

Summary
Tissue inhibitor of metalloproteinase 3 (TIMP-3) is an important regulator of extracellular matrix (ECM) turnover. TIMP-3 binds to sulfated ECM glycosaminoglycans or is endocytosed by cells via low-density lipoprotein receptor-related protein 1 (LRP-1). Here, we report that heparan sulfate (HS) and chondroitin sulfate E (CSE) selectively regulate postsecretory trafficking of TIMP-3 by inhibiting its binding to LRP-1. HS and CSE also increased TIMP-3 affinity for glycan-binding metalloproteinases, such as adamalysin-like metalloproteinase with thrombospondin motifs 5 (ADAMTS-5), by reducing the dissociation rate constants. The sulfation pattern was crucial for these activities because monosulfated or truncated heparin had a reduced ability to bind to TIMP-3 and increase its affinity for ADAMTS-5. Therefore, sulfation of ECM glycans regulates the levels and inhibitory activity of TIMP-3 and modulates ECM turnover, and small mimicries of sulfated glycans may protect the tissue from the excess destruction seen in diseases such as osteoarthritis, cancer, and atherosclerosis.
Graphical Abstract

Highlights
-
•
The metalloprotease inhibitor TIMP-3 binds to sulfated extracellular glycans
-
•
This inhibits cellular uptake of TIMP-3 by the endocytic receptor LRP-1
-
•
Glycans also increase TIMP-3 affinity for selected target proteases
-
•
The sulfation of matrix glycans therefore modulates TIMP-3 activity and ECM turnover
Tissue inhibitor of metalloproteinase (TIMP)-3 regulates extracellular matrix turnover. Troeberg et al. show that TIMP-3 levels are regulated by the balance between its binding to matrix glycans and receptor-mediated endocytosis and that glycan sulfation regulates this equilibrium.
Introduction
Tissue inhibitor of metalloproteinases 3 (TIMP-3) is unique among the family of mammalian TIMPs in that it binds to the extracellular matrix (ECM), whereas the other TIMPs (TIMP-1, TIMP-2, and TIMP-4) diffuse freely in the extracellular environment (Blenis and Hawkes, 1983, Brew and Nagase, 2010). This ECM-binding ability is thought to position TIMP-3 as a major inhibitor of ECM turnover, and Timp3-null mice exhibit a range of phenotypes associated with increased ECM degradation. For example, the mice show increased collagen degradation in the lungs, leading to increased alveolar size, impaired lung function, and reduced life span (Leco et al., 2001). Additional phenotypes associated with increased ECM degradation include elevated epithelial apoptosis upon mammary gland involution (Fata et al., 2001), impaired bronchiole branching morphogenesis (Gill et al., 2006), cardiac hypertrophy (Fedak et al., 2004), increased cardiac remodeling after myocardial infarction (Tian et al., 2007), and accelerated spontaneous osteoarthritis (Sahebjam et al., 2007).
Although ECM binding is a distinguishing feature of TIMP-3, the molecular mechanism and functional consequences of ECM binding remain unknown. Yu et al. (2000) showed that TIMP-3 can be solubilized from uterine tissue by adding sulfated glycosaminoglycans such as heparin, heparan sulfate, and chondroitin sulfates. TIMP-3 has also been shown to bind to heparin in vitro, leading the authors to suggest that TIMP-3 binds to sulfated glycosaminoglycans (sGAGs) in the ECM. This was supported by Lee et al. (2007), who identified a highly basic region on the surface of TIMP-3 that, they postulated, interacts with negatively charged sulfated ECM components. By mutation of this basic region, they generated a soluble mutant of TIMP-3 that did not bind to the ECM. However, although it is generally accepted that TIMP-3 binds to sGAG in the ECM, it is not known to which specific sGAG TIMP-3 binds and whether specific proteoglycans carry the TIMP-3-binding sGAG.
TIMP-3 secreted from the cell can be cleared from the extracellular environment through cellular uptake and degradation mediated by the endocytic receptor low-density lipoprotein receptor-related protein 1 (LRP-1) (Scilabra et al., 2013, Troeberg et al., 2008). It is unclear what determines postsecretory trafficking of TIMP-3 to the ECM or to endocytosis and how this balance is regulated in health and disease. Although several factors regulate TIMP-3 transcription, posttranslational regulatory mechanisms appear to be central in determining TIMP-3 availability (Troeberg et al., 2008). Reduced tissue levels of TIMP-3 are associated with several pathological conditions, such as osteoarthritis (Morris et al., 2010), cancer (Cruz-Munoz and Khokha, 2008), and atherosclerosis (Cardellini et al., 2009). Therefore, identification of the molecular determinants that regulate TIMP-3 trafficking in the extracellular environment is important to understand both physiological and pathological ECM degradation.
We set out to identify to which ECM glycosaminoglycans TIMP-3 binds and to determine their effects on TIMP-3 trafficking and affinity for target metalloproteinases.
Results
TIMP-3 Binds to Heparan Sulfate and Chondroitin Sulfate E
Using a solid-phase binding assay, we investigated TIMP-3 binding to several glycosaminoglycans. TIMP-3 bound strongly to highly sulfated heparin (12.5 kDa, KD = 19 ± 4 nM) and to pentosan polysulfate (PPS, 4.7 kDa, KD = 3 ± 0.4 nM), a highly sulfated polyxylan generated by chemical sulfation of cellulose (Figure 1). TIMP-3 had a comparable affinity for the natural matrix glycan heparan sulfate (HS, 22 kDa), with a KD of 29 ± 18 nM. TIMP-3 also bound strongly to chondroitin sulfate E (CSE), a 4,6-O-disulfated glycan. TIMP-3 had a higher affinity for CSE from squid (50 kDa, KD = 10 ± 1 nM) than for CSE from bonito (8.7 kDa, KD = 39 ± 6 nM) (Figure 1). No binding to dermatan sulfate, aggrecan, hyaluronic acid, chondroitin-4-sulfate (C-4-S), or chondroitin-6-sulfate (C-6-S) was detected.
Figure 1.
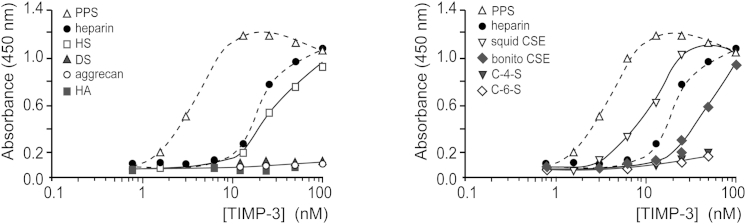
TIMP-3 Binds to Highly Sulfated Glycosaminoglycans
Glycosaminoglycan-binding multiwell plates were coated with 10 μg/ml PPS, heparin, HS, DS, aggrecan, hyaluronic acid (HA), squid CSE, bonito CSE, C-4-S, or C-6-S. Wells were then incubated with TIMP-3-FLAG, and bound TIMP-3 was detected using M2 anti-FLAG primary antibody and a horseradish peroxidase-coupled secondary antibody.
HS and CSE Block TIMP-3 Endocytosis by LRP-1
The effect of HS and CSE on TIMP-3 endocytosis by LRP-1 was investigated by adding recombinant TIMP-3 to HTB94 chondrosarcoma cells and following its disappearance from the medium by immunoblotting. TIMP-3 was taken up by the cells with a half-life of 5 hr, and this endocytosis was inhibited by an LRP ligand binding antagonist, receptor-associated protein (RAP) (Figures 2A and 2B). Endocytosis was also inhibited by preincubation of TIMP-3 with heparin, HS, or bonito CSE, whereas C-4-S had no effect (Figures 2A and 2B).
Figure 2.

Sulfated Glycosaminoglycans Block LRP-1-Mediated Endocytosis of TIMP-3
(A) HTB94 chondrosarcoma cells were incubated in serum-free DMEM with recombinant TIMP-3-FLAG (0.5 nM) either alone (control) or in the presence of RAP (1 μM), heparin (200 μg/ml), HS (200 μg/ml), C-4-S (200 μg/ml), or bonito CSE (200 μg/ml). TIMP-3 remaining in the medium was detected by immunoblotting using M2 anti-FLAG antibody.
(B) TIMP-3 remaining in the medium of cells shown in (A) was quantified using Phoretix 1D software. Symbols indicate TIMP-3 alone (○) or with C-4-S (▼), HS (□), heparin (●), bonito CSE (♦), or RAP (▲).
(C) HTB94 chondrosarcoma cells were incubated in serum-free DMEM containing RAP (0.5 and 0.75 μM), heparin (100 and 200 μg/ml), HS (100 and 200 μg/ml), C-4-S (100 and 200 μg/ml), or bonito CSE (100 and 200 μg/ml). Accumulation of endogenous TIMP-3 in the medium was detected by immunoblotting using a rabbit anti-TIMP-3 antibody.
(D) LRP-1 (5 nM) was coated onto a microtiter plate and wells blocked with 3% (m/v) bovine serum albumin in TIMP buffer. Wells were then incubated with TIMP-3-FLAG (control) or with TIMP-3-FLAG preincubated for 1 hr at 37°C with heparin, HS, bonito CSE, or squid CSE (200 μg/ml). Bound TIMP-3-FLAG was detected using M2 anti-FLAG antibody.
Little endogenous TIMP-3 was found in the culture medium of HTB94 cells, but TIMP-3 accumulated in the medium when cells were treated with CSE or HS (Figure 2C), indicating that they blocked endocytosis of endogenous TIMP-3. The amount of TIMP-3 detected was comparable with that seen in the presence of heparin and RAP. No TIMP-3 accumulated in the medium of cells treated with C-4-S.
Recombinant TIMP-3 bound to the ectodomain of LRP-1 with a KD value of 28 ± 6 nM (Figure 2D). This binding was completely inhibited by preincubation of TIMP-3 with squid CSE and partially inhibited by bonito CSE, heparin, and HS.
HS and CSE Increase TIMP-3 Affinity for ADAMTS-5
We evaluated the effect of HS and CSE on TIMP-3 affinity for ADAMTS-5. In the absence of sGAGs, a Ki value of 1.36 ± 0.02 nM was calculated for TIMP-3 inhibition of ADAMTS-5 (Figure 3A), in agreement with previous reports (Troeberg et al., 2009, Troeberg et al., 2012). Addition of PPS (0.4 μg/ml, equivalent to 100 nM of 4.7 kDa polysaccharide or 2 μM disaccharide) improved the affinity between ADAMTS-5 and TIMP-3 so that even 0.5 nM ADAMTS-5 was fully inhibited by 0.5 nM TIMP-3 (Figure 3A), indicating a substantial affinity increase. We were unable to accurately determine the improved Ki because we could not measure the activity of lower enzyme concentrations, but, by fitting the data to the tight-binding equation (Bieth, 1995), we estimated that Ki is approximately 1 pM, a 1000-fold improvement.
Figure 3.

sGAGs Increase TIMP-3 Affinity for ADAMTS-5
(A) ADAMTS-5 (0.5 nM) was incubated with TIMP-3 (0.5–10 nM, 1 hr, 37°C) in TIMP buffer alone (○) or with HS (□, 1 μM of 8 kDa polysaccharide and 13 μM disaccharide), heparin (●, 100 nM of 12.5 kDa polysaccharide and 2 μM disaccharide), or PPS (▵, 100 nM of 4.7 kDa polysaccharide and 0.8 μM disaccharide), and the residual activity against fluorescent peptide substrate was determined.
(B) ADAMTS-5 (0.5 nM) was incubated with TIMP-3 (0.5–10 nM, 1 hr, 37°C) in TIMP buffer alone (○) or with bonito CSE (♦, 100 nM of 8.7 kDa polysaccharide and 1.5 μM disaccharide) or squid CSE (▿, 100 nM of 50 kDa polysaccharide and 8.3 μM disaccharide), and the residual activity against the fluorescent peptide substrate was determined.
(C) TIMP-3 (0.5 nM) was incubated with ADAMTS-5 (0.5 nM) and various concentrations of HS (10−8–10−4 M disaccharide), heparin (10−8–10−5 M disaccharide), or PPS (10−9–10−4 M disaccharide) (1 hr, 37°C). The residual activity against the fluorescent peptide substrate was determined.
(D) TIMP-3 (0.5 nM) was incubated with ADAMTS-5 (0.5 nM) and various concentrations of bonito CSE (10−11–10−5 M disaccharide), squid CSE fraction 1 (10−8–10−5 M disaccharide), squid CSE fraction 2 (10−12–10−5 M disaccharide), or squid CSE fraction 3 (10−12–10−5 M disaccharide) (1 hr, 37°C). The residual activity against the fluorescent peptide substrate was determined.
Ki was similarly improved by addition of HS (8 μg/ml, equivalent to 1 μM of 22 kDa polysaccharide or 13 μM disaccharide) or heparin (1 μg/ml, equivalent to 100 nM of 12.5 kDa polysaccharide or 2 μM disaccharide) (Figure 3A) and also by CSE from bonito (1 μg/ml, equivalent to 100 nM of 8.7 kDa polysaccharide or 1.5 μM disaccharide) or squid (8 μg/ml, equivalent to 100 nM of 50 kDa polysaccharide or 8.3 μM disaccharide) (Figure 3B).
To compare their efficacies, increasing concentrations of HS and CSE were added to a fixed concentration of TIMP-3 and ADAMTS-5. Concentrations are expressed as molarity of disaccharide to allow comparison between HS and CSE of greatly differing lengths. At concentrations near a Ki of 1.36 nM, TIMP-3 only partially inhibited ADAMTS-5. Therefore, 0.5 nM TIMP-3 inhibited only 20% of the activity of 0.5 nM ADAMTS-5 (Figure 3A). At concentrations above 0.1 μM disaccharide, PPS improved the affinity between TIMP-3 and ADAMTS-5 so that 0.5 nM TIMP-3 completely inhibited 0.5 nM ADAMTS-5 (Figure 3C). The affinity was increased similarly by heparin and HS at disaccharide concentrations above 10 and 100 μM, respectively. CSE was similarly effective above 10 μM disaccharide, although bonito CSE (8.7 kDa) was less effective than squid CSE of 50, 200, or 800 kDa at lower concentrations.
HS and CSE Reduce the Dissociation Rate of the TIMP-3-ADAMTS-5 Complex
Biolayer interferometry was used to further explore the mechanism by which sGAGs increase ADAMTS-5-TIMP-3 affinity. ADAMTS-5 bound to immobilized biotinylated TIMP-3 with a KD of 72 ± 7 nM (Figure 4A). In accordance with previous experiments (Troeberg et al., 2002), this is higher than the Ki calculated in solution, most likely because of the effects of TIMP-3 immobilization. The koff value was determined as 23 ± 0.2 × 0−3 s−1 and the kon rate as 1.41 × 104 M-1.s-1.
Figure 4.
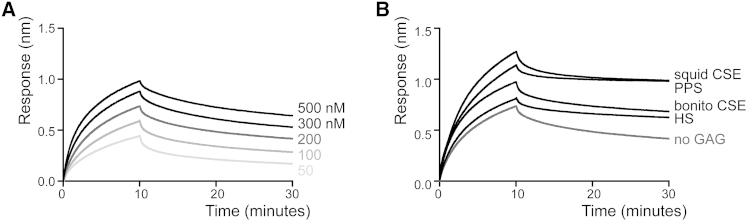
Biolayer Interferometry Analysis Showing that sGAGs Reduce Dissociation of the TIMP-3-ADAMTS-5 Complex
(A) Streptavidin tips were coated with biotinylated TIMP-3, and binding of ADAMTS-5 (20–500 nM) was analyzed by biolayer interferometry.
(B) Streptavidin tips coated with biotinylated TIMP-3 were incubated with ADAMTS-5 (200 nM) and TIMP buffer (no GAG), HS (100 μM), bonito CSE (10 μM), PPS (100 nM), or squid CSE (10 μM) in TIMP buffer. Binding was analyzed by biolayer interferometry.
Addition of sGAG markedly reduced the dissociation rate of the ADAMTS-5-TIMP-3 complex (Figure 4B) so that it was not possible to determine a value for koff using this method. Squid CSE and PPS stabilized the complex to the extent that no dissociation was observed over 20 min, whereas minimal dissociation was observed with bonito CSE and HS.
sGAG had a lesser effect on association of TIMP-3 and ADAMTS-5, with the association rate increasing from 2.13 ± 0.02 M-1.s-1 in the absence of sGAG to 4.81 ± 0.03 M-1.s-1 in the presence of HS and 5.68 ± 0.04 M-1.s-1 in the presence of squid CSE.
Heparin of dp14 and Longer Increases Affinity
We investigated the minimum chain length required for sGAG activity using heparin of differing sizes. TIMP-3 bound strongly to heparin with a degree of polymerization of 36 (dp36, 12.5 kDa) as well as to heparin of dp14 (5 kDa) and dp9 (3 kDa), with KD values of 2 nM for all three heparin sizes (Figure 5A). TIMP-3 binding to dp5 heparin (1.7 kDa) was undetectable. Similarly, ADAMTS-5 bound strongly to heparin of dp36, dp14, and dp9, with KD values of 10 nM for all three sizes (Figure 5B), and binding to dp5 heparin was undetectable.
Figure 5.

Size and Sulfation Affect the Heparin-Mediated Increase in TIMP-3 Affinity for ADAMTS-5
(A and B) Heparin (60 μM disaccharide) of dp36 (12.5 kDa), dp14 (5 kDa), dp9 (3 kDa), or dp5 (1.7 kDa) was immobilized on glycosaminoglycan-binding multiwell plates. The wells were then incubated with FLAG-tagged TIMP-3 (0.08–10 nM) (A) or FLAG-tagged ADAMTS-5 (0.3–20 nM) (B). Bound protein was detected using M2 anti-FLAG primary antibody and a horseradish peroxidase-coupled secondary antibody.
(C) TIMP-3 (0.5 nM) and ADAMTS-5 (0.5 nM) were incubated (1 hr, 37°C) with various concentrations (1–2000 nM disaccharide) of heparin with dp36, dp14, dp9, or dp5. The residual activity against the fluorescent peptide substrate was determined.
(D and E) Heparin (60 μM disaccharide) of dp36 (12.5 kDa) either normally sulfated (untreated), N-desulfated (N-deS), 6-O-desulfated (6-O-deS), or 2-O-desulfated (2-O-deS) was immobilized on glycosaminoglycan-binding multiwell plates. The wells were then incubated with FLAG-tagged TIMP-3 (0.08–10 nM) (D) or FLAG-tagged ADAMTS-5 (0.16–20 nM) (E). Bound protein was detected using M2 anti-FLAG primary antibody and a horseradish peroxidase-coupled secondary antibody.
(F) TIMP-3 (0.5 nM) and ADAMTS-5 (0.5 nM) were incubated (1 hr, 37°C) with various concentrations (10–2000 nM disaccharide) of dp36 (12.5 kDa) heparin, either normally sulfated (untreated), N-desulfated, 6-O-desulfated, or 2-O-desulfated The residual activity against the fluorescent peptide substrate was determined.
(G and H) Heparin of dp36 (untreated) or glycol-split dp36 heparin (60 μM disaccharide) were immobilized on glycosaminoglycan-binding multiwell plates, and the wells were then incubated with FLAG-tagged TIMP-3 (0.08–10 nM) (G) or FLAG-tagged ADAMTS-5 (0.16–20 nM) (H). Bound protein was detected using M2 anti-FLAG primary antibody and a horseradish peroxidase-coupled secondary antibody.
(I) TIMP-3 (0.5 nM) and ADAMTS-5 (0.5 nM) were incubated (1 hr, 37°C) with various concentrations (10–2000 nM disaccharide) of dp36 heparin (untreated) or glycol-split dp36 heparin. The residual activity against the fluorescent peptide substrate was determined.
Heparin of dp36 and dp14 effectively increased TIMP-3-ADAMTS-5 affinity, whereas heparins of dp9 and dp5 were ineffective at disaccharide concentrations of 1 μM (Figure 5C). Although dp9 heparin bound strongly to both proteins, it was unable to increase their affinities. This is most likely due to the lack of sufficient length to bridge the binding sites on the two proteins to form a high-affinity trimolecular complex.
The ability of heparin to block interaction between TIMP-3 and LRP-1 also depended on length. Heparins of dp36, dp14, and dp9 caused the accumulation of endogenous TIMP-3 in HTB94 cells, whereas dp5 heparin was ineffective (data not shown).
Heparin Sulfation Is Required for Affinity Increase and Endocytosis
Using heparin of dp36, we examined the influence of sGAG sulfation. TIMP-3 bound to heparin with an estimated KD of 2 nM. 2-O-desulfation and N-desulfation increased the KD to 4 and 5 nM, respectively. 6-O-desulfation decreased affinity the most, with a KD too high to be determined (Figure 5D). ADAMTS-5 bound to heparin with an estimated KD of 9 nM. 2-O-desulfation had little effect on binding (KD of 12 nM), whereas N-desulfation and 6-O-desulfation decreased affinity the most, and the KD could not be determined (Figure 5E). Therefore, 6-O-desulfation had the greatest effect on binding to both proteins, and 2-O-desulfation had the least effect.
N-desulfation, 2-O-desulfation, and 6-O-desulfation all ablated the ability of heparin to increase the affinity between TIMP-3 and ADAMTS-5, with no improvement in affinity seen at up to 2 μM disaccharide. For 6-O-desulfated heparin, this correlated with a substantial reduction in binding to both proteins. 2-O-desulfated heparin had only a small reduction in binding to each protein (KD reduced from 2 to 4 nM for TIMP-3 and from 9 to 12 nM for ADAMTS-5) but greatly affected the ability to increase the affinity between the two proteins (Figure 5F). This indicates that disulfation is critical for this trimolecular interaction.
TIMP-3 accumulated in the medium of cells treated with 2-O-desulfated and N-desulfated heparin but not in those treated with 6-O-desulfated heparin (data not shown). Therefore, the ability to block TIMP-3 endocytosis by LRP-1 correlated with the ability to bind to the inhibitor.
Effect of Heparin Ring Integrity on Affinity Increase
Periodate-treated, glycol-split dp36 heparin bound to TIMP-3 and ADAMTS-5 as well as untreated heparin (Figures 5G and 5H) and was also effective at increasing ADAMTS-5-TIMP-3 affinity (Figure 5I) and blocking TIMP-3 endocytosis (data not shown).
TIMP-3 Colocalizes with Perlecan
We stained for TIMP-3 in cartilage, a tissue with an ECM rich in multiple sGAGs. TIMP-3 colocalized with the HS-bearing proteoglycan perlecan in the pericellular matrix of femoral head cartilage (Figure 6A). This staining was absent in Timp3−/− mice, confirming the specificity of the immunolocalization. Perlecan colocalization with TIMP-3 was also observed in the pericellular matrix of knee cartilage from adult mice (Figure 6B).
Figure 6.

TIMP-3 Colocalizes with Perlecan in the Pericellular Matrix of Cartilage
(A) Hip cartilage from Timp3+/+ and Timp3−/− mice were cryosectioned (8 μm). Sections were air-dried, fixed in ice-cold acetone, and blocked with 5% (v/v) goat serum and 3% (w/v) bovine serum albumin in PBS. TIMP-3 was detected using rabbit anti-TIMP-3 and anti-rabbit Alexa Fluor 468. Perlecan was detected using rat anti-perlecan and anti-rat Alexa Fluor 568. Nuclei were stained with DAPI. Scale bar, 23 μm.
(B) Knee cartilage from wild-type mice was sectioned and stained as in (A).
Cartilage Contains CSE
Perlecan is well established to be an HS-containing proteoglycan. We investigated whether CS containing E-type disaccharide units is also present in cartilage by analyzing the disaccharide composition of cartilage CS. Both human and porcine adult cartilage contained the N-acetylgalactosamine 4,6-O-disulfate (ΔDi-diSE) unit characteristic of CSE (Table 1). As expected, given the abundance of aggrecan in cartilage, the majority of CS was either 4-sulfated (27% in human cartilage, 56% in porcine cartilage) or 6-sulfated (63% in human cartilage, 33% in porcine cartilage). The E-type disaccharide unit made up 3% of the total CS in human cartilage and 1% in porcine cartilage.
Table 1.
Chondroitin Sulfate Disaccharide Composition of Human and Porcine Cartilage
| ΔDiS | Human Cartilage |
Porcine Cartilage |
||
|---|---|---|---|---|
| nmol/mg | % | nmol/mg | % | |
| ΔDi-0S | 0.006 | 1 | 0.029 | 4 |
| ΔDi-4S | 0.188 | 27 | 0.424 | 56 |
| ΔDi-6S | 0.436 | 63 | 0.246 | 33 |
| ΔDi-diSE | 0.019 | 3 | 0.009 | 1 |
| ΔDi-diSD | 0.047 | 7 | 0.043 | 6 |
Sulfated glycosaminoglycans were isolated from human or porcine articular cartilage, fractionated by ion exchange chromatography, and digested with chondroitinase ABC. The resulting unsaturated disaccharides were analyzed by HLPC to determine the relative abundance of the various sulfated species.
Discussion
TIMP-3 has long been known to bind to the ECM, with this localization thought to position it optimally for inhibition of ECM-degrading metalloproteinases. In this study, we show that the matrix not only serves to maintain a reservoir of TIMP-3 but that it directly modulates TIMP-3 inhibitory activity and blocks its endocytic uptake and cellular degradation.
TIMP-3 is thought to bind to sGAG in the ECM (Yu et al., 2000), but the specific TIMP-3-binding glycosaminoglycans and the proteoglycan(s) bearing these moieties have remained unidentified, largely because of the difficulties associated with expression, purification, and biochemical analysis of TIMP-3. Following our elucidation that the inhibitor is cleared rapidly from the medium of cultured cells by LRP-1-mediated endocytosis (Troeberg et al., 2008, Scilabra et al., 2013), we used a strategy of blocking endocytosis to purify sufficient recombinant TIMP-3 for such analyses and showed that HS and CS chains containing E-type disaccharide units are likely to be the physiological TIMP-3-binding sGAGs in the ECM. In cartilage, we found that TIMP-3 colocalizes with perlecan in the pericellular ECM, indicating that, in this tissue, perlecan is likely to be the proteoglycan responsible for TIMP-3 retention. Perlecan is primarily thought of as an HS proteoglycan (Hassell et al., 1980), but it can also bear CS (SundarRaj et al., 1995), dermatan sulfate (DS) (Couchman et al., 1996), or keratan sulfate (Knox et al., 2005). Of particular interest, perlecan has been reported to carry CSE in bovine cartilage (Kvist et al., 2006). In this study, we found that the E-type CS disaccharide unit is also present in adult human and porcine cartilage. Perlecan may also mediate TIMP-3 binding to the ECM in other locations, such as in the basement membrane, and other HS- and CSE-bearing proteoglycans may bind TIMP-3 in other tissues. In cartilage, perlecan is exclusively localized in the pericellular matrix (Iozzo et al., 1994), where it binds several growth factors and regulatory molecules. For example, FGF-2 bound to the HS chains of perlecan transduces mechanical stimuli to chondrocytes (Vincent et al., 2007). Perlecan expression increases in osteoarthritis (OA) cartilage (Tesche and Miosge, 2004), but its sulfation status in disease is unknown.
TIMP-3 levels are primarily regulated posttranslationally, and the inhibitor is cleared from the extracellular environment by the endocytic receptor LRP-1 (Scilabra et al., 2013). Heparin blocks TIMP-3 binding to LRP-1 and inhibits cellular endocytosis, suggesting that the LRP-1 binding site overlaps with the ECM-binding site, identified by Lee et al. (2007) as an extended patch of basic residues. Here we show that HS and CSE similarly block TIMP-3 binding to LRP-1 and inhibit its endocytosis. Extracellular trafficking of TIMP-3 is therefore regulated by the balance between matrix binding and endocytosis, and factors altering either matrix sulfation or the endocytic capacity of the cell will shift this balance. A number of other LRP-1 ligands also bind to sGAGs. For example, midkine, connective tissue growth factor, MMP-13, MMP-2, MMP-9, ADAMTS-4, and ADAMTS-5 are all LRP-1 ligands (Lillis et al., 2008, Yamamoto et al., 2013, Yamamoto et al., 2014) that have been shown to bind to heparin in vitro. This suggests that the model outlined here may not be unique to TIMP-3 and that extracellular levels of many bioactive molecules that bind to the ECM may be regulated by the interplay between matrix binding and LRP-1-mediated endocytosis. This positions ECM sulfation as a potential key regulator of the ECM proteome.
In addition to blocking LRP-1-mediated endocytosis, HS and CSE increased TIMP-3 affinity for ADAMTS-5. We estimate that affinity was increased by 1000-fold, indicating that the sGAGs are likely to have induced a conformational change in one or both of the proteins. PPS similarly increases TIMP-3 affinity for ADAMTS-4 (Troeberg et al., 2012), so HS and CSE are likely to increase TIMP-3 affinity for other target metalloproteinases that bind to sGAG. Such enzymes are also likely to interact with the ECM, indicating that this mechanism selectively augments TIMP-3 inhibition of enzymes it is likely to encounter in the ECM environment. Target metalloproteinases that do not bind sGAG, including many MMPs, would escape this increase in affinity.
HS and CSE are the two most highly sulfated among the ECM sGAGs we tested, and sulfation is likely to be critical for their binding ability. Indeed, only disulfated CSE bound to TIMP-3, whereas monosulfated C-4-S and C-6-S did not. Although 2-O-desulfation did not greatly affect heparin binding to TIMP-3 or ADAMTS-5, it abolished the ability of heparin to increase the affinity between the two proteins. This indicates that the effects of the ECM on TIMP-3 may be altered under conditions that change sulfation. In cartilage, changes in sulfation have primarily focused on the most abundant proteoglycan, aggrecan. No data are available on changes in HS or CSE sulfation in cartilage with age or disease, but several potential mechanisms for eliciting such changes have been proposed. For example, expression of several HS-synthesizing enzymes is altered during inflammation (Krenn et al., 2008), and cartilage expression of the HS sulfatases Sulf-1 and Sulf-2 is increased with aging and in OA (Otsuki et al., 2008), with Sulf-null mice developing OA more rapidly (Otsuki et al., 2010). Studies on HS and CSE sulfation in cartilage are clearly warranted, given their important role in regulating the amount and activity of TIMP-3 as well as other regulatory proteins, such as FGF-2, CTGF, midkine, ADAMTS-4, and ADAMTS-5. Interestingly, TIMP-3 staining in the ECM of renal and pulmonary blood vessels increases with age (Macgregor et al., 2009), with the authors postulating that the TIMP-3-binding capacity of the ECM may be increased by age-related changes in glycosylation.
CSE, containing the disulfated N-acetylgalactosamine 4,6-O-disulfate residue, was initially isolated from squid cartilage (Kawai et al., 1966) but has since been isolated from numerous mammalian sources. Its disulfated status confers CSE with a binding activity more similar to heparin than to monosulfated C-4-S or C-6-S. CSE has been shown to interact with a number of growth factors, including midkine (Deepa et al., 2002), pleiotrophin (Deepa et al., 2002), several fibroblast growth factors (Asada et al., 2009, Deepa et al., 2002), heparin-binding epidermal growth factor (Deepa et al., 2002), brain-derived neurotrophic factor, and bone morphogenetic protein 4 (Miyazaki et al., 2008). CSE is particularly abundant in the brain (Ueoka et al., 2000), where it is thought to play a role in brain development (Purushothaman et al., 2007), neurite outgrowth (Sotogaku et al., 2007), and neuronal plasticity (Dick et al., 2013). It is intriguing to consider whether E-type CS disaccharide units may modulate TIMP-3 levels and activity in the brain. Our study implies that, rather than TIMP-3 exerting a protective effect on an inert extracellular matrix, the two have a symbiotic relationship, with changes in one bringing about changes in the other. This concept is likely to be of importance not only in cartilage but also in other physiological and pathological settings, such as wound healing, atherosclerosis, fibrosis, and cancer, with TIMP-3 and the sulfation pattern of glycosaminoglycans being important modulators of ECM turnover.
Significance
Elucidation of the molecular mechanisms by which sulfated glycans affect protein activity is likely to be beneficial in understanding and modulating the behavior of many ECM-binding growth factors and biologically active proteins in a range of pathological conditions. In this study, we demonstrated that catabolism of the ECM is modulated by its own sulfation because this alters the extracellular levels and inhibitory activity of TIMP-3, a pivotal inhibitor of matrix-degrading metalloproteinase. HS and CSE selectively increase TIMP-3 levels in the matrix by inhibiting its binding to the endocytic receptor LRP-1 and subsequent degradation. These sulfated glycans also increase TIMP-3 affinity for a subset of ECM-degrading target metalloproteinases by reducing dissociation rate constants of the TIMP-3-metalloproteinase complexes. Our study suggests that synthetic glycan mimetics that similarly increase the TIMP-3 amount and activity in the extracellular environment would be potential lead compounds for the development of therapies to treat osteoarthritis and other diseases associated with increased ECM-degrading metalloproteinase activities.
Experimental Procedures
Materials
C-terminally FLAG-tagged human TIMP-3 (Troeberg et al., 2009) and ADAMTS-5 lacking the C-terminal thrombospondin domain (Gendron et al., 2007) were prepared as before.
All glycan masses reported are average values. PPS was from Bene-PharmaChemie. Porcine mucosal HS from Iduron had an average mass of 22 kDa and contained highly sulfated 9 kDa and less sulfated 35 kDa species. C-4-S, C-6-S, porcine mucosal 12.5 kDa heparin, 5 kDa low molecular mass mucosal heparin, and 3 kDa heparin were purchased from Sigma-Aldrich. Heparin pentasaccharide (fondaparinux sodium) was from GlaxoSmithKline.
Desulfated heparins were prepared from 12.5 kDa porcine mucosal heparin. N-desulfated heparin was prepared as described by Inoue and Nagasawa (1976). 2-O-desulfated heparin was prepared by dissolving heparin in 0.2 M NaOH and then lyophilizing (Jaseja et al., 1989). 6-O-desulfated heparin was prepared by hydrolysis with N-methyl-N-(trimethylsilyl)trifluoroacetamide (Kariya et al., 2000). Glycol-split heparin was prepared as described previously (Casu et al., 2002).
CSE was purified from squid cartilage on diethylaminoethyl (DEAE)-Sephadex (Habuchi et al., 1977). The 1.5 M NaCl eluate from this resin was further fractionated by gel filtration on Sepharose CL-4B equilibrated in 0.2 M NaCl. The eluate was pooled into three fractions, with average estimated molecular masses of 800 kDa (fraction 1), 200 kDa (fraction 2), and 50 kDa (fraction 3). Fractions were lyophilized, and the glycosaminoglycan content was quantified using the Elson-Morgan color reaction. Chondroitin sulfate E from bonito was a gift from Seikagaku. The disaccharide composition of the CSE preparations is shown in Table S1.
M2 anti-FLAG antibody, Tween 20, DNase I, and proteinase K were from Sigma-Aldrich. Rabbit polyclonal anti-TIMP-3 and rat anti-perlecan were from Millipore. Anti-mouse horseradish peroxidase was from Dako. Anti-mouse and anti-rabbit alkaline phosphatases were from Promega. 3,3′,5,5′-Tetramethylbenzidine substrate was from KPL.
Glycosaminoglycan Binding Assay
Glycosaminoglycans (10 μg/ml in PBS) were applied overnight at 25°C onto heparin binding plates (BD Life Sciences). Wells were washed in TIMP buffer (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 10 mM CaCl2, and 0.05% Brij-35) containing 0.1% (v/v) Tween 20 between each subsequent incubation. Wells were blocked with 0.2% (m/v) gelatin in TIMP buffer and then incubated with recombinant C-terminally FLAG-tagged TIMP-3 or ADAMTS-5 (0.08–100 nM in blocking solution, 3 hr, 37°C). Bound proteins were detected using M2 anti-FLAG antibody (3 hr, 37°C), followed by an anti-mouse antibody coupled to horseradish peroxidase (1 hr, 37°C). Hydrolysis of 3,3′,5,5′-tetramethylbenzidine substrate was measured at 450 nm using a FLUOstar Omega (BMG Labtech).
Analysis of TIMP-3 Endocytosis
HTB94 chondrosarcoma cells (6 × 105 cells/well in Dulbecco's modified Eagle's medium [DMEM] with 10% fetal calf serum [FCS]) were washed twice in serum-free DMEM and warmed for 1 hr at 37°C in 1 ml of DMEM with 0.1% FCS. For RAP-treated wells, RAP (1 μM) was added to cells during this incubation period. Recombinant TIMP-3-FLAG (0.5 nM) was incubated (1 hr, 37°C) either alone or with 200 μg/ml of heparin, HS, C-4-S, or bonito CSE and then added to cells. Conditioned media were precipitated by addition of 5% (v/v) trichloroacetic acid (TCA) (4°C, 18 h). After centrifugation (13,000 rpm, 10 min, 4°C), pellets were suspended in reducing SDS sample buffer, electrophoresed on a 10% polyacrylamide gel, and immunoblotted onto polyvinylidene fluoride. After blocking in 5% (m/v) BSA in Tris-buffered saline, TIMP-3-FLAG was detected using M2 anti-FLAG antibody and an alkaline phosphatase-linked secondary antibody.
To measure the accumulation of endogenous TIMP-3 in the presence of sGAG, HTB94 chondrosarcoma cells (6 × 105 cells/well in DMEM with 10% FCS) were washed twice in serum-free DMEM and incubated for 30 hr in 1 ml of serum-free DMEM containing RAP (0.5 and 0.75 μM), heparin, HS, C-4-S, or bonito CSE (100 and 200 μg/ml). Proteins were precipitated from media by addition of 5% (v/v) TCA, electrophoresed, and immunoblotted as described above. TIMP-3 was detected using polyclonal rabbit anti-TIMP-3 and an alkaline phosphatase-linked secondary antibody.
TIMP-3 Binding to LRP-1
LRP-1 ectodomain (5 nM, BioMac) was applied (18 hr, 4°C) onto Costar 96-well immunosorbent assay plates in TIMP buffer without Brij-35. Wells were washed in TIMP buffer containing 0.1% (v/v) Tween 20 between each subsequent incubation. Wells were blocked (90 min, 37°C) in 3% (m/v) bovine serum albumin in TIMP buffer. TIMP-3 (1.5–80 nM) was preincubated (90 min, 37°C) with 200 μg/ml heparin, HS, bonito CSE, or squid CSE and then added to wells (37°C, 3 hr). Bound TIMP-3 was detected using M2 anti-FLAG antibody (2 hr, 37°C) and anti-mouse antibody coupled to horseradish peroxidase (1 hr, 37°C). Hydrolysis of 3,3′,5,5′-tetramethylbenzidine substrate was measured at 450 nm using a FLUOstar Omega (BMG Labtech).
Analysis of TIMP-3 Affinity for ADAMTS-5
The affinity between TIMP-3 and ADAMTS-5 was measured using a steady-state enzyme kinetic analysis (Bieth, 1995, Troeberg et al., 2012). TIMP-3 (0.5–2 nM) and ADAMTS-5 (0.5 nM) were incubated in TIMP buffer (1 hr, 37°C). Residual ADAMTS-5 activity was measured (18 hr, 37°C) using the fluorescent peptide substrate ortho-aminobenzoyl-Thr-Glu-Ser-Glu∼Ser-Arg-Gly-Ala-Ile-Tyr-(N-3-[2,4-dinitrophenyl]-L-2,3-diaminopropionyl)-Lys-Lys-NH2 (20 μM, Bachem). Endpoint fluorescence was measured using a Gemini microplate spectrofluorometer (Molecular Devices), and Ki was determined from steady-state velocities by fitting the data to the tight-binding equation using Prism software (GraphPad).
Biolayer Interferometry
TIMP-3 (200 nM) was biotinylated by incubation with EZ-Link succinimidyl 6-(biotinamido)hexanoate (10 μM, 30 min, 25°C, Thermo Fisher Scientific), and excess biotin was removed using a PD-10 desalting column (GE Healthcare). Biotinylated TIMP-3 was immobilized on streptavidin-coated biosensors (20 min, 25°C) using an Octet RED384 biolayer interferometer (Pall ForteBio). To determine the affinity between TIMP-3 and ADAMTS-5, TIMP-3-coated biosensors were incubated with ADAMTS-5 (50–500 nM in TIMP buffer), and association was observed for 10 min at 25°C. Tips were then incubated in TIMP buffer for a further 20 min at 25°C to observe dissociation of the complex. No binding of ADAMTS-5 to uncoated biosensors was observed. To determine the effect of sGAG on the affinity between TIMP-3 and ADAMTS-5, TIMP-3-coated biosensors were incubated with ADAMTS-5 (200 nM) and either PPS (100 nM), HS (100 μM), bonito CSE (10 μM), or squid CSE (10 μM) in TIMP buffer. Association was observed for 10 min at 25°C, after which tips were incubated in TIMP buffer for 20 min at 25°C to observe dissociation of the complex. Kinetic constants were calculated using a 1:1 binding model in ForteBio OctetRED evaluation data analysis software. KD was calculated from equilibrium responses, and koff was calculated by determining the first-order rate constant. Observed association rate constant (kobs) values were calculated by determining the second-order rate constant using GraphPad Prism, and kon was calculated from linear regression of kobs on ADAMTS-5 concentration.
Immunofluorescence
Timp3−/− mice were provided by Professor Rama Khokha (Ontario Cancer Institute). Cartilage was avulsed from the femoral heads of 6-week-old wild-type and Timp3−/− mice (Leco et al., 2001) using forceps. The tissue was frozen in optimal cutting temperature (OCT) medium embedding matrix (CellPath) and cryosectioned (8 μm). Sections were air-dried (1 hr, ambient temperature) and fixed in ice-cold acetone (10 min, −20°C). Sections were blocked with 5% (v/v) goat serum and 3% (w/v) bovine serum albumin in PBS (1 hr, room temperature). TIMP-3 was detected using polyclonal rabbit anti-TIMP-3 (1 μg/ml) and anti-rabbit Alexa Fluor 488 (Molecular Probes). Perlecan was detected using rat anti-perlecan (1 μg/ml) and anti-rat Alexa Fluor 568 (Molecular Probes). Nuclei were stained with DAPI. Sections were viewed with a charge-coupled device camera-equipped microscope (Nikon TE-2000) with a ×40 objective lens.
Disaccharide Composition Analysis of Cartilage
Human articular cartilage was obtained from knees of patients undergoing amputation because of soft tissue sarcoma or osteosarcoma. Patients provided informed consent following approval by the Riverside Ethics Committee. For the analysis shown, cartilage was pooled from three patients (two males, one female, 40–51 years of age). Porcine articular cartilage was dissected from metacarpophalangeal joints of commercially slaughtered pigs.
Lyophilized cartilage (30 mg dry weight) was suspended in 0.4 N KOH and stirred for 18 hr at room temperature. After neutralization with acetic acid, samples were digested sequentially with DNase I (10 hr, 37°C) and proteinase K (40 hr, 45°C). The enzymes were heat-inactivated (100°C, 5 min), the samples were centrifuged (13,000 rpm, 10 min, 4°C), and the supernatant was precipitated by addition of 3 volumes of cold ethanol containing 1.3% (m/v) potassium acetate (on ice, 30 min). Pellets were collected by centrifugation (13,000 rpm, 10 min, 4°C), dissolved in deionized water, and ethanol-precipitated a further two times before being dissolved in deionized water. The glycosaminoglycans were then applied to a DEAE-Sephacel resin (GE Healthcare) equilibrated in 20 mM Tris-HCl (pH 7.2). The column was washed in equilibration buffer containing 0.2 M NaCl and glycosaminoglycans eluted in equilibration buffer containing 2 M NaCl. The eluate was desalted by centrifugation on a Nanosep centrifugation filter unit (3 kDa molecular weight cutoff, Pall Life Sciences).
For CS analysis, chondroitinase ABC (5 milliunits, Seikagaku) was added to the filter cup in chondroitinase buffer (50 mM Tris-HCl [pH 7.2] and 1 mM CaCl2). After incubation for 1 hr at 37°C, unsaturated disaccharides were eluted by centrifugation (13,000 rpm, 20 min, 4°C) and analyzed by high-performance liquid chromatography (HPLC) on a Sensyu Pak Docosil column, with elution monitored by a fluorescence detector (model RF-10AxL, Shimadzu). HPLC was carried out as described by Toyoda et al. (2000), with a slightly modified elution condition. Briefly, the disaccharides were labeled after elution with 2-cyanoacetoamide (Wako Pure Chemicals Industries) (Anower-E-Khuda et al., 2013).
Author Contributions
L.T. designed and performed the experiments, analyzed the data, and wrote the manuscript. C.F. generated the heparin variants. C.L. performed the enzyme kinetic experiments. O.F. performed biolayer interferometry. M.F.A.E.K., H.H., and O.H. prepared and analyzed CSE. K.K. and H.N. analyzed the data and wrote the manuscript.
Acknowledgments
We thank Rama Khokha for the Timp3−/− mice. L.T. is an Arthritis Research UK Career Development Fellow (Grant 19466). This work was also supported by grants from Arthritis Research UK (Arthritis Research UK Centre for Osteoarthritis Pathogenesis, Grant 20205) and from the National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS) (Grant AR40994). O.F. acknowledges support from the Structural Genomics Consortium, a registered charity (number 1097737) that receives funds from the Canadian Institute for Health Research, the Canada Foundation for Innovation, Genome Canada, GlaxoSmithKline, Pfizer, Eli Lilly, Takeda, AbbVie, Bayer, the Novartis Research Foundation, the Ontario Ministry of Research and Innovation, and the Wellcome Trust (092809/Z/10/Z).
Published: August 28, 2014
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/3.0/).
Supplemental Information includes one table and can be found with this article online at http://dx.doi.org/10.1016/j.chembiol.2014.07.014.
Supplemental Information
References
- Anower-E-Khuda M.F., Matsumoto K., Habuchi H., Morita H., Yokochi T., Shimizu K., Kimata K. Glycosaminoglycans in the blood of hereditary multiple exostoses patients: Half reduction of heparan sulfate to chondroitin sulfate ratio and the possible diagnostic application. Glycobiology. 2013;23:865–876. doi: 10.1093/glycob/cwt024. [DOI] [PubMed] [Google Scholar]
- Asada M., Shinomiya M., Suzuki M., Honda E., Sugimoto R., Ikekita M., Imamura T. Glycosaminoglycan affinity of the complete fibroblast growth factor family. Biochim. Biophys. Acta. 2009;1790:40–48. doi: 10.1016/j.bbagen.2008.09.001. [DOI] [PubMed] [Google Scholar]
- Bieth J.G. Theoretical and practical aspects of proteinase inhibition kinetics. Methods Enzymol. 1995;248:59–84. doi: 10.1016/0076-6879(95)48007-2. [DOI] [PubMed] [Google Scholar]
- Blenis J., Hawkes S.P. Transformation-sensitive protein associated with the cell substratum of chicken embryo fibroblasts. Proc. Natl. Acad. Sci. USA. 1983;80:770–774. doi: 10.1073/pnas.80.3.770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brew K., Nagase H. The tissue inhibitors of metalloproteinases (TIMPs): an ancient family with structural and functional diversity. Biochim. Biophys. Acta. 2010;1803:55–71. doi: 10.1016/j.bbamcr.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardellini M., Menghini R., Martelli E., Casagrande V., Marino A., Rizza S., Porzio O., Mauriello A., Solini A., Ippoliti A. TIMP3 is reduced in atherosclerotic plaques from subjects with type 2 diabetes and increased by SirT1. Diabetes. 2009;58:2396–2401. doi: 10.2337/db09-0280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casu B., Guerrini M., Naggi A., Perez M., Torri G., Ribatti D., Carminati P., Giannini G., Penco S., Pisano C. Short heparin sequences spaced by glycol-split uronate residues are antagonists of fibroblast growth factor 2 and angiogenesis inhibitors. Biochemistry. 2002;41:10519–10528. doi: 10.1021/bi020118n. [DOI] [PubMed] [Google Scholar]
- Couchman J.R., Kapoor R., Sthanam M., Wu R.R. Perlecan and basement membrane-chondroitin sulfate proteoglycan (bamacan) are two basement membrane chondroitin/dermatan sulfate proteoglycans in the Engelbreth-Holm-Swarm tumor matrix. J. Biol. Chem. 1996;271:9595–9602. doi: 10.1074/jbc.271.16.9595. [DOI] [PubMed] [Google Scholar]
- Cruz-Munoz W., Khokha R. The role of tissue inhibitors of metalloproteinases in tumorigenesis and metastasis. Crit. Rev. Clin. Lab. Sci. 2008;45:291–338. doi: 10.1080/10408360801973244. [DOI] [PubMed] [Google Scholar]
- Deepa S.S., Umehara Y., Higashiyama S., Itoh N., Sugahara K. Specific molecular interactions of oversulfated chondroitin sulfate E with various heparin-binding growth factors. Implications as a physiological binding partner in the brain and other tissues. J. Biol. Chem. 2002;277:43707–43716. doi: 10.1074/jbc.M207105200. [DOI] [PubMed] [Google Scholar]
- Dick G., Tan C.L., Alves J.N., Ehlert E.M.E., Miller G.M., Hsieh-Wilson L.C., Sugahara K., Oosterhof A., van Kuppevelt T.H., Verhaagen J. Semaphorin 3A binds to the perineuronal nets via chondroitin sulfate type E motifs in rodent brains. J. Biol. Chem. 2013;288:27384–27395. doi: 10.1074/jbc.M111.310029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fata J.E., Leco K.J., Voura E.B., Yu H.Y.E., Waterhouse P., Murphy G., Moorehead R.A., Khokha R. Accelerated apoptosis in the Timp-3-deficient mammary gland. J. Clin. Invest. 2001;108:831–841. doi: 10.1172/JCI13171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedak P.W., Smookler D.S., Kassiri Z., Ohno N., Leco K.J., Verma S., Mickle D.A., Watson K.L., Hojilla C.V., Cruz W. TIMP-3 deficiency leads to dilated cardiomyopathy. Circulation. 2004;110:2401–2409. doi: 10.1161/01.CIR.0000134959.83967.2D. [DOI] [PubMed] [Google Scholar]
- Gendron C., Kashiwagi M., Lim N.H., Enghild J.J., Thøgersen I.B., Hughes C., Caterson B., Nagase H. Proteolytic activities of human ADAMTS-5: comparative studies with human ADAMTS-4. J. Biol. Chem. 2007;282:18294–18306. doi: 10.1074/jbc.M701523200. [DOI] [PubMed] [Google Scholar]
- Gill S.E., Pape M.C., Leco K.J. Tissue inhibitor of metalloproteinases 3 regulates extracellular matrix—cell signaling during bronchiole branching morphogenesis. Dev. Biol. 2006;298:540–554. doi: 10.1016/j.ydbio.2006.07.004. [DOI] [PubMed] [Google Scholar]
- Habuchi O., Sugiura K., Kawai N. Glucose branches in chondroitin sulfates from squid cartilage. J. Biol. Chem. 1977;252:4570–4576. [PubMed] [Google Scholar]
- Hassell J.R., Robey P.G., Barrach H.J., Wilczek J., Rennard S.I., Martin G.R. Isolation of a heparan sulfate-containing proteoglycan from basement membrane. Proc. Natl. Acad. Sci. USA. 1980;77:4494–4498. doi: 10.1073/pnas.77.8.4494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue Y., Nagasawa K. Selective N-desulfation of heparin with dimethyl sulfoxide containing water or methanol. Carbohydr. Res. 1976;46:87–95. doi: 10.1016/s0008-6215(00)83533-8. [DOI] [PubMed] [Google Scholar]
- Iozzo R.V., Cohen I.R., Grässel S., Murdoch A.D. The biology of perlecan: the multifaceted heparan sulphate proteoglycan of basement membranes and pericellular matrices. Biochem. J. 1994;302:625–639. doi: 10.1042/bj3020625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaseja M., Rej R.N., Sauriol F., Perlin A.S. Novel regio-selective and stereoselective modifications of heparin in alkaline solution – nuclear magnetic resonance spectroscopic evidence. Can. J. Chem. 1989;67:1449–1456. [Google Scholar]
- Kariya Y., Kyogashima M., Suzuki K., Isomura T., Sakamoto T., Horie K., Ishihara M., Takano R., Kamei K., Hara S. Preparation of completely 6-O-desulfated heparin and its ability to enhance activity of basic fibroblast growth factor. J. Biol. Chem. 2000;275:25949–25958. doi: 10.1074/jbc.M004140200. [DOI] [PubMed] [Google Scholar]
- Kawai Y., Seno N., Anno K. Chondroitin polysulfate of squid cartilage. J. Biochem. 1966;60:317–321. doi: 10.1093/oxfordjournals.jbchem.a128438. [DOI] [PubMed] [Google Scholar]
- Knox S., Fosang A.J., Last K., Melrose J., Whitelock J. Perlecan from human epithelial cells is a hybrid heparan/chondroitin/keratan sulfate proteoglycan. FEBS Lett. 2005;579:5019–5023. doi: 10.1016/j.febslet.2005.07.090. [DOI] [PubMed] [Google Scholar]
- Krenn E.C., Wille I., Gesslbauer B., Poteser M., van Kuppevelt T.H., Kungl A.J. Glycanogenomics: a qPCR-approach to investigate biological glycan function. Biochem. Biophys. Res. Commun. 2008;375:297–302. doi: 10.1016/j.bbrc.2008.07.144. [DOI] [PubMed] [Google Scholar]
- Kvist A.J., Johnson A.E., Mörgelin M., Gustafsson E., Bengtsson E., Lindblom K., Aszódi A., Fässler R., Sasaki T., Timpl R., Aspberg A. Chondroitin sulfate perlecan enhances collagen fibril formation. Implications for perlecan chondrodysplasias. J. Biol. Chem. 2006;281:33127–33139. doi: 10.1074/jbc.M607892200. [DOI] [PubMed] [Google Scholar]
- Leco K.J., Waterhouse P., Sanchez O.H., Gowing K.L., Poole A.R., Wakeham A., Mak T.W., Khokha R. Spontaneous air space enlargement in the lungs of mice lacking tissue inhibitor of metalloproteinases-3 (TIMP-3) J. Clin. Invest. 2001;108:817–829. doi: 10.1172/JCI12067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M.H., Atkinson S., Murphy G. Identification of the extracellular matrix (ECM) binding motifs of tissue inhibitor of metalloproteinases (TIMP)-3 and effective transfer to TIMP-1. J. Biol. Chem. 2007;282:6887–6898. doi: 10.1074/jbc.M610490200. [DOI] [PubMed] [Google Scholar]
- Lillis A.P., Van Duyn L.B., Murphy-Ullrich J.E., Strickland D.K. LDL receptor-related protein 1: unique tissue-specific functions revealed by selective gene knockout studies. Physiol. Rev. 2008;88:887–918. doi: 10.1152/physrev.00033.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macgregor A.M., Eberhart C.G., Fraig M., Lu J., Halushka M.K. Tissue inhibitor of matrix metalloproteinase-3 levels in the extracellular matrix of lung, kidney, and eye increase with age. J. Histochem. Cytochem. 2009;57:207–213. doi: 10.1369/jhc.2008.952531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki T., Miyauchi S., Tawada A., Anada T., Matsuzaka S., Suzuki O. Oversulfated chondroitin sulfate-E binds to BMP-4 and enhances osteoblast differentiation. J. Cell. Physiol. 2008;217:769–777. doi: 10.1002/jcp.21557. [DOI] [PubMed] [Google Scholar]
- Morris K.J., Cs-Szabo G., Cole A.A. Characterization of TIMP-3 in human articular talar cartilage. Connect. Tissue Res. 2010;51:478–490. doi: 10.3109/03008201003686958. [DOI] [PubMed] [Google Scholar]
- Otsuki S., Taniguchi N., Grogan S.P., D’Lima D., Kinoshita M., Lotz M. Expression of novel extracellular sulfatases Sulf-1 and Sulf-2 in normal and osteoarthritic articular cartilage. Arthritis Res. Ther. 2008;10:R61. doi: 10.1186/ar2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otsuki S., Hanson S.R., Miyaki S., Grogan S.P., Kinoshita M., Asahara H., Wong C.-H., Lotz M.K. Extracellular sulfatases support cartilage homeostasis by regulating BMP and FGF signaling pathways. Proc. Natl. Acad. Sci. USA. 2010;107:10202–10207. doi: 10.1073/pnas.0913897107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purushothaman A., Fukuda J., Mizumoto S., ten Dam G.B., van Kuppevelt T.H., Kitagawa H., Mikami T., Sugahara K. Functions of chondroitin sulfate/dermatan sulfate chains in brain development. Critical roles of E and iE disaccharide units recognized by a single chain antibody GD3G7. J. Biol. Chem. 2007;282:19442–19452. doi: 10.1074/jbc.M700630200. [DOI] [PubMed] [Google Scholar]
- Sahebjam S., Khokha R., Mort J.S. Increased collagen and aggrecan degradation with age in the joints of Timp3(-/-) mice. Arthritis Rheum. 2007;56:905–909. doi: 10.1002/art.22427. [DOI] [PubMed] [Google Scholar]
- Scilabra S.D., Troeberg L., Yamamoto K., Emonard H., Thøgersen I., Enghild J.J., Strickland D.K., Nagase H. Differential regulation of extracellular tissue inhibitor of metalloproteinases-3 levels by cell membrane-bound and shed low density lipoprotein receptor-related protein 1. J. Biol. Chem. 2013;288:332–342. doi: 10.1074/jbc.M112.393322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotogaku N., Tully S.E., Gama C.I., Higashi H., Tanaka M., Hsieh-Wilson L.C., Nishi A. Activation of phospholipase C pathways by a synthetic chondroitin sulfate-E tetrasaccharide promotes neurite outgrowth of dopaminergic neurons. J. Neurochem. 2007;103:749–760. doi: 10.1111/j.1471-4159.2007.04849.x. [DOI] [PubMed] [Google Scholar]
- SundarRaj N., Fite D., Ledbetter S., Chakravarti S., Hassell J.R. Perlecan is a component of cartilage matrix and promotes chondrocyte attachment. J. Cell Sci. 1995;108:2663–2672. doi: 10.1242/jcs.108.7.2663. [DOI] [PubMed] [Google Scholar]
- Tesche F., Miosge N. Perlecan in late stages of osteoarthritis of the human knee joint. Osteoarthritis Cartilage. 2004;12:852–862. doi: 10.1016/j.joca.2004.07.004. [DOI] [PubMed] [Google Scholar]
- Tian H., Cimini M., Fedak P.W., Altamentova S., Fazel S., Huang M.L., Weisel R.D., Li R.K. TIMP-3 deficiency accelerates cardiac remodeling after myocardial infarction. J. Mol. Cell. Cardiol. 2007;43:733–743. doi: 10.1016/j.yjmcc.2007.09.003. [DOI] [PubMed] [Google Scholar]
- Toyoda H., Kinoshita-Toyoda A., Selleck S.B. Structural analysis of glycosaminoglycans in Drosophila and Caenorhabditis elegans and demonstration that tout-velu, a Drosophila gene related to EXT tumor suppressors, affects heparan sulfate in vivo. J. Biol. Chem. 2000;275:2269–2275. doi: 10.1074/jbc.275.4.2269. [DOI] [PubMed] [Google Scholar]
- Troeberg L., Tanaka M., Wait R., Shi Y.E., Brew K., Nagase H. E. coli expression of TIMP-4 and comparative kinetic studies with TIMP-1 and TIMP-2: insights into the interactions of TIMPs and matrix metalloproteinase 2 (gelatinase A) Biochemistry. 2002;41:15025–15035. doi: 10.1021/bi026454l. [DOI] [PubMed] [Google Scholar]
- Troeberg L., Fushimi K., Khokha R., Emonard H., Ghosh P., Nagase H. Calcium pentosan polysulfate is a multifaceted exosite inhibitor of aggrecanases. FASEB J. 2008;22:3515–3524. doi: 10.1096/fj.08-112680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troeberg L., Fushimi K., Scilabra S.D., Nakamura H., Dive V., Thøgersen I.B., Enghild J.J., Nagase H. The C-terminal domains of ADAMTS-4 and ADAMTS-5 promote association with N-TIMP-3. Matrix Biol. 2009;28:463–469. doi: 10.1016/j.matbio.2009.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troeberg L., Mulloy B., Ghosh P., Lee M.H., Murphy G., Nagase H. Pentosan polysulfate increases affinity between ADAMTS-5 and TIMP-3 through formation of an electrostatically driven trimolecular complex. Biochem. J. 2012;443:307–315. doi: 10.1042/BJ20112159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueoka C., Kaneda N., Okazaki I., Nadanaka S., Muramatsu T., Sugahara K. Neuronal cell adhesion, mediated by the heparin-binding neuroregulatory factor midkine, is specifically inhibited by chondroitin sulfate E. Structural ans functional implications of the over-sulfated chondroitin sulfate. J. Biol. Chem. 2000;275:37407–37413. doi: 10.1074/jbc.M002538200. [DOI] [PubMed] [Google Scholar]
- Vincent T.L., McLean C.J., Full L.E., Peston D., Saklatvala J. FGF-2 is bound to perlecan in the pericellular matrix of articular cartilage, where it acts as a chondrocyte mechanotransducer. Osteoarthritis Cartilage. 2007;15:752–763. doi: 10.1016/j.joca.2007.01.021. [DOI] [PubMed] [Google Scholar]
- Yamamoto K., Troeberg L., Scilabra S.D., Pelosi M., Murphy C.L., Strickland D.K., Nagase H. LRP-1-mediated endocytosis regulates extracellular activity of ADAMTS-5 in articular cartilage. FASEB J. 2013;27:511–521. doi: 10.1096/fj.12-216671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto K., Owen K., Parker A.E., Scilabra S.D., Dudhia J., Strickland D.K., Troeberg L., Nagase H. Low density lipoprotein receptor-related protein 1 (LRP1)-mediated endocytic clearance of a disintegrin and metalloproteinase with thrombospondin motifs-4 (ADAMTS-4): functional differences of non-catalytic domains of ADAMTS-4 and ADAMTS-5 in LRP1 binding. J. Biol. Chem. 2014;289:6462–6474. doi: 10.1074/jbc.M113.545376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W.H., Yu S., Meng Q., Brew K., Woessner J.F., Jr. TIMP-3 binds to sulfated glycosaminoglycans of the extracellular matrix. J. Biol. Chem. 2000;275:31226–31232. doi: 10.1074/jbc.M000907200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.