyze this possibility, we generated a phosphospecific antibody against Acn-pS641, one of the two conserved AKT1 target sites. To confirm its specificity, we used S2 cells to express AKT1 together with either Myc-tagged Acnwt or AcnS641,731A, which eliminates both potential AKT1 target sites. After immunoprecipitation using anti-Myc antibodies, both Acn proteins were detected by anti-Acn antibodies on Western blots. The phosphospecific Acn-pS641 antibody, however, detected only wild-type Acn but not dephosphomimetic AcnS641,731A (Fig. 6 J). Furthermore, in lysates of Myc-Acnwt–expressing S2 cells, treatment with calf intestinal phosphatase substantially reduced the signals obtained with Acn-pS641 on Western blots but not those of anti-Acn or Actin control antibodies (Fig. 6 K). Together, these data indicate that Acn is phosphorylated at serine641 and that our Acn-pS641 antibody specially recognizes phosphorylated Acn.
To explore the role of Acn phosphorylation, we generated flies in which endogenous Acn was replaced by Myc-AcnS641,731A or Myc-AcnS641,731D substituting either an inert alanine or a phosphomimetic aspartate residue for the two potential AKT1 phosphorylation sites. Both mutant Acn proteins were expressed from genomic transgenes inserted at the same 96F3 landing site as described previously for Acnwt and AcnD527A (Fig. 1 A), and both rescued lethality and endocytic trafficking defects of acn-null mutations (Fig. S1, A–F). Acn-null flies rescued by the phosphomimetic AcnS641,731,D or the dephosphomimetic AcnS641,731A did not display externally visible phenotypes.
We used these flies to explore phosphorylation of Acn at serine641 in eye discs. In cone cells, Acnwt is more consistently expressed at high levels than in the dynamically changing expression of photoreceptors (Haberman et al., 2010). Cone cells (Fig. 6 L, marked CC) also show strong staining with Acn-pS641 antibodies. This staining is dramatically reduced in larvae expressing only dephosphomimetic AcnS641,731A (Fig. 6 M), although anti-Myc antibodies detect AcnS641,731A expression in cone cells (Fig. 6 M′). To investigate whether this phosphorylation is caused by AKT1 activity, we generated AKT1 knockdown clones in eye discs marked by RFP. In such clones, Acn-pS641 antibody staining was reduced compared with surrounding wild-type tissue (Fig. 6 N), consistent with AKT1 phosphorylating Acn.
The aforementioned results suggest that phosphorylation of the AKT1 target sites in Acn is the main basis for the genetic interactions between AKT1 and Acn. To further test this possibility, we expressed Acnwt or the AKT1 target site mutants AcnS641,731D and AcnS641,731A from UAS-controlled transgenes. GMR-Gal4–driven coexpression of AKT1 enhanced the rough eye phenotype of the Acnwt transgene (Fig. 7, A, B, and G) but not roughness of either of the transgenes with mutated AKT1 target sites (Fig. 7, C–G).
Figure 7.

AKT1 phosphorylation regulates Acn levels. (A–F) SEMs of eyes expressing the indicated UAS-Acn transgenes with or without UAS-AKT1 under control of GMR-Gal4. Bar, 50 µm. (G) Number of flies with the indicated degree of roughness for genotypes as in A–F. (H) Western blot of lysates from fed 75-h larvae with the indicated Acn transgenes probed for Acn. Bands of full-length and cleaved Acn are indicated. FL, full length; wt, wild type. (I and J) Acn expression in developing photoreceptors visualized in projections of confocal sections of eye discs from acnP-Myc-AcnS641,731D (I) or acnP-Myc-AcnS641,731A (J) larvae stained for Myc and Acn. Bar, 10 µm. (K and L) Quantification of Acn and Myc levels in early R3/4 and R2/5/8 cells from ≥120 ommatidia from three eye discs. (K) Box and whisker graphs of integrated intensities of Acn and Myc immunoreactivity for individual groups of R3/4 and R2/5/8 cells in discs with the genomic transgenes (genTgs) Myc-AcnS641,731A and Myc-AcnS641,731D. Box and whisker graphs show boxes with medians, 25 and 75 percentiles, and whiskers extending to minimal and maximal values. arb units, arbitrary units. (L) Intensity blots of normalized integrated intensities of Acn and Myc immunoreactivity for individual groups of R3/4 and R2/5/8 cells in Myc-AcnS641,731A and AcnS641,731D discs and calculated Pearson correlation coefficients r. flour. int., fluorescence intensities. Detailed genotypes are given in Table S2.
Next, we tested whether this phosphorylation affects Acn levels by comparing AcnS641,731A and AcnS641,731D flies. Western blotting of whole 96-h larvae or fat bodies revealed that, compared with Acnwt, the level of dephosphomimetic AcnS641,731A was reduced, and phosphomimetic AcnS641,731D was increased similar to the cleavage-resistant AcnD527A (Fig. 2, B and C; and Fig. 3, D and E). This accumulation was not yet detectable in 75-h larval lysates (Fig. 7 H). However, in these early larvae, an intermediate cleavage product detectable for Acnwt and AcnS641,731A was drastically reduced for phosphomimetic AcnS641,731D, similar to caspase cleavage-resistant AcnD527A (Fig. 7 H, arrow). These findings suggest that the AKT-mediated phosphorylation of Acn stabilizes the protein against proteolytic degradation. Stabilization of phosphomimetic AcnS641,731D was also evident in developing photoreceptors in third instar discs (Fig. 7, I–L). Similar to the effect of caspase cleavage-resistant AcnD527A (Fig. 2), phosphomimetic AcnS641,731D exhibited higher levels of staining with Acn and Myc antibodies (Fig. 7, I and K), and the correlation of staining for N-terminal Myc and C-terminal Acn epitopes was increased (Fig. 7 L). Dephosphomimetic AcnS641,731A (Fig. 7 J) displayed low levels of Acn and Myc staining, but the remaining protein displayed elevated levels of correlation between Acn and Myc staining (Fig. 7 L) when compared with wild type (Fig. 2 H). Together, these data support the notion that in fat bodies and developing photoreceptor cells, Acn stability is modified by AKT1-dependent phosphorylation.
As increased levels of Acn can enhance autophagy, we examined Acn proteins with mutated AKT1 target sites for their effect on autophagy. Compared with Acnwt (Fig. 3 A) or dephosphomimetic AcnS641,731A (Fig. 8, A and C), eye discs expressing AcnS641,731D at endogenous levels exhibited increased numbers of autophagosomes (Fig. 8, B and C). Furthermore, fat bodies of fed AcnS641,731D larvae exhibited significantly enhanced basal autophagy at early and late stages of the autophagy pathway as detected by increased GFP-Atg8a punctae, punctae of antibody-stained endogenous Atg8a (Fig. 8, D–I), and LysoTracker staining (Fig. S2, D and E). In addition, TEM revealed larger and more abundant autolysosomes (Fig. 8, J–L). Interestingly, these effects closely resembled those observed in larvae expressing stabilized AcnD527A resistant to Dcp-1 cleavage (compare with Fig. 3). In contrast, basal levels of autophagy were unaltered for destabilized AcnS641,731A (Fig. 8, A–I; and Fig. S2 C). Nevertheless, AcnS641,731A and AcnS641,731D both supported significant increases in autophagy upon starvation (Fig. 8, D–I; and Fig. S2, C′ and D′), arguing against a significant contribution of AKT1-mediated phosphorylation of Acn to the regulation of starvation-induced autophagy. Together, these data indicate that AKT1-mediated phosphorylation regulates Acn stability and, thereby, the basal levels of autophagy.
Figure 8.

AKT1-mediated phosphorylation of Acn modulates basal autophagy. (A and B) Micrographs of fed AcnS641,731A or AcnS641,731D eye discs stained for Atg8a. (C) Quantification of Atg8a punctae in eye discs from five larvae. (D–E′ and G–H′) Micrographs of AcnS641,731A or AcnS641,731D fat bodies encompassing six to eight cells from 96-h fed or starved size-matched larvae. Bars, 40 µm. (D–E′) GFP-Atg8a fluorescence detected in live fat bodies. (F) Quantification of GFP-Atg8a punctae averaged from five larvae from one representative experiment out of three repeats. (G–H′) Fat bodies stained for Atg8a. (I) Quantification of Atg8a punctae averaged from five larvae from a single representative experiment out of three repeats. (J) TEM images of fed 96-h fat bodies expressing AcnS641,731D. Examples of dense lysosomes (arrowheads) and membrane-enriched autolysosomes (arrows) are shown at higher magnification (inset). (K) Quantification of diameters of ≥100 lysosomes and autolysosomes diameters per genotype. (L) Quantification of percentage of autolysosomal area averaged from 25 images per genotype. Wild-types images are shown in Fig. 3 L. Detailed genotypes are given in Table S2. wt, wild type. Error bars show means ± SD. **, P < 0.01; ***, P < 0.001.
Acn gain of function enhances quality control autophagy and improves life span
Next, we explored the physiological consequences of altered Acn regulation and the resulting changes in autophagy. The most striking effect of stabilized Acn proteins was the significant increase in basal autophagy (Figs. 3 and 8), which is important for quality control and maintenance of muscles (Demontis and Perrimon, 2010; Bai et al., 2013) and neurons (Hara et al., 2006; Komatsu et al., 2006; Mizushima et al., 2008). Neurodegeneration in flies can be efficiently followed by electroretinograms (ERGs) as a sensitive measure of the health of photoreceptor neurons (Williamson et al., 2010). At 1 or 2 wk of age, wild-type flies exhibit ERGs with a mean sustained depolarization of ∼10 mV. Acn knockdown significantly reduced depolarization after 1 wk and even further after 2 wk (Fig. 9, A and B). Expression of the Huntingtin-derived HttQ93 protein in photoreceptor cells using GMR-Gal4 triggers strong degeneration in flies with only a small detectable depolarization remaining after 1 wk (Fig. 9, A and B). This was further reduced by Acn knockdown (Fig. 9, A and B), prompting us to test whether increasing autophagy through stabilized versions of Acn could improve retinal degeneration induced by HttQ93. We found that in 2-wk-old flies, one copy of the genomic AcnD527A transgene significantly improved depolarization compared with Acnwt (Fig. 9, C and D) and also reduced the load of aggregated HttQ93 protein as measured by dot blots (Fig. 9 E). AcnS641,731D also lowered aggregated HttQ93 protein levels (Fig. 9 E) but not sufficiently to reduce neurodegeneration (Fig. 9 D).
Figure 9.

Acn regulates quality control autophagy and longevity. (A and B) ERGs of 1- or 2-wk-old flies expressing the indicated UAS transgenes (Tgs) under control of GMR-Gal4. (B) Quantification of sustained negative photoreceptor response averaged from three replicate experiments including at least 64 traces from eight flies. (C and D) ERGs of 2-wk-old flies all expressing UAS-HttQ93 under control of GMR-Gal4 (GMR>HttQ93) and the indicated genomic Acn transgenes. (D) Quantification of sustained negative photoreceptor response from three replicate experiments including at least 64 traces from eight flies. Avg. sust., average sustained. (E) Quantification of dot blots measuring aggregated HttQ93 protein in fly heads expressing UAS-HttQ93 under control of GMR-Gal4 (GMR>HttQ93) and the indicated genomic Acn transgenes. Flies expressing neither indicate background level of dot blot measurements (n = 3). rel., relative; arb units, arbitrary units. (F) Starvation-induced mortality of flies expressing the indicated Acn proteins. In the parenthesis shown are the numbers of initial flies in a single representative experiment. (G) Survival curves of flies expressing the indicated Acn proteins. In the parenthesis shown are the numbers of initial flies in a single representative experiment. Detailed genotypes are given in Table S2. wt, wild type. Error bars show means ± SD. **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
To test whether the beneficial long-term effects of stabilized Acn extend beyond the eye, we measured survival of Acn transgenic flies. First, we tested a role of Acn in survival under starvation conditions (Fig. 9 F). When resistance to starvation was compared with Acnwt, flies expressing only stabilized AcnD527A (P < 0.0001 log rank) or AcnS641,731D (P < 0.0001 log rank) survived significantly longer in the absence of food. In contrast, survival of AcnS641,731A flies was not significantly different.
Next, we tested whether Acn stability can modify longevity. Compared with Acnwt or AcnS641,731A, we found significantly enhanced life spans for stabilized AcnD527A and AcnS641,731D (both P < 0.0001 log rank), with the median life span extended by 50% from 38 to 58 d for AcnD527A (Fig. 9 G). Together, these results are consistent with a model in which AKT1 phosphorylates Acn to inhibit its caspase-mediated cleavage, and resulting elevated levels of Acn enhance basal autophagy with beneficial effects for neural maintenance and prolonged life span.
Discussion
We have previously shown that Acn is a regulator of autophagy in Drosophila as loss or gain of Acn function results in reduced or exaggerated autophagy (Haberman et al., 2010). In this study, we examine the mechanisms by which Acn levels are fine-tuned in developing tissues to determine the level of basal autophagy. We find that Acn levels are negatively regulated by Dcp-1–mediated cleavage. Elevated levels of Acn, whether caused by loss of Dcp-1 function, mutations modifying its target site D527, or mimicking AKT1-mediated phosphorylation, enhanced basal autophagy. The requirement of basal autophagy for neuronal maintenance and normal longevity is well established in flies (e.g., Simonsen et al., 2007; Juhász and Neufeld, 2008). Corresponding benefits resulted when stabilized Acn proteins enhanced basal autophagy: such flies lived longer, were more resistant to starvation stress, and exhibited reduced neurodegeneration in a Drosophila Huntington’s disease model (Scherzinger et al., 1997). We cannot exclude that roles in other pathways, such as endocytic trafficking (Haberman et al., 2010) or RNA metabolism (Murachelli et al., 2012), may contribute to the benefits of enhanced Acn function. The importance of elevated basal autophagy is consistent, however, with the beneficial outcomes of its up-regulation in neurons or muscles by increased levels of Atg8a in flies (Simonsen et al., 2008; Bai et al., 2013) or Beclin 1 in mice (Spencer et al., 2009).
The window for beneficial effects is narrow, however. Eye-specific overexpression of Acn disrupts retinal development in an autophagy-dependent manner (this study), and systemic high-level expression can cause lethality as a result of excessive autophagy (Haberman et al., 2010). Detrimental consequences of autophagy also result from overexpression of Atg1 (Scott et al., 2007). In a more limited way, autophagy contributes to the elimination of cells of salivary glands or the midgut during metamorphosis (Berry and Baehrecke, 2007; Denton et al., 2009). A similarly ambivalent picture arises from genetic activation of autophagy in Caenorhabditis elegans (Kang et al., 2007) and from pharmacological studies in murine cells (Sarkar et al., 2009; Liu et al., 2013; Shoji-Kawata et al., 2013), with low levels of autophagy being protective for cells but high levels causing cell death. Intriguingly, changes in Acn levels, its phosphorylation, or epigenetic status have been observed in different cancer cells (Shu et al., 2006; Jang et al., 2008; Patwa et al., 2008) and linked to differential cell survival (Park et al., 2009).
Our data suggest that these links between cancer and Acn may reflect its conserved function in autophagy. In the past, however, much of these data have been attributed to a role of Acn in apoptosis caused in part, by the initial discovery of Acn as a factor promoting apoptotic DNA condensation or fragmentation after being activated by caspase-mediated cleavage (Sahara et al., 1999). We find that caspase-mediated cleavage of Acn as well as its stabilization by AKT-mediated phosphorylation (Hu et al., 2005) is conserved in flies, but our results differ in two aspects.
First, acn-null cells still display DNA fragmentation and condensation (Haberman et al., 2010), and here, we show that eye-specific overexpression of either of the fragments predicted from caspase-mediated cleavage at AcnD527 does not disrupt normal development. Furthermore, elevated levels of Acn in the eye, which are sufficient to induce increased autophagy, are not sufficient to increase levels of apoptosis (Fig. 3 B and not depicted). Moreover, developmental defects induced by GMR-Gal4–driven overexpression of Acn are suppressed by reduced function of core autophagy genes but not by reduced function of caspases, including the apical caspase Dronc (Figs. 4 and 6). Together, these data suggest that the role of Acn in DNA condensation or fragmentation in apoptotic mammalian cells (Sahara et al., 1999; Joselin et al., 2006) may not be conserved in Drosophila. In contrast, Acn function in autophagy is conserved between Drosophila and human cells (Haberman et al., 2010; Orvedahl et al., 2011).
Second, cleavage of Acn by Dcp-1 is not restricted to apoptotic cells but is found in developing photoreceptors and fat body cells. In apoptotic cells, caspases gain access to nuclear proteins upon breakdown of the nuclear envelope (Faleiro and Lazebnik, 2000). The cleavage of nuclear-enriched Acn by cytoplasmic Dcp-1 in nonapoptotic photoreceptor precursor cells thus suggests that Acn shuttles in and out of the nucleus. This is consistent with Acn being detected in the cytoplasm in a subset of eye discs cells (Haberman et al., 2010) and with a predicted nuclear export signal at amino acids 691–698 of Acn (la Cour et al., 2004).
Caspase-mediated cleavage in nonapoptotic cells is not unique to Acn, other examples include dSREBP (Drosophila sterol regulatory element-binding protein) activation by Drice (Amarneh et al., 2009) and local caspase activation during axon branching (Campbell and Okamoto, 2013). Subapoptotic activation of caspases contributes to tumor invasion and metastasis (Rudrapatna et al., 2013). Atg16l1 levels, which are critical for the regulation of autophagy in Paneth cells (Cadwell et al., 2008), are modulated by stress-induced, but subapoptotic, levels of Caspase-3 (Murthy et al., 2014). Mutations in Atg16l1 that enhance its cleavage by Caspase-3 thus impair autophagy and pathogen clearance and thereby contribute to Crohn’s disease (Hampe et al., 2007).
Other proautophagic targets of caspases include Atg4 (Betin and Lane, 2009) and Beclin 1 (Luo and Rubinsztein, 2010; Wirawan et al., 2010). Their cleavage by caspases also reduces autophagy and suppresses its cytoprotective function (Kroemer et al., 2010).
A surprising finding of our study is the proautophagy function that AKT1 can exert through its effect on Acn levels. This observation contrasts with previous results in flies and mammals, which firmly established an inhibitory role of AKT1 signaling in autophagy (Kroemer et al., 2010; Mariño et al., 2014). In the context of the canonical PtdIns3K–Akt–TOR pathway, AKT1 activates the mTOR complex directly by phosphorylation of Raptor and indirectly via the inhibitory phosphorylation of the TSC1/2 inhibitory complex (Neufeld, 2010). Furthermore, AKT1-mediated phosphorylation of Beclin 1 inhibits its proautophagy function (Wang et al., 2012). In contrast, our results imply a proautophagy role for AKT1 signaling as a result of its conserved phosphorylation of Acn and the resulting inhibition of its proteolytic cleavage (Hu et al., 2005; Park et al., 2009; this study).
How can we resolve the conundrum of these seemingly opposing AKT1 functions in the regulation of autophagy? A possible explanation is provided by the presence of distinct pools of AKT1 kinase that may be engaged in different contexts (Martelli et al., 2012). For example, although Beclin 1, mTOR, and TSC1/2 are primarily cytoplasmic proteins, Acn is mostly nuclear in flies and mammals (Schwerk et al., 2003; Haberman et al., 2010). Therefore, a distinct nuclear pool of AKT1 may be responsible for Acn regulation (Martelli et al., 2012). This is not without precedent. Nuclear AKT1 has been suggested to phosphorylate Foxo proteins (Martelli et al., 2012) and the coactivator p300 (Huang and Chen, 2005), which have been implicated in regulating autophagy (Demontis and Perrimon, 2010; Ferdous et al., 2010; Lo Ré et al., 2012). Consistent with the notion of two distinct AKT1 pools regulating separate modes of autophagy, we find that AKT1-dependent activation of Acn does not occlude additional enhancement of autophagy via the conventional starvation-induced mTOR pathway. Physiologically, the availability of independent pathways for the induction of autophagy may be advantageous for cells as they respond to distinct challenges. Acn activation may provide an alternative pathway to engage basal, starvation-independent autophagy without invoking the complex metabolic changes triggered by altered mTOR signaling (Laplante and Sabatini, 2012; Costa-Mattioli and Monteggia, 2013).
How elevated levels of Acn induce autophagy is not clear. It may increase the activity of the ASAP complex that Acn forms with SAP18 and RNPS1 (Schwerk et al., 2003) or reduce an inhibitory function of the alternative Pinin-containing ASAP complex, in which Acn is replaced by Pinin (Murachelli et al., 2012). These complexes have been implicated in the regulation of splicing, nuclear export, translation and decay of mRNAs through their interactions with spliceosomes, the exon junction complex, and messenger ribonucleoprotein particles (Tange et al., 2005; Joselin et al., 2006; Singh et al., 2010, 2012; Hayashi et al., 2014; Malone et al., 2014), but the primary function of ASAP or Pinin-containing ASAP complexes is still poorly understood. Alternatively, the regulation of autophagy by Acn may be independent of these complexes and instead relay on its interaction with other binding partners, such as AAC-11. Inhibition of their interaction reduced resistance to chemotherapeutic drugs possibly as a result of altered apoptosis or autophagy (Rigou et al., 2009). Chemotherapy is one of many stress situations in which it is important to understand how the proper balance between apoptosis and autophagy is regulated (White, 2012; Mariño et al., 2014). The mechanisms by which Acn contributes to that regulation will be an important goal of future research.
Materials and methods
Fly work
Flies were maintained using standard conditions. RNAi lines were obtained from Vienna Drosophila RNAi Center or the Bloomington Stock Center, which also provided Da-Gal4, Lsp2-Gal4, and GMR-Gal4 driver lines, w1118, UAS-GFP-KDEL, UAS-AKT, P[hs-FLP]1, y1 w1118; Dr1/TM3, Sb1, P[hs-FLP]1, w1118; Adv1/CyO, and w1118; P[GAL4-Act5C(FRT.CD2).P]S, P[UAS-RFP.W]3/TM3, Sb1. Other fly strains used were dcp-1 (contains a 40-bp partial P element insertion in the coding region of Dcp-1, resulting in a frame shift in Dcp-1 and an in-frame stop codon within the 40-bp insertion; Laundrie et al., 2003; gift from K. McCall, Boston University, Boston, MA), UAS-htt-exon1-Q93 (expresses Huntington’s disease exon1 peptide with 93 glutamines under UAS control; Steffan et al., 2001; gift from R. Hiesinger, University of Texas Southwestern Medical Center, Dallas, TX), and UAS-GFP-Atg8a (expresses GFP-tagged Atg8a protein under Gal4/UAS control; Juhász et al., 2008; gift from T. Neufeld, University of Minnesota, Saint Paul, MN). RNAi clones were generated by heat shocking larvae carrying hs-Flp; Actin>CD2>Gal4, UAS-RNAi, and UAS-RFP at 38°C for 1 h for a consecutive 2 d. Transgenic flies were generated by BestGene, Inc. DNA constructs in the context of genomic acn were generated by standard mutagenesis of the 4-kb Acn DNA fragment sufficient for genomic rescue (Haberman et al., 2010), confirmed by sequencing, cloned into an Attb vector, and inserted into the 96F3 attP landing site (Venken et al., 2006). Similarly, UAS-controlled wild-type and mutant Acn transgenes were generated by standard mutagenesis from full-length Acn cDNA, confirmed by sequencing, and inserted into pUAS vectors. An initial set of UAS-Acnwt and UAS-AcnD527A transgenes was generated by P element–mediated transformation. Subsequently, transgenes—including new lines of UAS-Acnwt and UAS-AcnD527A—were generated in a pUAS(t) variant with an added Attb site and inserted into the 53B2 or 28E7 landing sites (Venken et al., 2006) as indicated (Table S2).
Starvation resistance was measured in vials with 4–5-d-old virgins at 25°C at 6-h intervals. To measure life spans, males that emerged within a 2-d period were pooled and aged for an additional 3 d, and their survival at 25°C was recorded as described in Stenesen et al. (2013). In brief, 70–100 flies were placed in demography cages (∼1 liter). Every other day, food vials were changed, and dead flies were counted and removed. Initial life span measurements were performed with the acn transgenes in an acn1/acn27 background with both chromosomes containing background lethals (Haberman et al., 2010) and, in the absence of the TM6, Hu balancer. Results in multiple experiments in that background were qualitatively the same as for the experiments shown (Fig. 9 G) for which the chromosome carrying the acn27-null allele was cleaned from other mutations before crossing the different acn transgenes into the isogenized acn27 stock for the longevity measurements.
Biochemistry
Acn-pS641 antibody was raised in rabbits by Genemed Synthesis against the RSRSGS(p)PASKTKKC peptide and double affinity purified. For Western blots, five 96-h or ten 75-h larvae were crushed in 300 µl lysis buffer (10% SDS, 6 M urea, and 50 mM Tris-HCl, pH 6.8) at 95°C, boiled for 2 min, and spun for 10 min at 20,000 g to remove larval cuticle. 20 µl lysate was separated by SDS-PAGE. Proteins were transferred to nitrocellulose membranes or, for detection of Atg8 proteins, onto 0.2-µm polyvinylidene fluoride membranes and probed with mouse anti-Actin (1:2,000; JLA 20; Developmental Studies Hybridoma Bank), guinea pig anti-Acn (aa 423–599; 1:3,000; Haberman et al., 2010), mouse anti-Myc (1:2,000; 9E10; Covance), rabbit anti–Acn-pS641 (1:1,000), or rabbit anti-Atg8a (1:5,000; Nagy et al., 2014; gift from G. Juhàsz, Eötvös Loránd University, Budapest, Hungary). Bound antibodies were detected and quantified by comparison to Actin using fluorescence-labeled antibodies and the Odyssey scanner (LI-COR Biosciences). Prestained molecular weight markers were obtained from Invitrogen (HiMark) or New England Biolabs, Inc. (ColorPlus).
For expression in S2 cells, plasmids encoding Myc-tagged versions of Acnwt and AcnS641,731A under control of the metallothionein promoter were transfected using the TransIT-2020 reagent (Mirus Bio LLC) followed by induction of expression, cell lysis, and immunoprecipitation with anti-Myc beads (Pulipparacharuvil et al., 2005). For phosphatase treatment, Acnwt-transfected S2 cell lysates were treated with 400 U calf intestinal phosphatase (New England Biolabs, Inc.).
Larval fat bodies were dissected from 10 larvae in ice-cold 50 µl larval lysis buffer (25 mM Tris-HCl, 1 mM EDTA, 0.1 mM EGTA, 5 mM MgCl2, 150 mM NaCl, 2 mM Na3VO4, 10% glycerol, 1% NP-40, 1 mM DTT, and protease and phosphatase inhibitor mixes [Roche]). Dissected fat bodies were sonicated for 5 min on ice and mixed with 50 µl Freon. The mixture was centrifuged at 10,000 g for 10 min, and supernatants were collected for Western blots.
Transcript levels of Myc-tagged Acn transgenes were measured using quantitative RT-PCR as previously described (Akbar et al., 2011). In short, RNA was isolated using TRIZOL (Ambion) according to the manufacturer’s instructions. 2 µg RNA was reverse transcribed using High-Capacity cDNA Reverse Transcription kit (Applied Biosystems) using random hexamer primers. Quantitative PCR was performed using the Fast SYBR Green Master Mix in a real-time PCR system (Fast 7500; Applied Biosystems). Each data point was repeated three times and normalized with rp49 as an internal control. Primers were left, 5′-CTGGAGGAGCAGAAGCTGAT-3′, within the Myc and right, 5′-GGAGTCTCGACCTCGGTCTT-3′, within the Acn coding regions, and rp49 primers were left, 5′-ATCGGTTACGGATCGAACAA-3′, and right, 5′-GACAATCTCCTTGCGCTTCT-3′.
Histology
Micrographs of eyes were obtained on a microscope (SteREO Discovery.V12; Carl Zeiss) with a camera (AxioCam MRc 5; Carl Zeiss) using AxioVision image acquisition software (Carl Zeiss) at 72× magnification. Images of eyes are a composite of pictures taken at multiple z positions and compressed using CZFocus software.
For TEM, size-matched 96-h fed larvae were dissected, inverted to expose fat bodies, and fixed in 2% glutaraldehyde in 0.1 M cacodylate buffer, pH 7.2. Samples were postfixed with 2% OsO4 and 1.5% KFeCN in the same buffer. Samples were embedded in epoxy resin. A series of ultrathin sections (60–80 nm) containing fat body were cut, and sequential sections were collected on mesh and formvar-coated slot grids. Sections were stained with uranyl acetate and lead citrate to enhance contrast, examined with a transmission electron microscope (120 kV; Tecnai G2 Spirit BioTWIN; FEI), and images were captured with an 11-megapixel camera (Morada; Olympus). From TEMs, measurements of autolysosomal diameters and areas were obtained using Macnification software (Orbicule).
SEMs of fly eyes were obtained as previously described (Wolff, 2011). In short, eyes were fixed in 2% paraformaldehyde, 2% glutaraldehyde, 0.2% Tween 20, and 0.1 M cacodylate buffer, pH 7.4, for 2 h. Samples were processed in a series of four washes with increasing ethanol (25–100%) for 12 h, each followed by a series hexamethyldisilazane washes (25–100% in ethanol) for 1 h each. Flies air dried overnight were mounted on SEM stubs and coated in fast-drying silver paint on their bodies only. Flies were sputter coated with a gold/pallidum mixture for 90 s and imaged at 1,000× magnification, with extra high tension set at 3.0 kV on a scanning electron microscope (SIGMA; Carl Zeiss). The microscope was equipped with the InLens detector (Carl Zeiss).
Whole-mount tissues were prepared for immunofluorescence staining as previously described (Akbar et al., 2011). In short, dissected samples were fixed in periodate-lysine-paraformaldehyde, washed in PBS, permeabilized with 0.3% saponin in PBS (PBSS), blocked with 5% goat serum in PBSS, and stained with the indicated primary antibodies: guinea pig anti-Acn (1:1,000; Haberman et al., 2010), mouse anti-Myc (9E10; 1:1,000), rabbit anti-dAtg8a (1:500; Barth et al., 2011; Abcam; gift from K. Koehler, Eidgenössische Technische Hochschule Zürich, Zürich, Switzerland), rabbit anti-Boss (1:1;000), rabbit anti–activated Caspase (1:300; Cell Signaling Technology), or rabbit-GFP (1:500; Invitrogen), and secondary antibodies were labeled with Alexa Fluor 488, 568, or 647 (1:500; Molecular Probes) and mounted in Vectashield (Vector Laboratories). Fluorescence images were captured with 63×, NA 1.4 or 40×, NA1.3 Plan Apochromat lenses on an inverted confocal microscope (LSM 510 Meta; Carl Zeiss). Confocal z stacks of eye discs were collected at 1-µm step size.
To quantify Acn/Myc immunoreactivity in eye discs, confocal stacks were imported to ImageJ (National Institutes of Health), and apical slices containing nuclei of early photoreceptors were projected into a single slice by summation of pixel intensities. Masks for individual groups of R3/4 and R2/5/8 cells were generated in ImageJ based on nuclear GFP expression (Fig. 2 D′). Integrated pixel intensities for Acn and Myc immunoreactivity were determined for ≥120 ommatidia from at least three eye discs per genotype. To compare different eye discs and measure the correlation coefficients for individual R-cell groups, integrated densities for each R-cell group were normalized to the mean value of Acn intensities in R2/5/8 cells for a given eye disc. For statistical evaluation and computation of Pearson correlation coefficients, data were imported into Prism software (GraphPad Software).
LysoTracker (GFP-Certified Lyso-ID red lysosomal detection kit; Enzo Life Sciences) staining of size-matched 90–96-h fat bodies from fed and starved larvae was performed as previously described (Rusten et al., 2004; Scott et al., 2004). In brief, larvae were dissected in Schneider’s Drosophila media (Gibco), inverted to expose fat bodies, and incubated in 100 µM LysoTracker Red DND-99 for 1 min. Inverted carcasses were then washed in PBS and placed onto a droplet of Vectashield (Vector Laboratories) for fat body separation and mounting. Samples were imaged immediately using 63×, NA 1.4 Plan Apochromat lens on an inverted confocal microscope (LSM 510 Meta). LysoTracker and Atg8a punctae in fat bodies were quantified using Imaris software (Bitplane) from z projections of three optical sections of fat body tissue, each 1 µm apart. The number of punctate was quantified either per fat body cell or per frame, in which one frame represents a volume of (471 µm)2 × 2 µm = 44 × 103 µm3. Digital images for display were imported into Photoshop (Adobe) and adjusted for gain, contrast, and γ settings.
ERGs
ERGs were recorded as previously described (Williamson et al., 2010). In brief, female flies were immobilized with nontoxic Glue-All (Elmer’s). Recording and reference electrodes containing 2 M NaCl were placed on the fly’s corneal surface and inserted into the thorax, respectively. Voltage measurements were filtered through an electrometer (IE-210; Warner Instruments), digitized with a Digidata 1440A and MiniDigi 1B system (Molecular Devices), and recorded using Clampex (version 10.2; Axon Instruments). 1-s light pulses were provided by a computer-controlled white light-emitting diode system (MC1500; Schott). Eight ERG traces from at least eight individual flies of each genotype were used for quantification with Clampfit software (Axon Instruments).
Poly-Q dot blot filter retardation assay
Polyglutamine aggregates were detected with a modified filter assay (Scherzinger et al., 1997). In brief, 25 heads were homogenized in 200 µl Cytoplasmic Extraction Reagent I buffer and fractionated using NE-PER Nuclear and Cytoplasmic Extraction Reagents following the manufacturer’s protocol (Thermo Fisher Scientific). Cytosolic fractions were adjusted to 1% SDS, incubated for 15 min at room temperature, denatured at 95°C for 5 min, and filtered through a 0.2-µm cellulose acetate membrane (Sterlitech Corporation) preequilibrated with 1% SDS. Filters were washed twice with 0.2% SDS and blocked in TBS (100 mM Tris-HCl, pH 7.4, and 150 mM NaCl) containing 3% nonfat dried milk before development with mouse 1C2 antibody (1:1,000; MAB1574; EMD Millipore) and detection and quantification with Odyssey scanner and software.
Statistical methods
Statistical significance was determined in Prism using log-rank for survival assays and one-way analysis of variance for multiple comparisons, followed by Tukey’s test. Pearson correlation coefficients for Acn and Myc double-stained eye discs were calculated in Prism from normalized integrated intensities measured in ImageJ. All bar graphs resulting from these comparisons show means ± SD. For quantifications of fluorescence images and Western blots, at least three independent experiments were used. Box and whisker graphs show boxes with median, 25 and 75 percentiles, and whiskers extending to minimal and maximal values. P-values smaller than 0.05 are considered significant, and values are indicated with one (<0.05), two (<0.01), three (<0.001), or four (<0.0001) asterisks.
Online supplemental material
Fig. S1 shows the characterization of genomic Acn transgenes and effectiveness of Acn-RNAi. Fig. S2 shows autophagy levels in different Acn transgenes as measured by LysoTracker staining of fat bodies or ATG8 Western blots. Table S1 shows the quantification of the roughness of eyes with different genotypes for which representative examples are shown in Figs. 2, 4, and 6. Table S2 shows the complete genotypes of flies used in different figures. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.201404028/DC1. Additional data are available in the JCB DataViewer at http://dx.doi.org/10.1083/jcb.201404028.dv.
Supplementary Material
Acknowledgments
We thank Dr. Beth Levine for critical reading of the manuscript, Drs. Thomas Neufeld, Kim McCall, Robin Hiesinger, and the Bloomington Stock Center (National Institutes of Health P40OD018537) for flies, Drs. Katja Koehler, Gàbor Juhàsz, and the Developmental Studies Hybridoma Bank at The University of Iowa, Iowa City for antibodies, Dr. Adrian Rothenfluh for help and advice with starvation resistance assays, and the Molecular and Cellular Imaging Facility at the University of Texas Southwestern for help with electron microscopy.
This work was supported by a National Institutes of Health grant (EY10199) to H. Krämer and National Science Foundation Graduate Research Fellowship (4900835401-36068) to L.K. Tyra.
The authors declare no competing financial interests.
Footnotes
Abbreviations used in this paper:
- Acn
- Acinus
- ASAP
- apoptosis- and splicing-associated protein
- Da
- Daughterless
- ERG
- electroretinogram
- GMR
- glass multimer reporter
- mTOR
- mammalian TOR
- SEM
- scanning EM
- TEM
- transmission EM
- TOR
- target of rapamycin
- UAS
- upstream activating sequence
References
- Akbar, M.A., Tracy C., Kahr W.H., and Krämer H.. 2011. The full-of-bacteria gene is required for phagosome maturation during immune defense in Drosophila. J. Cell Biol. 192:383–390. 10.1083/jcb.201008119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amarneh, B., Matthews K.A., and Rawson R.B.. 2009. Activation of sterol regulatory element-binding protein by the caspase Drice in Drosophila larvae. J. Biol. Chem. 284:9674–9682. 10.1074/jbc.M900346200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai, H., Kang P., Hernandez A.M., and Tatar M.. 2013. Activin signaling targeted by insulin/dFOXO regulates aging and muscle proteostasis in Drosophila. PLoS Genet. 9:e1003941. 10.1371/journal.pgen.1003941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barth, J.M., Szabad J., Hafen E., and Köhler K.. 2011. Autophagy in Drosophila ovaries is induced by starvation and is required for oogenesis. Cell Death Differ. 18:915–924. 10.1038/cdd.2010.157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry, D.L., and Baehrecke E.H.. 2007. Growth arrest and autophagy are required for salivary gland cell degradation in Drosophila. Cell. 131:1137–1148. 10.1016/j.cell.2007.10.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betin, V.M., and Lane J.D.. 2009. Caspase cleavage of Atg4D stimulates GABARAP-L1 processing and triggers mitochondrial targeting and apoptosis. J. Cell Sci. 122:2554–2566. 10.1242/jcs.046250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadwell, K., Liu J.Y., Brown S.L., Miyoshi H., Loh J., Lennerz J.K., Kishi C., Kc W., Carrero J.A., Hunt S., et al. 2008. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature. 456:259–263. 10.1038/nature07416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell, D.S., and Okamoto H.. 2013. Local caspase activation interacts with Slit-Robo signaling to restrict axonal arborization. J. Cell Biol. 203:657–672. 10.1083/jcb.201303072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa-Mattioli, M., and Monteggia L.M.. 2013. mTOR complexes in neurodevelopmental and neuropsychiatric disorders. Nat. Neurosci. 16:1537–1543. 10.1038/nn.3546 [DOI] [PubMed] [Google Scholar]
- Demontis, F., and Perrimon N.. 2010. FOXO/4E-BP signaling in Drosophila muscles regulates organism-wide proteostasis during aging. Cell. 143:813–825. 10.1016/j.cell.2010.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denton, D., Shravage B., Simin R., Mills K., Berry D.L., Baehrecke E.H., and Kumar S.. 2009. Autophagy, not apoptosis, is essential for midgut cell death in Drosophila. Curr. Biol. 19:1741–1746. 10.1016/j.cub.2009.08.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efeyan, A., Zoncu R., Chang S., Gumper I., Snitkin H., Wolfson R.L., Kirak O., Sabatini D.D., and Sabatini D.M.. 2013. Regulation of mTORC1 by the Rag GTPases is necessary for neonatal autophagy and survival. Nature. 493:679–683. 10.1038/nature11745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faleiro, L., and Lazebnik Y.. 2000. Caspases disrupt the nuclear-cytoplasmic barrier. J. Cell Biol. 151:951–960. 10.1083/jcb.151.5.951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferdous, A., Battiprolu P.K., Ni Y.G., Rothermel B.A., and Hill J.A.. 2010. FoxO, autophagy, and cardiac remodeling. J. Cardiovasc. Transl. Res. 3:355–364. 10.1007/s12265-010-9200-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Füllgrabe, J., Klionsky D.J., and Joseph B.. 2014. The return of the nucleus: transcriptional and epigenetic control of autophagy. Nat. Rev. Mol. Cell Biol. 15:65–74. 10.1038/nrm3716 [DOI] [PubMed] [Google Scholar]
- Groth, A.C., Fish M., Nusse R., and Calos M.P.. 2004. Construction of transgenic Drosophila by using the site-specific integrase from phage phiC31. Genetics. 166:1775–1782. 10.1534/genetics.166.4.1775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haberman, A.S., Akbar M.A., Ray S., and Krämer H.. 2010. Drosophila acinus encodes a novel regulator of endocytic and autophagic trafficking. Development. 137:2157–2166. 10.1242/dev.044230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampe, J., Franke A., Rosenstiel P., Till A., Teuber M., Huse K., Albrecht M., Mayr G., De La Vega F.M., Briggs J., et al. 2007. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat. Genet. 39:207–211. 10.1038/ng1954 [DOI] [PubMed] [Google Scholar]
- Hara, T., Nakamura K., Matsui M., Yamamoto A., Nakahara Y., Suzuki-Migishima R., Yokoyama M., Mishima K., Saito I., Okano H., and Mizushima N.. 2006. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 441:885–889. 10.1038/nature04724 [DOI] [PubMed] [Google Scholar]
- Hayashi, R., Handler D., Ish-Horowicz D., and Brennecke J.. 2014. The exon junction complex is required for definition and excision of neighboring introns in Drosophila. Genes Dev. 28:1772–1785. 10.1101/gad.245738.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, Y., Yao J., Liu Z., Liu X., Fu H., and Ye K.. 2005. Akt phosphorylates acinus and inhibits its proteolytic cleavage, preventing chromatin condensation. EMBO J. 24:3543–3554. 10.1038/sj.emboj.7600823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, W.C., and Chen C.C.. 2005. Akt phosphorylation of p300 at Ser-1834 is essential for its histone acetyltransferase and transcriptional activity. Mol. Cell. Biol. 25:6592–6602. 10.1128/MCB.25.15.6592-6602.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang, S.W., Yang S.J., Ehlén A., Dong S., Khoury H., Chen J., Persson J.L., and Ye K.. 2008. Serine/arginine protein-specific kinase 2 promotes leukemia cell proliferation by phosphorylating acinus and regulating cyclin A1. Cancer Res. 68:4559–4570. 10.1158/0008-5472.CAN-08-0021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jewell, J.L., Russell R.C., and Guan K.L.. 2013. Amino acid signalling upstream of mTOR. Nat. Rev. Mol. Cell Biol. 14:133–139. 10.1038/nrm3522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joselin, A.P., Schulze-Osthoff K., and Schwerk C.. 2006. Loss of Acinus inhibits oligonucleosomal DNA fragmentation but not chromatin condensation during apoptosis. J. Biol. Chem. 281:12475–12484. 10.1074/jbc.M509859200 [DOI] [PubMed] [Google Scholar]
- Juhász, G., and Neufeld T.P.. 2008. Drosophila Atg7: required for stress resistance, longevity and neuronal homeostasis, but not for metamorphosis. Autophagy. 4:357–358. 10.4161/auto.5572 [DOI] [PubMed] [Google Scholar]
- Juhász, G., Hill J.H., Yan Y., Sass M., Baehrecke E.H., Backer J.M., and Neufeld T.P.. 2008. The class III PI(3)K Vps34 promotes autophagy and endocytosis but not TOR signaling in Drosophila. J. Cell Biol. 181:655–666. 10.1083/jcb.200712051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang, C., You Y.J., and Avery L.. 2007. Dual roles of autophagy in the survival of Caenorhabditis elegans during starvation. Genes Dev. 21:2161–2171. 10.1101/gad.1573107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky, D.J., and Ohsumi Y.. 1999. Vacuolar import of proteins and organelles from the cytoplasm. Annu. Rev. Cell Dev. Biol. 15:1–32. 10.1146/annurev.cellbio.15.1.1 [DOI] [PubMed] [Google Scholar]
- Komatsu, M., Waguri S., Chiba T., Murata S., Iwata J., Tanida I., Ueno T., Koike M., Uchiyama Y., Kominami E., and Tanaka K.. 2006. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 441:880–884. 10.1038/nature04723 [DOI] [PubMed] [Google Scholar]
- Kroemer, G., Mariño G., and Levine B.. 2010. Autophagy and the integrated stress response. Mol. Cell. 40:280–293. 10.1016/j.molcel.2010.09.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- la Cour, T., Kiemer L., Mølgaard A., Gupta R., Skriver K., and Brunak S.. 2004. Analysis and prediction of leucine-rich nuclear export signals. Protein Eng. Des. Sel. 17:527–536. 10.1093/protein/gzh062 [DOI] [PubMed] [Google Scholar]
- Laplante, M., and Sabatini D.M.. 2012. mTOR signaling in growth control and disease. Cell. 149:274–293. 10.1016/j.cell.2012.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laundrie, B., Peterson J.S., Baum J.S., Chang J.C., Fileppo D., Thompson S.R., and McCall K.. 2003. Germline cell death is inhibited by P-element insertions disrupting the dcp-1/pita nested gene pair in Drosophila. Genetics. 165:1881–1888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang, X.H., Jackson S., Seaman M., Brown K., Kempkes B., Hibshoosh H., and Levine B.. 1999. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 402:672–676. 10.1038/45257 [DOI] [PubMed] [Google Scholar]
- Liu, Y., Shoji-Kawata S., Sumpter R.M. Jr, Wei Y., Ginet V., Zhang L., Posner B., Tran K.A., Green D.R., Xavier R.J., et al. 2013. Autosis is a Na+,K+-ATPase-regulated form of cell death triggered by autophagy-inducing peptides, starvation, and hypoxia-ischemia. Proc. Natl. Acad. Sci. USA. 110:20364–20371. 10.1073/pnas.1319661110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Ré, A.E., Fernández-Barrena M.G., Almada L.L., Mills L.D., Elsawa S.F., Lund G., Ropolo A., Molejon M.I., Vaccaro M.I., and Fernandez-Zapico M.E.. 2012. Novel AKT1-GLI3-VMP1 pathway mediates KRAS oncogene-induced autophagy in cancer cells. J. Biol. Chem. 287:25325–25334. 10.1074/jbc.M112.370809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo, S., and Rubinsztein D.C.. 2010. Apoptosis blocks Beclin 1-dependent autophagosome synthesis: an effect rescued by Bcl-xL. Cell Death Differ. 17:268–277. 10.1038/cdd.2009.121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malone, C.D., Mestdagh C., Akhtar J., Kreim N., Deinhard P., Sachidanandam R., Treisman J., and Roignant J.Y.. 2014. The exon junction complex controls transposable element activity by ensuring faithful splicing of the piwi transcript. Genes Dev. 28:1786–1799. 10.1101/gad.245829.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariño, G., Niso-Santano M., Baehrecke E.H., and Kroemer G.. 2014. Self-consumption: the interplay of autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 15:81–94. 10.1038/nrm3735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martelli, A.M., Tabellini G., Bressanin D., Ognibene A., Goto K., Cocco L., and Evangelisti C.. 2012. The emerging multiple roles of nuclear Akt. Biochim. Biophys. Acta. 1823:2168–2178. 10.1016/j.bbamcr.2012.08.017 [DOI] [PubMed] [Google Scholar]
- Meléndez, A., Tallóczy Z., Seaman M., Eskelinen E.L., Hall D.H., and Levine B.. 2003. Autophagy genes are essential for dauer development and life-span extension in C. elegans. Science. 301:1387–1391. 10.1126/science.1087782 [DOI] [PubMed] [Google Scholar]
- Mizushima, N., Levine B., Cuervo A.M., and Klionsky D.J.. 2008. Autophagy fights disease through cellular self-digestion. Nature. 451:1069–1075. 10.1038/nature06639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima, N., Yoshimori T., and Ohsumi Y.. 2011. The role of Atg proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol. 27:107–132. 10.1146/annurev-cellbio-092910-154005 [DOI] [PubMed] [Google Scholar]
- Murachelli, A.G., Ebert J., Basquin C., Le Hir H., and Conti E.. 2012. The structure of the ASAP core complex reveals the existence of a Pinin-containing PSAP complex. Nat. Struct. Mol. Biol. 19:378–386. 10.1038/nsmb.2242 [DOI] [PubMed] [Google Scholar]
- Murthy, A., Li Y., Peng I., Reichelt M., Katakam A.K., Noubade R., Roose-Girma M., DeVoss J., Diehl L., Graham R.R., and van Lookeren Campagne M.. 2014. A Crohn’s disease variant in Atg16l1 enhances its degradation by caspase 3. Nature. 506:456–462. 10.1038/nature13044 [DOI] [PubMed] [Google Scholar]
- Nagy, P., Kárpáti M., Varga A., Pircs K., Venkei Z., Takáts S., Varga K., Erdi B., Hegedűs K., and Juhász G.. 2014. Atg17/FIP200 localizes to perilysosomal Ref(2)P aggregates and promotes autophagy by activation of Atg1 in Drosophila. Autophagy. 10:453–467. 10.4161/auto.27442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakai, A., Yamaguchi O., Takeda T., Higuchi Y., Hikoso S., Taniike M., Omiya S., Mizote I., Matsumura Y., Asahi M., et al. 2007. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat. Med. 13:619–624. 10.1038/nm1574 [DOI] [PubMed] [Google Scholar]
- Neufeld, T.P.2010. TOR-dependent control of autophagy: biting the hand that feeds. Curr. Opin. Cell Biol. 22:157–168. 10.1016/j.ceb.2009.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orvedahl, A., Sumpter R. Jr, Xiao G., Ng A., Zou Z., Tang Y., Narimatsu M., Gilpin C., Sun Q., Roth M., et al. 2011. Image-based genome-wide siRNA screen identifies selective autophagy factors. Nature. 480:113–117. 10.1038/nature10546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, H.S., Yun Y., Kim C.S., Yang K.H., Jeong M., Ahn S.K., Jin Y.W., and Nam S.Y.. 2009. A critical role for AKT activation in protecting cells from ionizing radiation-induced apoptosis and the regulation of acinus gene expression. Eur. J. Cell Biol. 88:563–575. 10.1016/j.ejcb.2009.05.004 [DOI] [PubMed] [Google Scholar]
- Patwa, T.H., Wang Y., Miller F.R., Goodison S., Pennathur S., Barder T.J., and Lubman D.M.. 2008. A novel phosphoprotein analysis scheme for assessing changes in premalignant and malignant breast cell lines using 2D liquid separations, protein microarrays and tandem mass spectrometry. Proteomics Clin. Appl. 3:51–66. 10.1002/prca.200800097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulipparacharuvil, S., Akbar M.A., Ray S., Sevrioukov E.A., Haberman A.S., Rohrer J., and Krämer H.. 2005. Drosophila Vps16A is required for trafficking to lysosomes and biogenesis of pigment granules. J. Cell Sci. 118:3663–3673. 10.1242/jcs.02502 [DOI] [PubMed] [Google Scholar]
- Rigou, P., Piddubnyak V., Faye A., Rain J.C., Michel L., Calvo F., and Poyet J.L.. 2009. The antiapoptotic protein AAC-11 interacts with and regulates Acinus-mediated DNA fragmentation. EMBO J. 28:1576–1588. 10.1038/emboj.2009.106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudrapatna, V.A., Bangi E., and Cagan R.L.. 2013. Caspase signalling in the absence of apoptosis drives Jnk-dependent invasion. EMBO Rep. 14:172–177. 10.1038/embor.2012.217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusten, T.E., Lindmo K., Juhász G., Sass M., Seglen P.O., Brech A., and Stenmark H.. 2004. Programmed autophagy in the Drosophila fat body is induced by ecdysone through regulation of the PI3K pathway. Dev. Cell. 7:179–192. 10.1016/j.devcel.2004.07.005 [DOI] [PubMed] [Google Scholar]
- Sahara, S., Aoto M., Eguchi Y., Imamoto N., Yoneda Y., and Tsujimoto Y.. 1999. Acinus is a caspase-3-activated protein required for apoptotic chromatin condensation. Nature. 401:168–173. 10.1038/43678 [DOI] [PubMed] [Google Scholar]
- Sarkar, S., Ravikumar B., Floto R.A., and Rubinsztein D.C.. 2009. Rapamycin and mTOR-independent autophagy inducers ameliorate toxicity of polyglutamine-expanded huntingtin and related proteinopathies. Cell Death Differ. 16:46–56. 10.1038/cdd.2008.110 [DOI] [PubMed] [Google Scholar]
- Scherzinger, E., Lurz R., Turmaine M., Mangiarini L., Hollenbach B., Hasenbank R., Bates G.P., Davies S.W., Lehrach H., and Wanker E.E.. 1997. Huntingtin-encoded polyglutamine expansions form amyloid-like protein aggregates in vitro and in vivo. Cell. 90:549–558. 10.1016/S0092-8674(00)80514-0 [DOI] [PubMed] [Google Scholar]
- Schwerk, C., Prasad J., Degenhardt K., Erdjument-Bromage H., White E., Tempst P., Kidd V.J., Manley J.L., Lahti J.M., and Reinberg D.. 2003. ASAP, a novel protein complex involved in RNA processing and apoptosis. Mol. Cell. Biol. 23:2981–2990. 10.1128/MCB.23.8.2981-2990.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott, R.C., Schuldiner O., and Neufeld T.P.. 2004. Role and regulation of starvation-induced autophagy in the Drosophila fat body. Dev. Cell. 7:167–178. 10.1016/j.devcel.2004.07.009 [DOI] [PubMed] [Google Scholar]
- Scott, R.C., Juhász G., and Neufeld T.P.. 2007. Direct induction of autophagy by Atg1 inhibits cell growth and induces apoptotic cell death. Curr. Biol. 17:1–11. 10.1016/j.cub.2006.10.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Settembre, C., Zoncu R., Medina D.L., Vetrini F., Erdin S., Erdin S., Huynh T., Ferron M., Karsenty G., Vellard M.C., et al. 2012. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 31:1095–1108. 10.1038/emboj.2012.32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoji-Kawata, S., Sumpter R., Leveno M., Campbell G.R., Zou Z., Kinch L., Wilkins A.D., Sun Q., Pallauf K., MacDuff D., et al. 2013. Identification of a candidate therapeutic autophagy-inducing peptide. Nature. 494:201–206. 10.1038/nature11866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu, Y., Iijima T., Sun W., Kano J., Ishiyama T., Okubo C., Anami Y., Tanaka R., Fukai S., and Noguchi M.. 2006. The ACIN1 gene is hypermethylated in early stage lung adenocarcinoma. J. Thorac. Oncol. 1:160–167. 10.1097/01243894-200602000-00010 [DOI] [PubMed] [Google Scholar]
- Simonsen, A., Cumming R.C., and Finley K.D.. 2007. Linking lysosomal trafficking defects with changes in aging and stress response in Drosophila. Autophagy. 3:499–501. 10.4161/auto.4604 [DOI] [PubMed] [Google Scholar]
- Simonsen, A., Cumming R.C., Brech A., Isakson P., Schubert D.R., and Finley K.D.. 2008. Promoting basal levels of autophagy in the nervous system enhances longevity and oxidant resistance in adult Drosophila. Autophagy. 4:176–184. 10.4161/auto.5269 [DOI] [PubMed] [Google Scholar]
- Singh, G., Kucukural A., Cenik C., Leszyk J.D., Shaffer S.A., Weng Z., and Moore M.J.. 2012. The cellular EJC interactome reveals higher-order mRNP structure and an EJC-SR protein nexus. Cell. 151:750–764. 10.1016/j.cell.2012.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh, K.K., Erkelenz S., Rattay S., Dehof A.K., Hildebrandt A., Schulze-Osthoff K., Schaal H., and Schwerk C.. 2010. Human SAP18 mediates assembly of a splicing regulatory multiprotein complex via its ubiquitin-like fold. RNA. 16:2442–2454. 10.1261/rna.2304410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer, B., Potkar R., Trejo M., Rockenstein E., Patrick C., Gindi R., Adame A., Wyss-Coray T., and Masliah E.. 2009. Beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in α-synuclein models of Parkinson’s and Lewy body diseases. J. Neurosci. 29:13578–13588. 10.1523/JNEUROSCI.4390-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffan, J.S., Bodai L., Pallos J., Poelman M., McCampbell A., Apostol B.L., Kazantsev A., Schmidt E., Zhu Y.Z., Greenwald M., et al. 2001. Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila. Nature. 413:739–743. 10.1038/35099568 [DOI] [PubMed] [Google Scholar]
- Stenesen, D., Suh J.M., Seo J., Yu K., Lee K.S., Kim J.S., Min K.J., and Graff J.M.. 2013. Adenosine nucleotide biosynthesis and AMPK regulate adult life span and mediate the longevity benefit of caloric restriction in flies. Cell Metab. 17:101–112. 10.1016/j.cmet.2012.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tange, T.O., Shibuya T., Jurica M.S., and Moore M.J.. 2005. Biochemical analysis of the EJC reveals two new factors and a stable tetrameric protein core. RNA. 11:1869–1883. 10.1261/rna.2155905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vellai, T., Takács-Vellai K., Sass M., and Klionsky D.J.. 2009. The regulation of aging: does autophagy underlie longevity? Trends Cell Biol. 19:487–494. 10.1016/j.tcb.2009.07.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venken, K.J., He Y., Hoskins R.A., and Bellen H.J.. 2006. P[acman]: a BAC transgenic platform for targeted insertion of large DNA fragments in D. melanogaster. Science. 314:1747–1751. 10.1126/science.1134426 [DOI] [PubMed] [Google Scholar]
- Wang, R.C., Wei Y., An Z., Zou Z., Xiao G., Bhagat G., White M., Reichelt J., and Levine B.. 2012. Akt-mediated regulation of autophagy and tumorigenesis through Beclin 1 phosphorylation. Science. 338:956–959. 10.1126/science.1225967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White, E.2012. Deconvoluting the context-dependent role for autophagy in cancer. Nat. Rev. Cancer. 12:401–410. 10.1038/nrc3262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson, W.R., Wang D., Haberman A.S., and Hiesinger P.R.. 2010. A dual function of V0-ATPase a1 provides an endolysosomal degradation mechanism in Drosophila melanogaster photoreceptors. J. Cell Biol. 189:885–899. 10.1083/jcb.201003062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirawan, E., Vande Walle L., Kersse K., Cornelis S., Claerhout S., Vanoverberghe I., Roelandt R., De Rycke R., Verspurten J., Declercq W., et al. 2010. Caspase-mediated cleavage of Beclin-1 inactivates Beclin-1-induced autophagy and enhances apoptosis by promoting the release of proapoptotic factors from mitochondria. Cell Death Dis. 1:e18. 10.1038/cddis.2009.16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolff, T.2011. Preparation of Drosophila eye specimens for scanning electron microscopy. Cold Spring Harb. Protoc. 2011:1383–1385 [DOI] [PubMed] [Google Scholar]
- Wolff, T., and Ready D.F.. 1993. Pattern formation in the Drosophila retina. The Development of Drosophila melanogaster. Bate M. and Martinez-Arias A., . Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. 1277–1325 [Google Scholar]
- Yan, L., and Lamb R.F.. 2012. Amino acid sensing and regulation of mTORC1. Semin. Cell Dev. Biol. 23:621–625. 10.1016/j.semcdb.2012.02.001 [DOI] [PubMed] [Google Scholar]
- Yang, Z., and Klionsky D.J.. 2010. Mammalian autophagy: core molecular machinery and signaling regulation. Curr. Opin. Cell Biol. 22:124–131. 10.1016/j.ceb.2009.11.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.