

Regulatory brain cytoplasmic RNAs cooperate with eukaryotic initiation factor 4B to couple translation to receptor activation in support of long-term plastic changes in neurons.
Abstract
In neurons, translational regulation of gene expression has been implicated in the activity-dependent management of synapto-dendritic protein repertoires. However, the fundamentals of stimulus-modulated translational control in neurons remain poorly understood. Here we describe a mechanism in which regulatory brain cytoplasmic (BC) RNAs cooperate with eukaryotic initiation factor 4B (eIF4B) to control translation in a manner that is responsive to neuronal activity. eIF4B is required for the translation of mRNAs with structured 5′ untranslated regions (UTRs), exemplified here by neuronal protein kinase Mζ (PKMζ) mRNA. Upon neuronal stimulation, synapto-dendritic eIF4B is dephosphorylated at serine 406 in a rapid process that is mediated by protein phosphatase 2A. Such dephosphorylation causes a significant decrease in the binding affinity between eIF4B and BC RNA translational repressors, enabling the factor to engage the 40S small ribosomal subunit for translation initiation. BC RNA translational control, mediated via eIF4B phosphorylation status, couples neuronal activity to translational output, and thus provides a mechanistic basis for long-term plastic changes in nerve cells.
Introduction
Translational control of gene expression is a major determinant of eukaryotic cellular form and function (Mathews et al., 2007). In neurons, locally regulated protein synthesis in postsynaptic microdomains is instrumental in the maintenance of long-term functionality and plasticity (Kindler et al., 2005; Miyashiro et al., 2009; Gkogkas et al., 2010; Darnell, 2011). Stringent control of local translation in neurons is essential to ensure that relevant proteins are synthesized only where needed and when needed (Job and Eberwine, 2001). Inadequate neuronal translational control precipitates excessive protein synthesis, neuronal hyperexcitability, and epileptiform activities in vitro and in vivo (Musumeci et al., 2000; Chuang et al., 2005; Dölen et al., 2007; Zhong et al., 2009, 2010). Neuronal translational regulators are typically repressors, which is consistent with the notion that protein synthesis is in a repressed state under basal steady-state (i.e., nonstimulated) conditions. Translational repressors that have been identified in neurons include the fragile X mental retardation protein (FMRP; Bhakar et al., 2012; Santoro et al., 2012) and regulatory RNAs of the brain cytoplasmic (BC) type (Iacoangeli and Tiedge, 2013).
Regulatory BC RNAs are non-protein-coding small cytoplasmic RNAs (scRNAs). BC RNAs, which include nonorthologous rodent BC1 RNA and primate BC200 RNA (Iacoangeli and Tiedge, 2013), regulate protein synthesis by targeting the initiation phase of the translation pathway (Wang et al., 2002, 2005; Kondrashov et al., 2005; Lin et al., 2008; Eom et al., 2011). They do so by specifically inhibiting the activities of eukaryotic initiation factors (eIFs) 4A and 4B (Iacoangeli and Tiedge, 2013). These two factors are required for the efficient initiation of mRNAs with structured 5′ UTRs (Dmitriev et al., 2003; Pestova et al., 2007; Shahbazian et al., 2010), a class of mRNAs that is prominently represented among synapto-dendritic mRNAs (unpublished data). eIF4A is an ATP-dependent RNA helicase that promotes initiation by unwinding higher-order structural content in mRNA 5′ UTRs (Pestova et al., 2007; Gkogkas et al., 2010). eIF4B is a plurifunctional factor that stimulates the helicase activity of eIF4A, interacts with eIF3, and binds to the small ribosomal subunit via 18S ribosomal RNA (rRNA; Méthot et al., 1996; Gingras et al., 1999; Kapp and Lorsch, 2004; Pestova et al., 2007). eIF4B mediates recruitment of the small ribosomal subunit (in form of the 43S preinitiation complex) to the mRNA in a mechanism that involves direct interactions of eIF4B with the small ribosomal subunit as well as indirect interactions via eIF3 (Méthot et al., 1996; Pestova et al., 2007; Walker et al., 2013).
A key component of the activity-dependent translational control model is the tenet that during basal steady-state (“resting”) conditions, translation is repressed, whereas upon neuronal activation, translation is activated (i.e., derepressed). This mechanism would allow a neuron to modulate synaptic strength in a stimulus-dependent manner. However, it is not understood how BC RNA translational control transits from a repressed to a permissive state after stimulation. Here we present a functional dissection of the activity-dependent modulation of BC RNA translational control. We show that dephosphorylation of eIF4B at serine 406 (S406), occurring rapidly after neuronal stimulation, allows the factor to disengage from BC RNA repression, thus enabling translation. Our data indicate that eIF4B acts as an activity-dependent molecular switch in BC RNA translational control.
Results
BC RNA binding to eIF4B is modulated by phosphorylation status
To achieve translational repression, BC RNAs need to engage in simultaneous dual interactions with eIF4A and eIF4B (Eom et al., 2011). The latter, but not the former, factor is a phosphoprotein that is subject to regulation via signaling pathways (Raught et al., 2004; Holz et al., 2005; Shahbazian et al., 2006; Raught and Gingras, 2007). For this reason, we investigated the reversibility of BC RNA interactions with eIF4B, asking whether binding is subject to modulation as a function of the factor’s phosphorylation status. To address this question, we took into consideration the eIF4B phosphorylation sites that may be relevant for BC RNA interactions and translational control.
Previous work has established serine 422 (S422) as an eIF4B phosphorylation site that can modulate factor function (Raught et al., 2004; Holz et al., 2005; Shahbazian et al., 2006). Working with phosphomimetic eIF4B point mutant S422E (Holz et al., 2005; Shahbazian et al., 2006) in electrophoretic mobility assays (EMSAs), we found that S422 phosphorylation status does not impact binding of BC1 RNA to the factor (Fig. 1 A). In contrast, eIF4B phosphorylation at S406 (represented by phosphomimetic eIF4B S406E) resulted in significantly increased binding of BC1 RNA (Fig. 1 A). In agreement with previous observations (Gingras et al., 1999), we also note that strong binding of BC1 RNA causes eIF4B to homo-oligomerize (Fig. 1 A). In contrast to phosphomimetic eIF4B S406E, dephosphomimetic eIF4B S406A (i.e., eIF4B that is not and cannot be phosphorylated) was found binding BC1 RNA at levels comparable to unmodified eIF4B (Fig. 1 B). In addition, using Western blot analysis, we established that recombinant unmodified eIF4B is indeed not phosphorylated at S406 (Fig. 1 C). We will therefore also refer to unmodified eIF4B as dephospho-S406 eIF4B.
Figure 1.

S406 phosphorylation status is a determinant of eIF4B binding affinity to BC1 RNA. (A) EMSA experiments were performed with 32P-labeled BC1 RNA and recombinant eIF4B proteins (unmodified eIF4B, eIF4B S406E, and eIF4B S422E). RNP complexes were formed in lanes 2–10 (in the presence of eIF4B proteins) but not in lane 1 (in the absence of eIF4B). Intensities of RNP bands were substantially increased in BC1 RNA–eIF4B S406E reactions (lanes 5–7), in comparison with basal levels in BC1 RNA–unmodified eIF4B reactions (lanes 2–4) or in BC1 RNA–eIF4B S422E reactions (lanes 8–10). Proteins were used at concentrations of 115 nM, 330 nM, and 445 nM, respectively. (B) To examine binding of BC1 RNA to eIF4B S406A, EMSA experiments were performed with 32P-labeled BC1 RNA and unmodified eIF4B or eIF4B S406A. RNP complexes were formed in the presence of unmodified eIF4B (lanes 2 and 3) and in the presence of eIF4B S406A (lanes 4 and 5), but not in the absence of eIF4B (lane 1). Intensities of RNP bands were similar in BC1 RNA–eIF4B reactions (lanes 2 and 3) and in BC1 RNA–eIF4B S406A reactions (lanes 4 and 5). Proteins were used at concentrations of 115 nM and 330 nM. (C) To examine the phosphorylation status of S406 in recombinant eIF4B, we performed Western blot analysis with unmodified eIF4B and phosphomimetic eIF4B S406E. Unmodified eIF4B and eIF4B S406E were both recognized by a pan-eIF4B antibody (left). In contrast, only eIF4B S406E was detected by an antibody specific for phospho-S406 eIF4B. (D) Quantitative analysis of a series of EMSA experiments was used to calculate equilibrium dissociation constants by fitting the data to the Hill equation (Ryder et al., 2008). For the interaction of BC1 RNA with unmodified (i.e., dephospho-S406; see Fig. S1 B) eIF4B (blue curve), an equilibrium dissociation constant of Kd = 760 nM was established, whereas for the interaction with phosphomimetic eIF4B S406E (red curve), the dissociation constant was Kd = 90 nM.
We subsequently performed quantitative EMSA analysis to establish equilibrium binding constants (Ryder et al., 2008; Muslimov et al., 2011) as a function of S406 phosphorylation status. The data obtained were fitted to the Hill equation, revealing that while dephospho-S406 eIF4B bound BC1 RNA with an equilibrium dissociation constant of Kd = 760 nM, phosphomimetic eIF4B S406E bound BC1 RNA with a significantly higher affinity of Kd = 90 nM (Fig. 1 D).
Protein kinase Mζ (PKMζ; Hernandez et al., 2003) phosphorylates eIF4B at S406 (Fig. S1 A). We took advantage of this observation to ask whether direct eIF4B phosphorylation at S406, rather than use of an eIF4B S406 phosphomimetic, also strengthens binding of BC1 RNA to the factor. Fig. S1 B shows that this is indeed the case. In contrast, eIF4B S406A cannot be S406-phosphorylated (Fig. S1 A) and is thus unable to increase its BC1 RNA binding capacity (Fig. S1 B).
In summary, we conclude that binding of BC1 RNA to eIF4B is modulated by S406 phosphorylation.
BC RNAs compete with 18S rRNA and with the 40S small ribosomal subunit
Previous work has shown that BC RNAs repress translation initiation by disrupting interactions of eIF4B with 18S rRNA, and that this competitive molecular mechanism is mediated by a noncanonical C-loop motif in the BC RNA 3′ stem-loop domain (Eom et al., 2011). Is this mechanism subject to modulation by the eIF4B S406 phosphorylation status? To address this question, we performed quantitative EMSA competition analysis with 18S rRNA and with purified 40S small ribosomal subunits.
We first asked whether the ability of BC RNAs to compete with 18S rRNA for access to eIF4B is dependent on the factor’s phosphorylation status. EMSA competition data revealed that 18S rRNA easily competed BC1 RNA off dephospho-S406 eIF4B, but not off phosphomimetic eIF4B S406E (Fig. 2, A and B). This result indicates that the ability of BC1 RNA to deny 18S rRNA access to eIF4B is dependent on S406 phosphorylation. We subsequently asked whether the 40S small ribosomal subunit is also able to compete with BC1 RNA for access to eIF4B, and whether such competition is analogously dependent on eIF4B S406 phosphorylation status. Similar to 18S rRNA, the 40S small ribosomal subunit did indeed compete with BC1 RNA for access to dephospho-S406 eIF4B (Fig. 2, C and D, left). The competition with BC1 RNA for eIF4B was specific to the 40S small ribosomal subunit, as the 60S large ribosomal subunit, used under identical conditions, failed to compete. In contrast to dephospho-S406 eIF4B, however, the ability of the 40S small ribosomal subunit to compete with BC1 RNA was significantly reduced with phosphomimetic eIF4B S406E (Fig. 2, C and D, right). Again, the 60S large ribosomal subunit was unable to compete.
Figure 2.

Competition between BC1 RNA and the 40S small ribosomal subunit for eIF4B is a function of S406 phosphorylation status. (A and B) Formation of RNP complexes between BC1 RNA and unmodified (dephospho-S406) eIF4B was effectively inhibited by increasing amounts of unlabeled competitor 18S rRNA (lanes 1–4). In contrast, significantly less inhibition of BC1 RNA binding by competitor 18S rRNA was observed with phosphomimetic eIF4B S406E (lanes 5–8). Note that strong RNA binding causes eIF4B to homo-oligomerize (Gingras et al., 1999). (B) Quantitative analysis confirmed differential competition between BC1 RNA and 18S rRNA for dephospho-S406 eIF4B versus phosphomimetic eIF4B S406E. While BC1 RNA was easily competed off dephospho-S406 eIF4B, phosphomimetic eIF4B S406E retained BC1 RNA at equivalent concentrations of competitor 18S rRNA. The signal of RNP complexes (bound) was calculated against total signal (bound + free). The signal in the absence of 18S rRNA was normalized to 1. n = 4. (C and D) Increasing amounts of the small ribosomal subunit effectively competed BC1 RNA off dephospho-S406 eIF4B (lanes 2–5). In contrast, the ability of the 40S small ribosomal subunit to compete with BC1 RNA for access to eIF4B was significantly reduced with phosphomimetic eIF4B S406E (lanes 10–13). The 60S large ribosomal subunit was unable to compete with BC1 RNA for access to either the dephospho or the phosphomimetic factor (lanes 6–8 and lanes 14–16). Strong RNA binding causes eIF4B to homo-oligomerize (Gingras et al., 1999). (D) Quantitative analysis confirmed differential competition between BC1 RNA and the 40S small ribosomal subunit for dephospho-S406 eIF4B versus phosphomimetic eIF4B S406E. Quantification was performed as described in B. n = 4. Error bars represent SEM.
We have previously reported that primate BC200 RNA and rodent BC1 RNA are functionally fully equivalent (Eom et al., 2011). This equivalence includes the ability to compete with 18S rRNA for access to eIF4B. We have expanded this analysis to establish that BC200 RNA competes with the 40 ribosomal subunit, but not with the 60 ribosomal subunit, for access to eIF4B, and that this competition is dependent on the S406 phosphorylation status (Fig. S2). The combined evidence further corroborates the notion (Eom et al., 2011) that primate BC200 RNA and rodent BC1 RNA, despite their distinct phylogenetic pedigrees, are functionally indistinguishable from each other.
In conclusion, the data indicate that the 40S small ribosomal subunit, and its constituent 18S rRNA, are able to compete effectively with BC RNAs for access to dephospho-S406 eIF4B. In contrast, BC RNAs are only minimally dislodged from phosphomimetic eIF4B S406E by the 40S small ribosomal subunit or by 18S rRNA. We interpret these results as indicating that the ability of BC RNAs to impose repression on eIF4B is a function of the factor’s S406 phosphorylation status.
BC RNA control is modulated by eIF4B phosphorylation status
Is the translational repression competence of BC RNAs dependent on the eIF4B S406 phosphorylation status? To address this question, we developed a cellular translation reporter system that is based on the fact that the 5′ UTR of PKMζ mRNA is highly structured (Hernandez et al., 2003) and will thus depend on eIF4B for efficient initiation (Dmitriev et al., 2003; Shahbazian et al., 2010). We fused the PKMζ mRNA 5′ UTR to the protein-coding region of EGFP (Fig. 3 A) and expressed this construct in murine Neuro-2A (N2a) cells (Schor et al., 2009) in which endogenous eIF4B has been knocked down (to <5% of normal levels) by species-specific siRNA (Fig. 3 B). BC RNA translational control was then ascertained after transfection with human eIF4B constructs, followed by Western blot analysis of EGFP expression. We had previously established that human eIF4B mRNA is resistant to targeted degradation because siRNAs were specifically directed against such sections of the murine eIF4B mRNA 3′ UTR and protein-coding region that are distinct from the respective sections in human eIF4B mRNA.
Figure 3.

BC RNA translational control is modulated by eIF4B S406 phosphorylation status. (A) Schematic representation of the reporter construct used. The 5′ UTR of PKMζ mRNA was fused to the EGFP protein-coding region. (B) Transfection of Neuro-2A cells with siRNA directed against murine eIF4B mRNA, but not transfection with nontargeting control (NC) siRNA, significantly reduced levels of endogenous eIF4B. Levels of endogenous γ-tubulin were not affected. (C) Western blot analysis was performed after transfection with unmodified eIF4B, eIF4B S406E, or eIF4B S406A constructs. All three eIF4B variants were expressed at similar levels. (D and E) BC1 RNA effectively repressed synthesis of EGFP in the presence of phosphomimetic eIF4B S406E and in the presence of unmodified eIF4B. BC1 RNA was not effective in repressing synthesis of EGFP in the presence of dephosphomimetic eIF4B S406A. (E) Quantitative analysis: one-way ANOVA, P < 0.001. Tukey post-hoc analysis, comparison with unmodified eIF4B: P < 0.01 for eIF4B S406A, P = 0.68 for eIF4B S406E. n = 4. **, P < 0.01. (F and G) In the absence of BC1 RNA, expression levels of the EGFP reporter were not significantly different from each other, regardless of the eIF4B phosphorylation variant used. (G) Quantitative analysis: one-way ANOVA, P = 0.8675. Tukey post-hoc analysis, comparison with unmodified eIF4B: P = 0.906 for eIF4B S406A, P = 0.997 for eIF4B S406E. n = 4. For E–G, EGFP expression levels were normalized against those of endogenous γ-tubulin. Error bars represent SEM.
We used this PKMζ reporter system to assess BC1 RNA translational repression competence as a function of eIF4B phosphorylation status. We transfected Neuro-2A cells with constructs encoding human unmodified eIF4B, phosphomimetic eIF4B S406E, or dephosphomimetic eIF4B S406A (we established that these proteins are expressed at equivalent levels after transfection; Fig. 3 C). Data obtained from these experiments indicate that BC1 RNA is a significantly more effective translational repressor with eIF4B in the S406 phospho state than with eIF4B in the S406 dephospho state (Fig. 3, D and E). Unmodified eIF4B is phosphorylated in the cellular milieu (see Figs. 5–7) and was therefore indistinguishable, vis-à-vis BC1 RNA, from eIF4B S406E. No translational repression was observed in the absence of BC1 RNA: in this case, no significant differences were observed in EGFP expression levels in cells transfected with unmodified eIF4B, with eIF4B S406A, or with eIF4B S406E (Fig. 3, F and G).
Figure 5.
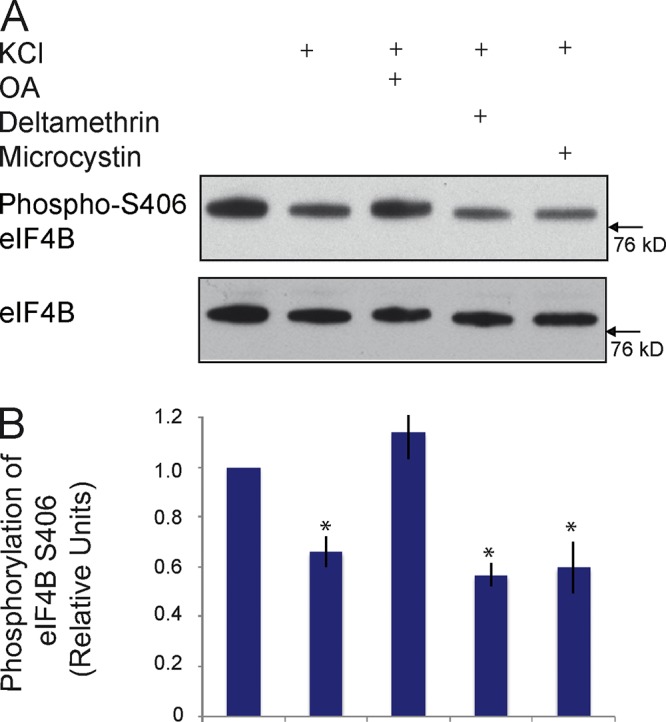
Dephosphorylation of eIF4B at S406 is mediated by protein phosphatase 2A. (A and B) Neuro-2A cells were depolarized using KCl, as described previously (Schor et al., 2009). Depolarization caused significant dephosphorylation of eIF4B at S406. The depolarization-induced dephosphorylation was blocked in the presence of OA, but not in the presence of deltamethrin or microcystin. The results indicate that PP2A, but not PP1 or PP2B, is responsible for eIF4B S406 dephosphorylation. (B) Quantitative analysis: one-way ANOVA, P < 0.001. Tukey post-hoc analysis, comparison with basal (no KCl): P < 0.05 for KCl, P < 0.05 for KCl/deltamethrin, P < 0.05 for KCl/microcystin, P = 0.97 for KCl/OA. n = 4. Error bars represent SEM. *, P < 0.05.
Figure 7.

Synapto-dendritic eIF4B is rapidly dephosphorylated at S406, in a PP2A-dependent manner, after activation of group I mGluRs in hippocampal neurons in primary culture. Neurons were double-labeled with an antibody against phospho-S406 eIF4B (green fluorescence) and with an antibody against the presynaptic marker synaptophysin (red fluorescence). (A and B) In the basal state, phospho-S406 eIF4B labeling appears in green puncta that often but not always are located in close apposition to red synaptophysin puncta, at times evidenced by yellow/orange overlap zones. (C) 2 min after activation of group I mGluRs with agonist DHPG, phospho-S406 eIF4B labeling (but not synaptophysin labeling) appears significantly reduced along the dendritic extent. (D) A reduction of synapto-dendritic phospho-S406 eIF4B labeling was not observed if group I mGluR activation occurred in the presence of the PP2A inhibitor OA. Insets show enlarged views of the boxed regions. Bar (shown in C): (A) 30 µm; (B–D) 10 µm; (insets) 4 µm. Number of cells analyzed: (A and B) 12 neurons, 36 dendrites; (C) 13 neurons, 39 dendrites; (D) 12 neurons, 36 dendrites. Number of cells examined for dendritic expression of total eIF4B: 13 neurons, 39 dendrites (basal); 12 neurons, 36 dendrites (DHPG); 13 neurons, 39 dendrites (DHPG/OA). Quantitative analysis: one-way ANOVA, P < 0.001. Tukey post-hoc analysis, comparison with basal: P < 0.001 for DHPG, P = 0.997 for DHPG/OA. Dendritic signal intensities of phospho-S406 eIF4B were normalized against dendritic signal intensities of total eIF4B (which did not significantly differ between the groups basal, DHPG, and DHPG/OA). Error bars represent SEM. ***, P < 0.001.
BC1 RNA translational repression was not observed when eIF4B was knocked down by siRNA (Fig. S3, A and B). In this experiment, translational efficiency was low even in the absence of BC1 RNA, which confirms previous observations that eIF4B is required for the translation of mRNAs with structured 5′ UTRs (Dmitriev et al., 2003; Shahbazian et al., 2010). This low translational efficiency was not further impacted by BC1 RNA (Fig. S3, A and B).
In additional experiments, we established that BC1 RNA translational control is ineffective if the mRNA features an unstructured 5′ UTR (Fig. S3, C and D). Furthermore, in the case of an unstructured 5′ UTR, translational efficiency in the presence of BC1 RNA was not a function of the phosphorylation status of eIF4B S406 (Fig. S3, E and F). These results support the notion that for the translation of mRNAs with unstructured 5′ UTRs, eIF4B is not required and translational repression by BC1 RNA, which is mediated through interactions with eIF4B, is therefore not effective.
The combined data indicate that BC1 RNA translational control is directed at mRNAs with structured 5′ UTRs, and that such control is in the repressive mode as long as eIF4B is phosphorylated at S406, but in the permissive mode if eIF4B S406 is dephosphorylated. This result is consistent with the data showing that binding of BC1 RNA to eIF4B (Fig. 1), and the ability of BC1 RNA to compete with 18S rRNA and the 40S ribosomal subunit for access to eIF4B (Fig. 2), are both dependent on the S406 phosphorylation status.
eIF4B S406 dephosphorylation is activity dependent
Our data indicate that phosphorylation of eIF4B S406 is a determinant of the ability of BC RNAs to bind the factor with high affinity, to compete for such binding with the 40S small ribosomal subunit, and thus to achieve translational repression. On the basis of these results, we hypothesized that changes in the phosphorylation status of eIF4B S406 serve as a molecular switch in BC RNA translational control. We therefore asked how this switch is activated in a manner that is responsive to neuronal stimulation. We used in vivo and in vitro approaches to address this question.
Noting that epileptogenesis is a form of neuronal plasticity (Chuang et al., 2005), we elicited neuronal activity in vivo by inducing epileptogenic responses. Two separate approaches were used to induce seizures in vivo. In the first approach, we induced audiogenic seizures in Fmr1−/− animals by auditory stimulation, as described previously (Zhong et al., 2010). Western blot analysis was subsequently performed with cortical brain material from experimental and control animals to establish levels of phospho-S406 eIF4B. As illustrated in Fig. 4 (A and B), seizure activity resulted in a significant reduction of phospho-S406 eIF4B levels in brain, in comparison with control littermates that had not undergone seizure activity. Total levels of eIF4B did not significantly differ between experimental and control littermates. eIF4B S406 dephosphorylation was rapid as it occurred within 2 min after induction of neuronal activity. The phosphorylation status of eIF4B S406 was indistinguishable between wild type (WT) animals and Fmr1−/− animals in the absence of auditory stimulation (Fig. 4, C and D). The data indicate that neurons maintain eIF4B S406 at a high relative level of phosphorylation under basal steady-state conditions but rapidly dephosphorylate this site upon stimulation.
Figure 4.

eIF4B S406 dephosphorylation is activity-dependent. (A and B) Audiogenic seizures were induced in Fmr1−/− animals by auditory stimulation (Zhong et al., 2010). Levels of eIF4B S406 phosphorylation were established in cortical brain material by Western blot analysis. 2 min after seizure induction (two left lanes), eIF4B S406 phosphorylation levels were significantly lower than in littermate brains that had not been subjected to auditory stimulation (two right lanes). Levels of total eIF4B did not significantly differ between stimulated and nonstimulated littermates. Quantitative analysis (B): Student’s t test, P < 0.001. Number of animals used for quantification: audiogenic seizure induction, 4; no seizure induction, 4. (C and D) Cortical eIF4B S406 phosphorylation was found to be indistinguishable between nonstimulated Fmr1−/− mice and WT mice. (D) Quantitative analysis: Student’s t test, P = 0.118. Four Fmr1−/− and four WT animals were used. (E and F) Seizures were induced by application of KA (two left lanes). Cortical levels of eIF4B S406 phosphorylation were established in comparison with those of littermates that were injected with vehicle (two right lanes). Levels of total eIF4B did not significantly differ between KA- and vehicle-injected littermates. (F) Quantitative analysis: Student’s t test, P < 0.001. Number of animals used for quantification: KA injection, 4; vehicle injection, 4. Error bars represent SEM. ***, P < 0.001.
These results were corroborated in a second in vivo approach in which epileptogenesis was induced chemically by application of kainic acid (KA; Zhong et al., 2006). Again, animals that experienced seizures exhibited significantly reduced cortical levels of eIF4B S406 phosphorylation, in comparison with littermate control animals that had been injected with vehicle (Fig. 4, E and F). We conclude that in two different in vivo epileptogenesis models, eIF4B S406 is dephosphorylated upon neuronal stimulation.
Activity-dependent eIF4B S406 dephosphorylation is mediated by protein phosphatase 2A (PP2A)
What is the mechanism underlying the activity-dependent dephosphorylation of eIF4B S406? To address this question, we first established that activity-dependent dephosphorylation can also be observed in cultured cells. We depolarized Neuro-2A cells using KCl, as described previously (Schor et al., 2009), and noted a significant decrease in eIF4B S406 phosphorylation levels (Fig. 5), a result that mirrors those obtained with in vivo stimulation (Fig. 4). We next used specific inhibitors to determine which of the key neuronal serine-threonine protein phosphatases (PPs) is responsible for the observed dephosphorylation. The inhibitors used were microcystin (specific for protein phosphatases PP1; may also activate or inhibit, depending on concentration, PP2A), okadaic acid (OA; specific for PP2A), and deltamethrin (specific for PP2B; Narayanan et al., 2007; Li et al., 2011, 2012). The depolarization-induced dephosphorylation of eIF4B S406 was completely blocked in cells that had been pretreated with the PP2A inhibitor OA (Fig. 5). In contrast, microcystin and deltamethrin were not effective. The data indicate that PP2A is specifically required for the activity-dependent dephosphorylation of eIF4B at S406.
We conclude that stimulation-induced S406 dephosphorylation of eIF4B is an actively mediated process that is performed by a specific protein phosphatase, PP2A.
Signaling through group I metabotropic glutamate receptors (mGluRs) couples to dephosphorylation of eIF4B S406 and to translational activation
Epileptogenesis is a form of neuronal plasticity that can be elicited by activation of group I mGluRs (Taylor et al., 1995; Zhao et al., 2004; Chuang et al., 2005). Consequently, given the data shown in Fig. 4, we hypothesized that the activity-dependent dephosphorylation of eIF4B S406 is mediated by signaling through group I mGluRs.
We used agonist 3,5-dihydroxyphenylglycine (DHPG) to activate group I mGluRs in Neuro-2A cells (Liu et al., 2002) in order to address this question. We found that, 2 min after DHPG stimulation, eIF4B S406 phosphorylation was significantly reduced (Fig. 6, A and B). This result led us to surmise that activation of group I mGluRs would also result in a reversal of BC1-mediated translational repression. To answer this question, we used the PKMζ reporter construct introduced in Fig. 3, cotransfected into Neuro-2A cells together with BC1 RNA. After stimulation of group I mGluRs with agonist DHPG, we observed a significant reduction in BC1-mediated translational repression (Fig. 6, C and D). This derepression did not occur when group I mGluRs were DHPG-stimulated in the presence of the PP2A inhibitor OA (Fig. S4). The data support the notion that activation of group I mGluRs causes a PP2A-dependent reversal of BC1-mediated translational repression via dephosphorylation of eIF4B at S406.
Figure 6.

Activation of group I mGluRs results in dephosphorylation of eIF4B at S406 and in up-regulation of translation. (A and B) 2 min after stimulation of Neuro-2A cells with the group I mGluR agonist DHPG, levels of eIF4B S406 phosphorylation, as determined by Western blot analysis, were significantly reduced. (B) Quantitative analysis: Student’s t test, P < 0.05. n = 4. (C and D) Neuro-2A cells were transfected with the PKMζ reporter construct (Fig. 3) and with BC1 RNA, and group I mGluRs were activated with DHPG. After 10 min, significantly higher levels of reporter EGFP were detected in DHPG-stimulated cells than in nonstimulated cells. (D) Quantitative analysis: Student’s t test, P < 0.001. n = 4. EGFP expression levels were normalized against those of endogenous γ-tubulin. (E and F) Group I mGluRs were activated with agonist DHPG in cortical neurons in primary culture. S406 eIF4B phosphorylation was significantly reduced 2 min after agonist application but recovered by 10 min after agonist application. No dephosphorylation was observed if DHPG was applied in the presence of OA. (F) Quantitative analysis: one-way ANOVA, P < 0.001. Tukey post-hoc analysis, comparison with basal: P = 0.495 for DHPG 10 min, P = 0.998 for DHPG/OA 10 min, P < 0.05 for DHPG 2 min, P = 0.998 for DHPG/OA 2 min. n = 4. (G and H) After transfection of Neuro-2A cells with siRNA directed against murine PP2A catalytic subunit α mRNA, phosphorylation of eIF4B S406 was not reduced upon application of DHPG. (H) Quantitative analysis: one-way ANOVA, P < 0.001. Tukey post-hoc analysis, comparison with basal: P < 0.01 for DHPG stimulation with NC siRNA, P = 0.761 for DHPG stimulation with siRNA directed against PP2A. n = 4. For A, B, and E–H, total levels of eIF4B were used for normalization. Error bars represent SEM. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
We next asked whether activation of group I mGluRs would also cause eIF4B S406 dephosphorylation in neurons. Working with cortical neurons in primary culture (Bassell et al., 1998), we examined the eIF4B S406 phosphorylation status after activation of group I mGluRs using DHPG. Fig. 6 (E and F) shows that eIF4B S406 was significantly dephosphorylated 2 min after application of DHPG but was rephosphorylated 8 min later (i.e., 10 min after DHPG application). An earlier paper (Narayanan et al., 2007) reported that, in neurons, DHPG stimulation of group I mGluRs results in a rapid increase of PP2A activity (within 60 s), followed by a suppression of PP2A activity beginning after 2 min. We therefore examined if PP2A was responsible for the rapid group I mGluR-dependent dephosphorylation of eIF4B S406. As is shown in Fig. 6 (E and F), application of the PP2A inhibitor OA completely blocked the DHPG-induced dephosphorylation of eIF4B S406. In a further experiment using Neuro-2A cells, we applied DHPG in the presence of siRNA directed against the PP2A catalytic subunit mRNA. We observed that in this case as well, the DHPG-induced dephosphorylation of eIF4B S406 was completely blocked (Fig. 6, G and H). The data confirm that PP2A is the phosphatase responsible for the rapid dephosphorylation of eIF4B S406 that is induced by activation of group I mGluRs.
Group I mGluR activation results in rapid eIF4B S406 dephosphorylation in synapto-dendritic domains
BC1 RNA is located at postsynaptic microdomains where it has been implicated in local translational regulation (Iacoangeli and Tiedge, 2013). Because our data indicate that BC1 RNA translational control switches from repressive to permissive upon activation of group I mGluRs and resultant dephosphorylation of eIF4B S406, we now asked if such dephosphorylation can also be observed in dendritic, particularly in postsynaptic, neuronal domains. We performed immunocytochemistry with hippocampal neurons in primary culture (Muslimov et al., 1998; Wang et al., 2002) in order to address this question. We observed (Fig. 7, A and B) that in the basal (nonstimulated) steady-state, phospho-S406 eIF4B was distributed along the entire dendritic extent. The labeling appeared to be punctate, a result that indicates a clustered distribution of the phosphorylated factor in dendrites. An antibody against pan-eIF4B revealed a labeling pattern indistinguishable from that obtained with the anti–phospho-S406 eIF4B antibody (not depicted). The experiments shown in Fig. 7 were performed in a double-labeling approach, using a second antibody that was directed against synaptophysin, a marker of presynaptic terminals (Muslimov et al., 1998; Wang et al., 2002). The results (Fig. 7, A and B) show that phospho-eIF4B immunolabeling appeared in the form of puncta that often but not always were in direct apposition to, or at times overlapping with, synaptophysin puncta along dendrites. We interpret these results to indicate that some but not all dendritic phospho-eIF4B is clustered in postsynaptic microdomains.
We next used agonist DHPG to activate group I mGluRs. We observed a rapid (≤2 min) and significant decrease of phopho-S406 eIF4B labeling along dendrites, with puncta now appearing smaller and less intense (Fig. 7 C). Punctate synaptophysin labeling remained unchanged (Fig. 7 C) as did pan-eIF4B labeling (not depicted). In contrast, no decrease of phospho-S406 eIF4B labeling was observed in dendrites when DHPG was applied in the presence of the PP2A inhibitor OA (Fig. 7 D). The combined data indicate that synapto-dendritic eIF4B is rapidly dephosphorylated at S406 upon group I mGluR activation, and that such dephosphorylation is dependent on the local action of PP2A (Fig. 7 E).
eIF4B S406 dephosphorylation causes BC1 RNA translational control to switch from repressive to permissive
We also addressed the question of whether dephosphorylation of eIF4B S406 is required for the activity-dependent induction (i.e., derepression) of BC1-repressed translation. To this end, we transfected Neuro-2A cells with siRNA against murine eIF4B mRNA (see also Fig. 3) with constructs to express human eIF4B (unmodified eIF4B or phosphomimetic eIF4B S406E), with BC1 RNA, and with the PKMζ reporter construct. Group I mGluRs were then activated with agonist DHPG. Fig. 8 (A and B) shows that in the presence of unmodified eIF4B, BC1 translational repression was reversed, i.e., translation induced, upon activation of group I mGluRs. In contrast, no such induction was observed in the presence of eIF4B S406E, i.e., of eIF4B that cannot be S406 dephosphorylated. Thus, as phosphomimetic eIF4B S406E blocked group I mGluR-induced reversal of BC1 translational repression, we conclude that dephosphorylation of eIF4B S406 is required for activity-dependent induction of translation.
Figure 8.

Dephosphorylation of eIF4B S406 is required for the activity-dependent reversal of BC1-mediated translational repression. (A and B) Neuro-2A cells were transfected with siRNA directed against murine eIF4B mRNA, with the PKMζ reporter construct (Fig. 3), with unmodified eIF4B or phosphomimetic eIF4B S406E, and with BC1 RNA. Group I mGluRs were activated with agonist DHPG. In the presence of unmodified eIF4B, application of DHPG resulted in a significant up-regulation of EGFP expression. In contrast, DHPG had no effect on EGFP levels in the presence of eIF4B S406E. (B) Quantitative analysis: one-way ANOVA, P < 0.001. Tukey post-hoc analysis, comparison with unmodified eIF4B in the absence of DHPG: P < 0.001 for unmodified eIF4B in the presence of DHPG, P = 0.723 for eIF4B S406E in the absence of DHPG, P = 0.995 for eIF4B S406E in the presence of DHPG. n = 4. (C and D) Group I mGluRs were DHPG-activated in cortical neurons in primary culture. Expression of PKMζ was significantly increased upon 10 min of DHPG stimulation but not upon DHPG stimulation in the presence of OA. (D) Quantitative analysis: one-way ANOVA, P < 0.001. Tukey post-hoc analysis, comparison with basal: P < 0.05 for DHPG, P = 0.967 for DHPG/OA. n = 4. For A–D, expression levels were normalized against those of endogenous γ-tubulin. Error bars represent SEM. *, P < 0.05; ***, P < 0.001.
We have shown (Fig. 6) that in Neuro-2A cells and in cortical neurons, activation of group I mGluRs results in the dephosphorylation of eIF4B S406, and, in Neuro-2A cells, in increased translation of a reporter mRNA with the highly structured PKMζ 5′ UTR. eIF4B dephosphorylation and translational stimulation were both PP2A dependent. We then asked whether DHPG stimulation of group I mGluRs also results in increased translation of an endogenous neuronal mRNA with a structured 5′ UTR. To address this question, we examined protein levels of endogenous PKMζ in cultured cortical neurons after application of DHPG. Fig. 8 (C and D) shows that 10 min after application of DHPG, PKMζ levels were significantly increased (and remained so for at least 30 min; not depicted). This increase was blocked if DHPG was applied in the presence of PP2A inhibitor OA (Fig. 8, C and D). We conclude that activation of group I mGluRs in cortical neurons causes rapid dephosphorylation of eIF4B S406 (Fig. 6 E and F) and consequentially increased translation of an endogenous neuronal mRNA with a structured 5′ UTR (Fig. 8, C and D).
PP2A-mediated dephosphorylation is required for the expression of long-term potentiation (LTP)
We previously reported that BC1 RNA and PKMζ mRNA colocalize in dendritic domains in the hippocampus (Muslimov et al., 2004). Together with the data presented in Fig. 8, the evidence led us to hypothesize that PKMζ mRNA may be subject to BC1 RNA translational control in hippocampal dendrites. As PKMζ has been implicated in hippocampal mechanisms of LTP (Pastalkova et al., 2006; but see Frankland and Josselyn, 2013), we asked if induction of LTP at hippocampal Schaffer collateral/commissural–CA1 synapses would trigger PP2A-dependent eIF4B S406 dephosphorylation and a consequential switch of BC1 RNA translational control from repressive to permissive. We show in Fig. 9 (A and B) that high-frequency stimulation (HFS) of Schaffer collaterals resulted in a rapid (≤2 min) dephosphorylation of eIF4B S406 in CA1. This dephosphorylation was blocked in the presence of OA (Fig. 9, A and B), which suggests that PP2A is activated during induction of LTP.
Figure 9.

PP2A-mediated dephosphorylation is required for PKMζ synthesis and is critical for the persistence of LTP in the CA1 field of hippocampus. (A and B) 2 min after delivery of HFS at the Schaffer collaterals, eIF4B S406 phosphorylation was significantly reduced. No such reduction was observed if HFS was delivered in the presence of OA. (B) Quantitative analysis. Signal intensities of phospho-S406 eIF4B were normalized against signal intensities of total eIF4B. Such signal intensities in HFS or OA HFS were again normalized to those in Control or OA Control. Student’s t test, P < 0.01 for HFS versus Control; P = 0.652 for OA HFS versus OA Control. n = 6. (C and D) 30 min after delivery of HFS at the Schaffer collaterals, levels of PKMζ were significantly increased. This increase was prevented in the presence of OA. (D) Quantitative analysis. PKMζ signal intensities were normalized against those of endogenous γ-tubulin. Such signal intensities in HFS or OA HFS were again normalized to those in Control or OA Control. Student’s t test, P < 0.05 for HFS versus Control; P = 0.266 for OA HFS versus OA Control. n = 4. (E) LTP was induced at CA3-CA1 synapses by HFS. Expression of LTP was blocked by OA. Sham stimulation (Control) was performed as described in the Materials and methods. Inset traces show superimposed representative fEPSPs recorded during the baseline period (top trace) and at 30 min after HFS (bottom trace), in a vehicle-treated control slice (left) and a slice treated with OA (right). Calibration: 1 mV, 20 ms. n = 7 for HFS, n = 4 for OA HFS, n = 6 for Control, n = 4 for OA Control. Quantitative analysis (one-way ANOVA): F3,17 = 11.20, P < 0.001. Tukey post-hoc analysis: HFS versus Control, P < 0.05; HFS versus OA Control, P < 0.05; HFS versus OA HFS, P < 0.05. Error bars represent SEM. *, P < 0.05; **, P < 0.01.
In CA1, PKMζ levels were significantly increased 30 min after delivery of HFS, a result consistent with previous work (Osten et al., 1996). This increase was not observed if HFS was applied in the presence of the PP2A inhibitor OA (Fig. 9, C and D). The question was thus raised of whether expression of LTP itself is also dependent on PP2A-mediated dephosphorylation. As we show in Fig. 9 E, inhibition of PP2A abolished expression of LTP after HFS of Schaffer collaterals. Thus, although the targets of PP2A remained undetermined in these experiments, the combined data support the notion that activity-dependent, PP2A-mediated translational derepression is promoting implementation of long-term plastic changes in neurons.
Discussion
Our work describes a novel mechanism that couples neuronal activity to RNA control of gene expression. In this mechanism, neuronal stimulation causes a rapid, PP2A-mediated dephosphorylation of eIF4B at S406. Dephosphorylation significantly decreases the binding affinity of BC RNAs to eIF4B, thus releasing the factor to engage with the 40S small ribosomal subunit and to recruit it for translation initiation. As a consequence, BC RNA control switches from a repressive to a permissive state, enabling translation.
BC RNAs achieve translational repression by interacting with eIFs 4A and 4B (Wang et al., 2002, 2005; Lin et al., 2008; Eom et al., 2011). In standard eukaryotic translation initiation, a heterotrimeric complex composed of eIFs 4A, 4E, and 4G recognizes the 5′ cap of an mRNA and interacts with the small ribosomal subunit (in the form of the 43S preinitiation complex) in the rate-limiting recruitment step (Kapp and Lorsch, 2004; Pestova et al., 2007; Parsyan et al., 2011). The role of eIF4B in this process has in recent years become the subject of increasing interest. eIF4B is required for the efficient initiation of mRNAs with structured 5′ UTRs (Dmitriev et al., 2003; Shahbazian et al., 2010), and eIF4A is assisted by eIF4B in the unwinding of such higher-order structural content (Pestova et al., 2007; Özeş et al., 2011). However, unlike mammalian eIF4B, yeast eIF4B does not stimulate the helicase activity of eIF4A (Altmann et al., 1993; Rajagopal et al., 2012). Nevertheless, deletion of the eIF4B gene in yeast results in severely reduced growth rates and impaired translation that can be corrected by mammalian eIF4B (Altmann et al., 1993; Walker et al., 2013). These data indicate that yeast and mammalian eIF4B share a function, other than stimulation of eIF4A, that is critical for cell viability.
Although their sequence identity is limited (<30%), mammalian and yeast eIF4B both interact directly with the small ribosomal subunit to promote the initiation recruitment step (Méthot et al., 1996; Walker et al., 2013). Mammalian eIF4B exhibits two binding sites for RNA, one that binds RNA (i.e., mRNA) independent of sequence content but with high affinity, and a second one that specifically binds 18S rRNA (Méthot et al., 1996; Fleming et al., 2003). eIF4B thus performs a bridging function that, by concomitantly interacting with mRNA and the small ribosomal subunit via 18S rRNA, mediates recruitment of the 43S preinitiation complex during 48S initiation complex formation.
In previous work (Eom et al., 2011), we showed that BC RNAs compete with 18S rRNA for access to eIF4B, and that a conserved noncanonical C-loop motif in the 3′ BC RNA stem-loop domain is responsible for this interaction. We now report that this interaction results in a direct and specific competition of BC RNAs with the 40S small ribosomal subunit. Significantly, this competition is strongly dependent on eIF4B phosphorylation at S406 (but, notably, not on eIF4B phosphorylation at S422). The equilibrium dissociation constant of BC1 RNA is Kd = 760 nM for dephospho-S406 eIF4B but Kd = 90 nM for phospho-S406 eIF4B. Given that the intracellular concentration of eIF4B has been estimated at 250–300 nM (Dmitriev et al., 2003), the data suggest that BC1 RNA is strongly associated with eIF4B, and thus effectively competing with the 40S ribosomal subunit, if the factor is in the phospho-S406 state, but not if it is in the dephospho-S406 state.
The eIF4B S406 phosphorylation status is therefore the basis for a molecular switch that modulates the ability of BC RNAs to engage with the factor and compete with the 40S ribosomal subunit for access. As a result, BC RNA translational control is in the repressive state with phospho-S406 eIF4B, but in the permissive state with dephospho-S406 eIF4B. This mechanism raises the question of how eIF4B S406 phosphorylation is regulated in neurons and, specifically, how it is controlled in a manner that is dependent on neuronal activity. We find that activation of group I mGluRs causes rapid dephosphorylation of eIF4B S406 and leads to a reversal of BC1-mediated translational repression. Dephosphorylation of eIF4B S406 is a requirement for the activity-dependent induction (i.e., derepression) of translation. In vivo, dephosphorylation of eIF4B S406 can be triggered by neuronal stimulation that elicits epileptogenic responses. Dephosphorylation is rapid, occurring within 2 min after neuronal stimulation, both in vivo and in cultured neurons. The BC RNA–eIF4B mechanism thus offers a means of neuronal translational modulation that can be implemented in a timely manner after changes in activity status.
We identified PP2A as the phosphatase responsible for activity-dependent dephosphorylation of eIF4B S406 (Fig. 10). Consequently, PP2A was found to be requisite for eIF4B dephosphorylation-mediated derepression of BC RNA controlled translation (Fig. 10). Our work describes a novel molecular mechanism in which neuronal stimulation causes rapid dephosphorylation of eIF4B S406, thus defining a window of opportunity of ∼10 min in which BC RNA translational control is permissive and neuronal mRNAs with structured 5′ UTRs become eligible for translation initiation (Fig. 10). This mechanism, our data suggest, may also be contributing to the manifestation of hippocampal LTP, with protein phosphatases playing multiple roles in its initial induction before or during tetanization (Woo et al., 2002) as well as in its protein synthesis–dependent phase after tetanization. We therefore posit, and invite further research to test this hypothesis, that activity-dependent dephosphorylation is a key strategic event in the long-term modulation of synaptic strength.
Figure 10.

Activation of group I mGluRs results in a transition of BC1 RNA translational control from repressive to permissive. PP2A rapidly dephosphorylates eIF4B at S406, causing the release of translational repressor BC1 RNA and freeing the factor to engage the 40S small ribosomal subunit and to initiate translation. Symbols are not drawn to scale.
Materials and methods
Constructs
eIF4B mutations were generated using the QuikChange site-directed mutagenesis kit (Agilent Technologies). The following primers were used (mutations are indicated in bold). eIF4B S406E: forward, 5′-CGGGAGAGACACCCAGAATGGCGAAGTGAAGA-3′; reverse, 5′-TCTTCACTTCGCCATTCTGGGTGTCTCTCCCG-3′. eIF4B S406A: forward, 5′-CGGGAGAGACACCCAGCCTGGCGAAGTGAAGA-3′; reverse, 5′-TCTTCACTTCGCCAGGCTGGGTGTCTCTCCCG-3′.
The PKMζ 5′ UTR was amplified by PCR. The following PCR product was inserted into EGFP-N1 (Takara Bio Inc.) using XhoI and BamHI: 5′-CTCGAGCCGTGTTTTAGCGCGTGCCTCAGCTGCCGGCCGTAGCTTCCCAGGGGAGCCGGGTACCACCCGGGCCTGGAGACATGAGGAGGAGGCAGAGATGTGAGAGGCGGGGAGCAGGAGAGCCAACCTTCTATTAGATGTCTGCTCCTCCAGCAGTAGCCCACCCTGCCCATTGTCGATCTGGAGACCCACCCTGCCCATTGTCGGTCCGGAGGCCAGGGGACGTAGCAGCATCGGCTCCCTTAAAGGGACGGAAGATGCCTCCTTGCACGCCCACCTTCGGTAGAGCATAAAGAATCCTGTGCTGAAGAGGAAGCAGAGAAAGCCGAGTCCATCTACCGCCGTGGAGCCAGAAGATGGAGGAAGCTGTACCGAGCCAACGGCCACCTCTTCCAAGCCAAGCGCTTTAACAGGGGAGCGTACTGCGGCCAGTGCAGCGAAAGGATATGGGGTCTCTCGAGGCAGGGCTACAGGTGCATCAACTGCAAGCTGCTGGTCCATAAACGCTGCCACGTCCTCGTCCCGCTGACCTGCAGGAGGCATATGGATCC-3′.
Phosphorylation of eIF4B by PKMζ
eIF4B or eIF4B S406A (660 nM) was incubated with recombinant PKMζ (0.5 µM) in 50 mM Hepes, pH 7.5, 150 mM NaCl, 10 mM MgCl2, 10% glycerol, 1 mM DTT, and proteinase and phosphatase inhibitors (complete Ultra tablets, mini, EDTA-free, EASY pack and phosSTOP; Roche) with or without ATP (1 mM) at 30°C for 30 min. Recombinant PKMζ was prepared as follows (Ling et al., 2002).
Spodoptera frugiperda (Sf9) cells were grown in SF-900 II Serum Free Medium (Gibco) containing 5 µg/ml gentamicin. Sf9 cells were spun and infected with the baculovirus-PKCζ virus stock. The cells were seeded and cultured for 3 d. Cells were harvested and washed with PBS. Cell pellets were sonicated in 65 ml of homogenization buffer (2 mM EDTA, 10 mM EGTA, 0.3% β-mercaptoethanol, 20 µg/ml aprotinin, 5 mM benzamidine, 0.1 mM leupeptin, 70 µg/ml 4-(2-aminoethyl) benzenesulfonylfluoride, and 20 mM Tris-HCl, pH 7.5). A two-step purification of PKMζ from PKCζ was performed using DEAE Fast Flow Sepharose and Superdex 75 10/300 GL columns (preparation grade; Pharmacia).
Purification of 40S and 60S ribosomal subunits
Rabbit reticulocyte lysate (RRL) was stored at −80°C. 40S and 60S ribosomal subunits were purified from 50–100 ml RRL as described previously (Pestova and Hellen, 2005), with the following modifications. Pefabloc (Sigma-Aldrich), a water-soluble irreversible inhibitor of serine proteases, was added to RRL at 0.5 mg/ml. Polyribosomes were precipitated by centrifugation of RRL at 45,000 rpm for 4.5 h in a 50.2 Ti rotor (Beckman Coulter) at 4°C. Pellets were resuspended in 4 ml of buffer A (50 mM KCl, 4 mM MgCl2, 2 mM DTT, and 20 mM Tris/HCl, pH 7.5) adjusted to a final A260 OD of 100–150 and stirred on ice for 30 min. Puromycin was added at 1 mM, and the sample was incubated for 10 min on ice and for 10 additional min at 37°C. KCl was added to a final concentration of 0.5 M. Aliquots of this suspension were loaded onto 10–30% sucrose density gradients and centrifuged at 22,000 rpm for 16 h in a rotor (SW 28; Beckman Coulter) at 4°C. Fractions were collected along the gradient, and the optical density of each fraction was measured in 1:10-dilution aliquots. To avoid cross-contamination of 40S and 60S subunits, care was taken to select only peak fractions. 60S subunit fractions were diluted with two volumes of buffer A, and 40S subunit fractions were diluted with one volume of buffer A. Samples were concentrated to a volume of 0.5–1 ml using an Amicon concentrator (Sigma-Aldrich), diluted with buffer B (50 mM KCl, 2 mM DTT, 250 mM sucrose, and 20 mM Tris/HCl, pH 7.5) to 4 ml, and concentrated to a final volume of 0.5 ml.
EMSA analysis
32P-labeled RNA probes were generated using riboprobe in vitro transcription systems (Promega) as described previously (Eom et al., 2011). Probes (1 nM; 50,000 cpm per reaction) were heated for 10 min at 70°C, allowed to cool to room temperature, and incubated with proteins in binding buffer (300 mM KCl, 5 mM magnesium acetate, 2 mM DTT, 5% glycerol, and 20 mM Hepes, pH 7.6) for 20 min at room temperature. Unlabeled RNAs (18S rRNA) or purified ribosomal subunits were used as competitors. RNA–protein complexes were separated on 5% polyacrylamide gels and analyzed by autoradiography.
Equilibrium dissociation constants were calculated on the basis of serial EMSA titration experiments (Ryder et al., 2008; Eom et al., 2011; Muslimov et al., 2011). EMSA was performed at 0, 33, 115, 330, 660, 990, 1,320, and 2,640 nM for eIF4B and 0, 28.4, 56.8, 142, 284, 568, 1,136, and 2,272 nM for eIF4B S406E. Intensities of bound RNA and free RNA signals were measured (ImageJ; NIH) as a function of eIF4B (or eIF4B S406E) concentrations. Fractions bound (%) were calculated by dividing intensities of bound RNA signals by intensities of total RNA signals (bound and free). Equilibrium dissociation constants were established by fitting the data to the Hill equation (Ryder et al., 2008; Eom et al., 2011; Muslimov et al., 2011). IGOR Pro software (WaveMetrics) was used to calculate Kd values. The software is based on the Levenberg-Marquardt algorithm (Ryder et al., 2008).
Western blot analysis
Proteins were extracted from Neuro-2A cell pellets, from neurons in primary culture, or from mouse forebrain using 1% Triton X-100 (Sigma-Aldrich). Protein extracts or recombinant proteins were resolved on 10% SDS-PAGE gels and transferred to PVDF membranes (EMD Millipore). Membranes were blocked (5% milk/5% BSA) and incubated with primary antibodies. Primary antibodies were directed against the following proteins (antibody species in parentheses): pan-eIF4B (rabbit), phospho-S406 eIF4B (rabbit), phospho-S422 eIF4B (rabbit; all from Cell Signaling Technology), PKMζ (rabbit; Hernandez et al., 2003), EGFP (mouse; Santa Cruz Biotechnology, Inc.), and γ-tubulin (mouse; Sigma-Aldrich and Santa Cruz Biotechnology, Inc.). After incubation with secondary antibodies (Cell Signaling Technology), the SuperSignal West Pico Chemiluminescent substrate (Thermo Fisher Scientific) was used for visualization. Restore Plus Western Blot Stripping Buffer (Thermo Fisher Scientific) was used for membrane stripping. For quantitative analysis, band intensities were normalized to those of γ-tubulin or total eIF4B.
Cell culture
Neuro-2A cells were plated onto 6-well dishes on day 1. The following day (day 2), cells were incubated with 60 mM KCl for 16 h (overnight; Schor et al., 2009). Cells were incubated with 100 µM DHPG for 2 min (to establish phosphorylation status) or 10 min (for reporter expression). Phosphatase inhibitors were applied before KCl as follows: 25 nM OA, 10 nM deltamethrin, and 5 nM microcystin. Proteins were extracted for Western blot analysis on day 3.
Timed-pregnant Sprague Dawley rats (Hilltop Lab Animals) were used to obtain embryonic day 18 embryos for the generation of hippocampal neurons in primary culture, as described previously (Muslimov et al., 1998; Wang et al., 2002). Cells were dissociated enzymatically (37°C, 20 min) in 1× Triple Express (Gibco) and mechanically in MEM (Gibco). Neurons were plated onto poly-l-lysine–treated glass coverslips in MEM containing 10% horse serum. Cells were plated at nominal densities of 50 cells/mm2. Cells attached to the surface 4 h after seeding. Coverslips were then transferred to dishes containing a monolayer of glial feeder cells and were maintained in a serum-free neuronal culture medium (NCM) at 37°C in an atmosphere of 5% CO2. NCM contained MEM (Gibco), N-2 supplement (Gibco), 0.5 mg/ml ovalbumin (Sigma-Aldrich), and 10 mM Hepes (Gibco). 5 µM cytosine-β-d-arabinofuranoside (Sigma-Aldrich) was added on the fourth day to reduce glial proliferation. Cells were used for immunocytochemistry after 3 wk in culture. They were washed with PBS, pH 7.4, and fixed in 4% formaldehyde (made from paraformaldehyde) and 4% sucrose in PBS (Gibco) at room temperature for 20 min.
To generate cortical neurons in primary culture, cerebral cortices were dissected from the brains of embryonic day 18 Sprague Dawley rat embryos. Cells were dissociated enzymatically (37°C, 20 min) in 1× Triple Express (Gibco) and mechanically in MEM (Gibco). Neurons were plated and cultured in 6-well plates coated with poly-l-lysine in neurobasal medium (Gibco) supplemented with 10% FBS (Gibco). After 3 h, the medium was replaced with neurobasal medium supplemented with 2% B27 (Gibco) and 1% Glutamax (Gibco). 5 µM cytosine-β-d-arabinofuranoside (Sigma-Aldrich) was added on the third day to reduce glial proliferation. Neurons were cultured for 10 d. After drug treatment, cells were washed with PBS, pH 7.4, and collected. Proteins were extracted using 1% Triton X-100 on ice.
For drug treatment, Neuro-2A cells or neurons in primary culture were incubated with 100 µM DHPG for 2 min (for phosphorylation status) or 10–30 min (for expression levels). 25 nM OA was applied to cells 5 min before application of DHPG.
Transfection
Neuro-2A cells were transfected with the various combinations of constructs and RNA using Lipofectamine 2000 (Invitrogen) as follows.
(1) To examine reporter translation, the following were transfected: the PKMζ reporter construct (PKMζ 5′ UTR fused to the EGFP-N1 protein-coding region), siRNA against murine eIF4B mRNA, human eIF4B constructs (unmodified eIF4B, eIF4B S406E, or eIF4B S406A), and BC1 RNA (15 pmol). siRNA was obtained from GE Healthcare (ON-TARGETplus SMARTpool). In control experiments, 15 pmol U6 RNA was transfected instead of 15 pmol BC1 RNA.
(2) To examine whether a long, structured 5′ UTR is necessary for translational repression by BC1 RNA, an EGFP-C1 construct containing a short, unstructured 5′ UTR (Takara Bio Inc.) and BC1 RNA (15 pmol) were transfected. To examine the relevance of eIF4B S406 phosphorylation in this setting, the same transfection was performed using cotransfection with siRNA against murine eIF4B mRNA and with human eIF4B constructs (unmodified eIF4B, eIF4B S406E, or eIF4B S406A).
(3) To examine whether dephosphorylation promotes derepression of translation upon DHPG stimulation, the PKMζ reporter construct and BC1 RNA were transfected.
(4) To examine expression levels of human eIF4B constructs, we transfected unmodified eIF4B, eIF4B S406E, or eIF4B S406A constructs.
(5) To knock down expression levels of PP2A, we transfected siRNA directed against murine PP2A catalytic subunit α mRNA. siRNA was obtained from GE Healthcare.
In vivo epileptogenesis
Audiogenic seizures were induced in Fmr1−/− mice (C57BL6/129SvJ/FVB, age 18–21 d). To be used with a standard plastic cage, a personal alarm device (TBO-Tech) was mounted on a Styrofoam cage cover. Animals were placed in the cage and subjected to auditory stimulation of 120 dB (Zhong et al., 2010). Animals were sacrificed after 2 min of auditory stimulation; generalized tonic-clonic seizures had fully materialized by this time. In addition, seizures were chemically induced in WT mice (C57BL/6J, age 2–3 mo) by i.p. injection of 25 mg/kg KA (Tocris Bioscience). After 30 min, mice were scored for seizure induction.
Forebrain cortices were dissected and homogenized in 1% Triton X-100 in PBS containing proteinase and phosphatase inhibitors (complete Ultra tablets, mini, EDTA-free, EASY pack, and phosSTOP; Roche) using a glass Dounce homogenizer, 10–20 strokes. Samples were centrifuged at 10,000 g for 5 min, and the supernatant was examined by Western blot analysis.
Mice were obtained from the Jackson Laboratory. Work with vertebrate animals was approved by the Institutional Animal Care and Use Committee of SUNY Downstate Medical Center.
Immunocytochemistry
Immunocytochemistry was performed with hippocampal neurons in primary culture as described previously (Muslimov et al., 1998; Wang et al., 2002). In brief, neurons were fixed in 4% formaldehyde (freshly made from paraformaldehyde) and 4% sucrose, and rinsed twice with PBS containing 5 mM MgCl2. Cells then were incubated in “superblock” (0.5 M Tris/HCl, pH 7.6, 0.1% gelatin, 10% BSA, 10% normal goat serum, and 0.05% sodium azide) in the presence of 0.25% Triton X-100 for 10 min at room temperature for permeabilization. Neurons were subsequently incubated overnight at 4°C with primary antibodies. Cells were washed twice in 0.5 M Tris-buffered saline, pH 7.6 (5 min each, room temperature) and incubated with species-specific secondary antibodies in 0.5 M Tris-buffered saline, pH 7.6, 0.1% gelatin, 1% BSA, and 0.05% sodium azide for 2 h at ambient temperature.
Primary antibodies were directed at the following antigens (antibody species and dilutions in parentheses): pan-eIF4B (rabbit, 1:500), phospho-S406 eIF4B (rabbit, 1:500), and synaptophysin (mouse, 1:1,000). Rabbit polyclonal anti-eIF4B and anti-phospho eIF4B antibodies were purchased from Cell Signaling Technology. The mouse monoclonal anti-synaptophysin antibody was purchased from Abcam. For double-labeling experiments, we used species-specific secondary antibodies conjugated with fluorescein or rhodamine derivatives (Jackson ImmunoResearch Laboratories, Inc.). Secondary antibodies were used as follows: anti–rabbit labeled with fluorescein isothiocyanate (Jackson ImmunoResearch Laboratories, Inc.; dilution 1:50) and anti–mouse labeled with rhodamine Red-X (Jackson ImmunoResearch Laboratories, Inc.; dilution 1:200). DHPG (100 µM) was applied for 2 min. OA (25 nM) was added 5 min before DHPG application. Background labeling was established by performing the procedure in the absence of the primary antibody. Neurons were fixed in 4% formaldehyde (freshly made from paraformaldehyde) and 4% sucrose.
Confocal images were acquired with a TCS SP5 II confocal laser scanning microscope system (Leica) attached to a DMI 6000CS microscope (Leica). Objective lens: 63×, oil, Leica HCX Pl Apo, ∞/0.17 NA/E. Image resolution: 512 × 512 pixels, 3.03 mm/pixel; 1,024 × 1,024 pixel, 1.52 mm/pixel; 2,048 × 2,048 pixel, 757.21 nm/pixel. A digital camera (DFC350FX; Leica) was used with digital zoom scan software (Leica). Fluorescence intensities were acquired from 4 µm2 dendritic regions of interest at distances of 100 µm from the respective neuronal cell body. Fluorescence intensities were quantified using ImageJ (NIH). Dendritic extent and morphology were found to be indistinguishable between basal, DHPG, and DHPG/OA experimental states, as indicated by phase-contrast microscopy. The person performing quantitative and statistical analysis was not cognizant of the nature of the performed experiment (i.e., basal, DHPG, or DHPG/OA). Quantitative/statistical analysis in conjunction with this and the other techniques was performed using STATISTICA software (StatSoft).
Hippocampal slice preparation and recording
Acute hippocampal slices (450 µm) were prepared from male Sprague Dawley rats (6–8 wk of age) as described previously (Tsokas et al., 2005). The rats were deeply anesthetized with isoflurane and decapitated, and the brain was rapidly removed and placed in ice-cold artificial cerebrospinal fluid (ACSF) containing the following (in mM): 118 NaCl, 3.5 KCl, 2.5 CaCl2, 1.3 MgSO4, 1.25 NaH2PO4, 24 NaHCO3, and 15 glucose, bubbled with 95% O2/5% CO2. The hippocampus was rapidly dissected out, and transverse slices were prepared on a tissue chopper (McIlwain) at 4°C. The slices were maintained in an interface chamber (ACSF and humidified 95% O2/5% CO2 atmosphere) at room temperature for at least 2 h after preparation. For recording, the slices were transferred to a submersion chamber preheated to 31–32°C, in which they were superfused on a nylon mesh with a recirculating volume of 10 ml ACSF, using a custom-made recirculation system using piezoelectric pumps (Bartels Mikrotechnik GmbH). The flow rate was 6–10 ml/min. Field EPSPs (fEPSPs) were recorded with a glass extracellular recording electrode filled with ACSF (2–5 MΩ), which was placed in the CA1 stratum radiatum. Concentric bipolar stimulating electrodes (Frederick Haer Company) were placed on either side of the recording electrode within CA3 or CA1, delivering monophasic, constant-current stimuli (100 µs). Pathway independence was confirmed by the absence of paired-pulse facilitation between the two pathways. The fEPSP was monitored by delivering stimuli at 0.0166 Hz, and the signals were low-pass filtered at 2 kHz and digitized at 20 kHz. fEPSPs were acquired, and the amplitudes and maximum initial slopes (10–90% of the rise of the fEPSP) were measured, using the WinLTP data acquisition program (Anderson et al., 2012). The high-frequency stimulation consisted of two 100-Hz, 1-s tetanic trains, at 70% of spike threshold, spaced 20 s apart. The spike threshold was typically 3–4 mV. In all experiments, HFS was delivered at least 60 min after transfer of the slices into the recording chamber when the basal EPSP had been stable for at least 45 min. In each experiment, two hippocampal slices were recorded concurrently: one slice was tetanized, whereas the other slice was a sham-stimulated control subjected to test stimuli only. OA was used as stock solutions in DMSO, diluted to 1 µM final concentration in ACSF (according to a previously published protocol for the use of OA with brain slices; Pei et al., 2003), and applied in the superfusate beginning 30 min before HFS and for the remainder of the experiment post-tetanization. Controls for these experiments were also preincubated and superfused in 0.4% DMSO.
As described previously (Tsokas et al., 2005), slices removed from the recording chamber were immediately frozen on glass on dry ice. Each CA1 region was excised in the cold room (4°C) and homogenized in 20 µl of ice-cold RIPA lysis buffer using a motorized homogenizer directly in the microcentrifuge tube (10 strokes, 1 stroke/s). The lysis buffer had the following composition (in mM, unless indicated otherwise): 25 Tris-HCl, pH 7.4, 150 NaCl, 6 MgCl2, 2 EDTA, 1.25% NP40, 0.125% SDS, 0.625% sodium deoxycholate, 26 NaF, 20 DTT, 10 β-glycerophosphate, 4 para-nitrophenylphosphate, 4 sodium tartrate dihydrate, 2 sodium pyrophosphate, 2 imidazole, 1 sodium orthovanadate, 1.5 sodium molybdate, 50 µm (-)-p-bromotetramisole oxalate, 10 µm cantharidin, 1 µm microcystin-LR, 1 phenylmethylsulfonyl fluoride, 20 µg/ml leupeptin, and 4 µg/ml aprotinin. Appropriate volumes of 4× NuPage LDS Sample Buffer (Invitrogen) and β-mercaptoethanol were added to the homogenates, and samples were boiled for 5 min followed by SDS-PAGE electrophoresis. After transfer at 4°C, nitrocellulose membranes (LI-COR Odyssey Blocking Buffer) were probed overnight at 4°C using primary antibodies dissolved in LI-COR Odyssey Blocking Buffer with 0.1% Tween 20 and 0.01% SDS. After washing in PBS, 0.1% Tween 20 (PBS-T; three washes, 5 min each), the membranes were incubated with IRDye (LI-COR) secondary antibodies. Proteins were visualized with the LI-COR Odyssey System. Densitometric analysis of the bands was performed using ImageJ, and values were normalized to γ-tubulin. LTP electrophysiology data were analyzed using one-way analysis of variance (ANOVA) followed by Tukey post-hoc tests, using Prism (GraphPad Software) and Origin (Origin Lab) software.
Online supplemental material
Fig. S1 provides evidence that PKMζ phosphorylates eIF4B at S406. Fig. S2 shows that primate BC200 RNA competes with the 40S ribosomal subunit, but not with the 60S ribosomal subunit, for access to eIF4B, and that phosphorylation of eIF4B at S406 favors binding of BC200 RNA at this site. Fig. S3 demonstrates that eIF4B is required for BC1 RNA translational control of mRNAs that feature structured 5′ UTRs. Fig. S4 shows a control experiment in which DHPG-induced translation of a reporter mRNA with a structured 5′ UTR was prevented in the presence of PP2A inhibitor OA. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.201401005/DC1.
Supplementary Material
Acknowledgments
We thank Dr. Tatyana Pestova (Department of Cell Biology) for advice concerning ribosomal subunit purification and Dr. Jeremy Weedon (Scientific Computing Center) for statistical consultation. V. Berardi acknowledges support from the Istituto Pasteur–Fondazione Cenci Bolognetti. T.C. Sacktor acknowledges support from the Lightfighter Organization. P. Tsokas has been a recipient of an Alexander S. Onassis Public Benefit Foundation award.
This work was supported by National Institutes of Health grants R37 MH057068 (to T.C. Sacktor), R01 MH53576 (to T.C. Sacktor), R01 DA034970 (to T.C. Sacktor), R01 NS046769 (to H. Tiedge), and R01 DA026110 (H. Tiedge).
The authors declare no competing financial interests.
Footnotes
Abbreviations used in this paper:
- ACSF
- artificial cerebrospinal fluid
- ANOVA
- analysis of variance
- BC
- brain cytoplasmic
- DHPG
- 3,5-dihydroxyphenylglycine
- eIF
- eukaryotic initiation factor
- EMSA
- electrophoretic mobility shift assay
- KA
- kainic acid
- LTP
- long-term potentiation
- mGluR
- metabotropic glutamate receptor
- NC
- nontargeting control
- OA
- okadaic acid
- PKMζ
- protein kinase Mζ
- PP2A
- protein phosphatase 2A
- RRL
- rabbit reticulocyte lysate
- rRNA
- ribosomal RNA
- WT
- wild type
References
- Altmann, M., Müller P.P., Wittmer B., Ruchti F., Lanker S., and Trachsel H.. 1993. A Saccharomyces cerevisiae homologue of mammalian translation initiation factor 4B contributes to RNA helicase activity. EMBO J. 12:3997–4003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson, W.W., Fitzjohn S.M., and Collingridge G.L.. 2012. Automated multi-slice extracellular and patch-clamp experiments using the WinLTP data acquisition system with automated perfusion control. J. Neurosci. Methods. 207:148–160. 10.1016/j.jneumeth.2012.04.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassell, G.J., Zhang H., Byrd A.L., Femino A.M., Singer R.H., Taneja K.L., Lifshitz L.M., Herman I.M., and Kosik K.S.. 1998. Sorting of β-actin mRNA and protein to neurites and growth cones in culture. J. Neurosci. 18:251–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhakar, A.L., Dölen G., and Bear M.F.. 2012. The pathophysiology of fragile X (and what it teaches us about synapses). Annu. Rev. Neurosci. 35:417–443. 10.1146/annurev-neuro-060909-153138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang, S.C., Zhao W., Bauchwitz R., Yan Q., Bianchi R., and Wong R.K.. 2005. Prolonged epileptiform discharges induced by altered group I metabotropic glutamate receptor-mediated synaptic responses in hippocampal slices of a fragile X mouse model. J. Neurosci. 25:8048–8055. 10.1523/JNEUROSCI.1777-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell, J.C.2011. Defects in translational regulation contributing to human cognitive and behavioral disease. Curr. Opin. Genet. Dev. 21:465–473. 10.1016/j.gde.2011.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dmitriev, S.E., Terenin I.M., Dunaevsky Y.E., Merrick W.C., and Shatsky I.N.. 2003. Assembly of 48S translation initiation complexes from purified components with mRNAs that have some base pairing within their 5′ untranslated regions. Mol. Cell. Biol. 23:8925–8933. 10.1128/MCB.23.24.8925-8933.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dölen, G., Osterweil E., Rao B.S., Smith G.B., Auerbach B.D., Chattarji S., and Bear M.F.. 2007. Correction of fragile X syndrome in mice. Neuron. 56:955–962. 10.1016/j.neuron.2007.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eom, T., Berardi V., Zhong J., Risuleo G., and Tiedge H.. 2011. Dual nature of translational control by regulatory BC RNAs. Mol. Cell. Biol. 31:4538–4549. 10.1128/MCB.05885-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming, K., Ghuman J., Yuan X., Simpson P., Szendröi A., Matthews S., and Curry S.. 2003. Solution structure and RNA interactions of the RNA recognition motif from eukaryotic translation initiation factor 4B. Biochemistry. 42:8966–8975. 10.1021/bi034506g [DOI] [PubMed] [Google Scholar]
- Frankland, P.W., and Josselyn S.A.. 2013. Neuroscience: Memory and the single molecule. Nature. 493:312–313. 10.1038/nature11850 [DOI] [PubMed] [Google Scholar]
- Gingras, A.-C., Raught B., and Sonenberg N.. 1999. eIF4 initiation factors: effectors of mRNA recruitment to ribosomes and regulators of translation. Annu. Rev. Biochem. 68:913–963. 10.1146/annurev.biochem.68.1.913 [DOI] [PubMed] [Google Scholar]
- Gkogkas, C., Sonenberg N., and Costa-Mattioli M.. 2010. Translational control mechanisms in long-lasting synaptic plasticity and memory. J. Biol. Chem. 285:31913–31917. 10.1074/jbc.R110.154476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez, A.I., Blace N., Crary J.F., Serrano P.A., Leitges M., Libien J.M., Weinstein G., Tcherapanov A., and Sacktor T.C.. 2003. Protein kinase M ζ synthesis from a brain mRNA encoding an independent protein kinase C ζ catalytic domain. Implications for the molecular mechanism of memory. J. Biol. Chem. 278:40305–40316. 10.1074/jbc.M307065200 [DOI] [PubMed] [Google Scholar]
- Holz, M.K., Ballif B.A., Gygi S.P., and Blenis J.. 2005. mTOR and S6K1 mediate assembly of the translation preinitiation complex through dynamic protein interchange and ordered phosphorylation events. Cell. 123:569–580. 10.1016/j.cell.2005.10.024 [DOI] [PubMed] [Google Scholar]
- Iacoangeli, A., and Tiedge H.. 2013. Translational control at the synapse: role of RNA regulators. Trends Biochem. Sci. 38:47–55. 10.1016/j.tibs.2012.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Job, C., and Eberwine J.. 2001. Localization and translation of mRNA in dendrites and axons. Nat. Rev. Neurosci. 2:889–898. 10.1038/35104069 [DOI] [PubMed] [Google Scholar]
- Kapp, L.D., and Lorsch J.R.. 2004. The molecular mechanics of eukaryotic translation. Annu. Rev. Biochem. 73:657–704. 10.1146/annurev.biochem.73.030403.080419 [DOI] [PubMed] [Google Scholar]
- Kindler, S., Wang H., Richter D., and Tiedge H.. 2005. RNA transport and local control of translation. Annu. Rev. Cell Dev. Biol. 21:223–245. 10.1146/annurev.cellbio.21.122303.120653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondrashov, A.V., Kiefmann M., Ebnet K., Khanam T., Muddashetty R.S., and Brosius J.. 2005. Inhibitory effect of naked neural BC1 RNA or BC200 RNA on eukaryotic in vitro translation systems is reversed by poly(A)-binding protein (PABP). J. Mol. Biol. 353:88–103. 10.1016/j.jmb.2005.07.049 [DOI] [PubMed] [Google Scholar]
- Li, T., Huang P., Liang J., Fu W., Guo Z., and Xu L.. 2011. Microcystin-LR (MCLR) induces a compensation of PP2A activity mediated by α4 protein in HEK293 cells. Int. J. Biol. Sci. 7:740–752. 10.7150/ijbs.7.740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, T., Ying L., Wang H., Li N., Fu W., Guo Z., and Xu L.. 2012. Microcystin-LR induces ceramide to regulate PP2A and destabilize cytoskeleton in HEK293 cells. Toxicol. Sci. 128:147–157. 10.1093/toxsci/kfs141 [DOI] [PubMed] [Google Scholar]
- Lin, D., Pestova T.V., Hellen C.U., and Tiedge H.. 2008. Translational control by a small RNA: dendritic BC1 RNA targets the eukaryotic initiation factor 4A helicase mechanism. Mol. Cell. Biol. 28:3008–3019. 10.1128/MCB.01800-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling, D.S., Benardo L.S., Serrano P.A., Blace N., Kelly M.T., Crary J.F., and Sacktor T.C.. 2002. Protein kinase Mζ is necessary and sufficient for LTP maintenance. Nat. Neurosci. 5:295–296. 10.1038/nn829 [DOI] [PubMed] [Google Scholar]
- Liu, F., Virshup D.M., Nairn A.C., and Greengard P.. 2002. Mechanism of regulation of casein kinase I activity by group I metabotropic glutamate receptors. J. Biol. Chem. 277:45393–45399. 10.1074/jbc.M204499200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews, M.B., Sonenberg N., and Hershey J.W.B.. 2007. Translational control in biology and medicine. Cold Spring Harbor Laboratory Press, Cold Spring Harbor. 934 pp [Google Scholar]
- Méthot, N., Pickett G., Keene J.D., and Sonenberg N.. 1996. In vitro RNA selection identifies RNA ligands that specifically bind to eukaryotic translation initiation factor 4B: the role of the RNA remotif. RNA. 2:38–50 [PMC free article] [PubMed] [Google Scholar]
- Miyashiro, K.Y., Bell T.J., Sul J.Y., and Eberwine J.. 2009. Subcellular neuropharmacology: the importance of intracellular targeting. Trends Pharmacol. Sci. 30:203–211. 10.1016/j.tips.2009.01.005 [DOI] [PubMed] [Google Scholar]
- Muslimov, I.A., Banker G., Brosius J., and Tiedge H.. 1998. Activity-dependent regulation of dendritic BC1 RNA in hippocampal neurons in culture. J. Cell Biol. 141:1601–1611. 10.1083/jcb.141.7.1601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muslimov, I.A., Nimmrich V., Hernandez A.I., Tcherepanov A., Sacktor T.C., and Tiedge H.. 2004. Dendritic transport and localization of protein kinase Mzeta mRNA: implications for molecular memory consolidation. J. Biol. Chem. 279:52613–52622. 10.1074/jbc.M409240200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muslimov, I.A., Patel M.V., Rose A., and Tiedge H.. 2011. Spatial code recognition in neuronal RNA targeting: role of RNA-hnRNP A2 interactions. J. Cell Biol. 194:441–457. 10.1083/jcb.201010027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musumeci, S.A., Bosco P., Calabrese G., Bakker C., De Sarro G.B., Elia M., Ferri R., and Oostra B.A.. 2000. Audiogenic seizures susceptibility in transgenic mice with fragile X syndrome. Epilepsia. 41:19–23. 10.1111/j.1528-1157.2000.tb01499.x [DOI] [PubMed] [Google Scholar]
- Narayanan, U., Nalavadi V., Nakamoto M., Pallas D.C., Ceman S., Bassell G.J., and Warren S.T.. 2007. FMRP phosphorylation reveals an immediate-early signaling pathway triggered by group I mGluR and mediated by PP2A. J. Neurosci. 27:14349–14357. 10.1523/JNEUROSCI.2969-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osten, P., Valsamis L., Harris A., and Sacktor T.C.. 1996. Protein synthesis-dependent formation of protein kinase Mζ in long-term potentiation. J. Neurosci. 16:2444–2451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Özeş, A.R., Feoktistova K., Avanzino B.C., and Fraser C.S.. 2011. Duplex unwinding and ATPase activities of the DEAD-box helicase eIF4A are coupled by eIF4G and eIF4B. J. Mol. Biol. 412:674–687. 10.1016/j.jmb.2011.08.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsyan, A., Svitkin Y., Shahbazian D., Gkogkas C., Lasko P., Merrick W.C., and Sonenberg N.. 2011. mRNA helicases: the tacticians of translational control. Nat. Rev. Mol. Cell Biol. 12:235–245. 10.1038/nrm3083 [DOI] [PubMed] [Google Scholar]
- Pastalkova, E., Serrano P., Pinkhasova D., Wallace E., Fenton A.A., and Sacktor T.C.. 2006. Storage of spatial information by the maintenance mechanism of LTP. Science. 313:1141–1144. 10.1126/science.1128657 [DOI] [PubMed] [Google Scholar]
- Pei, J.J., Gong C.X., An W.L., Winblad B., Cowburn R.F., Grundke-Iqbal I., and Iqbal K.. 2003. Okadaic-acid-induced inhibition of protein phosphatase 2A produces activation of mitogen-activated protein kinases ERK1/2, MEK1/2, and p70 S6, similar to that in Alzheimer’s disease. Am. J. Pathol. 163:845–858. 10.1016/S0002-9440(10)63445-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pestova, T.V., and Hellen C.U.. 2005. Reconstitution of eukaryotic translation elongation in vitro following initiation by internal ribosomal entry. Methods. 36:261–269. 10.1016/j.ymeth.2005.04.004 [DOI] [PubMed] [Google Scholar]
- Pestova, T., Lorsch J.R., and Hellen C.U.T.. 2007. The mechanism of translation initiation in eukaryotes. Translational Control in Biology and Medicine. Mathews M.B., Sonenberg N., and Hershey J.W.B., . Cold Spring Harbor Laboratory Press, Cold Spring Harbor. 87–128 [Google Scholar]
- Rajagopal, V., Park E.H., Hinnebusch A.G., and Lorsch J.R.. 2012. Specific domains in yeast translation initiation factor eIF4G strongly bias RNA unwinding activity of the eIF4F complex toward duplexes with 5′-overhangs. J. Biol. Chem. 287:20301–20312. 10.1074/jbc.M112.347278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raught, B., and Gingras A.-C.. 2007. Signaling to translation initiation. Translational Control in Biology and Medicine. Mathews M.B., Sonenberg N., and Hershey J.W.B., . Cold Spring Harbor Laboratory Press, Cold Spring Harbor. 369–400 [Google Scholar]
- Raught, B., Peiretti F., Gingras A.C., Livingstone M., Shahbazian D., Mayeur G.L., Polakiewicz R.D., Sonenberg N., and Hershey J.W.. 2004. Phosphorylation of eucaryotic translation initiation factor 4B Ser422 is modulated by S6 kinases. EMBO J. 23:1761–1769. 10.1038/sj.emboj.7600193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryder, S.P., Recht M.I., and Williamson J.R.. 2008. Quantitative analysis of protein-RNA interactions by gel mobility shift. Methods Mol. Biol. 488:99–115. 10.1007/978-1-60327-475-3_7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro, M.R., Bray S.M., and Warren S.T.. 2012. Molecular mechanisms of fragile X syndrome: a twenty-year perspective. Annu. Rev. Pathol. 7:219–245. 10.1146/annurev-pathol-011811-132457 [DOI] [PubMed] [Google Scholar]
- Schor, I.E., Rascovan N., Pelisch F., Alló M., and Kornblihtt A.R.. 2009. Neuronal cell depolarization induces intragenic chromatin modifications affecting NCAM alternative splicing. Proc. Natl. Acad. Sci. USA. 106:4325–4330. 10.1073/pnas.0810666106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahbazian, D., Roux P.P., Mieulet V., Cohen M.S., Raught B., Taunton J., Hershey J.W., Blenis J., Pende M., and Sonenberg N.. 2006. The mTOR/PI3K and MAPK pathways converge on eIF4B to control its phosphorylation and activity. EMBO J. 25:2781–2791. 10.1038/sj.emboj.7601166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahbazian, D., Parsyan A., Petroulakis E., Hershey J., and Sonenberg N.. 2010. eIF4B controls survival and proliferation and is regulated by proto-oncogenic signaling pathways. Cell Cycle. 9:4106–4109. 10.4161/cc.9.20.13630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor, G.W., Merlin L.R., and Wong R.K.. 1995. Synchronized oscillations in hippocampal CA3 neurons induced by metabotropic glutamate receptor activation. J. Neurosci. 15:8039–8052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsokas, P., Grace E.A., Chan P., Ma T., Sealfon S.C., Iyengar R., Landau E.M., and Blitzer R.D.. 2005. Local protein synthesis mediates a rapid increase in dendritic elongation factor 1A after induction of late long-term potentiation. J. Neurosci. 25:5833–5843. 10.1523/JNEUROSCI.0599-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker, S.E., Zhou F., Mitchell S.F., Larson V.S., Valasek L., Hinnebusch A.G., and Lorsch J.R.. 2013. Yeast eIF4B binds to the head of the 40S ribosomal subunit and promotes mRNA recruitment through its N-terminal and internal repeat domains. RNA. 19:191–207. 10.1261/rna.035881.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, H., Iacoangeli A., Popp S., Muslimov I.A., Imataka H., Sonenberg N., Lomakin I.B., and Tiedge H.. 2002. Dendritic BC1 RNA: functional role in regulation of translation initiation. J. Neurosci. 22:10232–10241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, H., Iacoangeli A., Lin D., Williams K., Denman R.B., Hellen C.U.T., and Tiedge H.. 2005. Dendritic BC1 RNA in translational control mechanisms. J. Cell Biol. 171:811–821. 10.1083/jcb.200506006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo, N.H., Abel T., and Nguyen P.V.. 2002. Genetic and pharmacological demonstration of a role for cyclic AMP-dependent protein kinase-mediated suppression of protein phosphatases in gating the expression of late LTP. Eur. J. Neurosci. 16:1871–1876. 10.1046/j.1460-9568.2002.02260.x [DOI] [PubMed] [Google Scholar]
- Zhao, W., Bianchi R., Wang M., and Wong R.K.. 2004. Extracellular signal-regulated kinase 1/2 is required for the induction of group I metabotropic glutamate receptor-mediated epileptiform discharges. J. Neurosci. 24:76–84. 10.1523/JNEUROSCI.4515-03.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong, J., Zhang T., and Bloch L.M.. 2006. Dendritic mRNAs encode diversified functionalities in hippocampal pyramidal neurons. BMC Neurosci. 7:17. 10.1186/1471-2202-7-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong, J., Chuang S.C., Bianchi R., Zhao W., Lee H., Fenton A.A., Wong R.K., and Tiedge H.. 2009. BC1 regulation of metabotropic glutamate receptor-mediated neuronal excitability. J. Neurosci. 29:9977–9986. 10.1523/JNEUROSCI.3893-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong, J., Chuang S.C., Bianchi R., Zhao W., Paul G., Thakkar P., Liu D., Fenton A.A., Wong R.K., and Tiedge H.. 2010. Regulatory BC1 RNA and the fragile X mental retardation protein: convergent functionality in brain. PLoS ONE. 5:e15509. 10.1371/journal.pone.0015509 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.