

Abstract
Objective
Genome-wide association studies have identified several genetic variants associated with coronary heart disease (CHD). The aim of this study was to evaluate the genetic risk discrimination and reclassification and apply the results for a two-stage population risk screening strategy for CHD.
Approach and Results
We genotyped 28 genetic variants in 24 124 participants in four Finnish population-based, prospective cohorts (recruitment years 1992-2002). We constructed a multi-locus genetic risk score and evaluated its association with incident cardiovascular disease events. During the median follow-up time of 12 years (IQR 8.75–15.25 years), we observed 1093 CHD, 1552 cardiovascular disease (CVD) and 731 acute coronary syndrome (ACS) events. Adding genetic information to conventional risk factors and family history improved risk discrimination of CHD (C-index 0.856 vs. 0.851, P=0.0002) and other end points (CVD: C-index 0.840 vs. 0.837, P=0.0004; ACS: C-index 0.859 vs. 0.855, P=0.001). In a standard population of 100 000 individuals, additional genetic screening of subjects at intermediate risk for CHD would reclassify 2144 (12%) subjects into high risk category. Statin allocation for these subjects is estimated to prevent 135 CHD cases over 14 years. Similar results were obtained by external validation, where the effects were estimated from a training dataset and applied for a test dataset.
Conclusions
Genetic risk score improves risk prediction of CHD and helps to identify individuals at high risk for the first CHD event. Genetic screening for individuals at intermediate cardiovascular risk could help to prevent future cases through better targeting of statins.
Keywords: Cardiovascular genomics, Genetic association, Genetic epidemiology, Risk factor, Risk prediction
INTRODUCTION
Coronary heart disease (CHD) is a complex disorder with the risk modified by both environmental and genetic factors. Currently, the established risk factors, such as high cholesterol and blood pressure, explain only a fraction of the variability in disease risk. This has motivated a search for new predictors, including genetic markers. Several genome-wide association studies have identified many novel genetic susceptibility loci for CHD 1-3. Although causal variants and biological function are still unknown for many of the loci, their potential for better identifying the high risk individuals has been studied. Genetic risk scores (GRSs) based on the subsets of the most strongly associated single-nucleotide polymorphisms (SNPs) in the identified loci have been associated with future CHD events in samples of European origin 4-7, but studies so far have shown little or no additional value for GRS’s ability to predict future cases of CHD over the traditional risk factors.
Recently, the Emerging Risk Factors Collaboration 8, 9 has evaluated lipid-related and inflammatory markers by their ability to improve risk classification in a two-stage population screening strategy. In this approach, a population of 100 000 individuals is first screened for the traditional cardiovascular risk factors. In a second stage, additional screening based on a novel risk marker is conducted for the subjects at the intermediate risk category (10-year risk 10–20%). The guidelines from NICE 10 and ATP-III 11 among others recommend that statin treatment should be allocated for the individuals with 10-year absolute risk of cardiovascular disease >20%. Thus, identification of individuals at the intermediate risk category, who would benefit from long term statin medication, could have both clinical and population health benefits.
In this study, we genotyped 28 previously identified genetic risk variants for CHD 2, 3, 12, 13 in four Finnish prospective cohorts (n=24 124) with up to 19 years of follow-up. We set out to evaluate the genetic risk discrimination of CHD, acute coronary syndrome (ACS) and combined CHD and stroke events (CVD), and estimate the improved risk classification of CHD in a two-stage population screening strategy.
RESULTS
Background Characteristics of Study Cohorts
Characteristics of study cohorts are shown in Table 1. In total, 24,124 subjects from FINRISK 1992, FINRISK 1997, FINRISK 2002 and Health 2000 cohorts were included in the analysis. We observed 1093 CHD (5%), 1552 CVD (6%) and 731 ACS (3%) cases during the median follow-up time of 12 years (IQR 8.75–15.25 years).
Table 1. Characteristics of study cohorts.
| FINRISK 1992 (N=5104) | FINRISK 1997 (N=6567) | FINRISK 2002 (N=7330) | Health 2000 (N=5123) | |
|---|---|---|---|---|
| Follow-up – yrs | 19 | 14 | 9 | 8 |
| Sex – no. (%) | ||||
| Men | 2287 (44.8) | 3005 (45.8) | 3299 (45.0) | 2371 (46.3) |
| Women | 2817 (55.2) | 3562 (54.2) | 4031 (55.0) | 2752 (53.7) |
| Age – yrs * | 43.9±11.3 | 46.8±12.9 | 47.5±13.0 | 50.0±11.7 |
| Cholesterol – mmol/l * | ||||
| Total | 5.6±1.1 | 5.5±1.1 | 5.6±1.1 | 5.9±1.1 |
| LDL | 3.5±1.0 | 3.5±0.9 | 3.4±0.9 | 3.8±1.2 |
| HDL | 1.4±0.3 | 1.4±0.4 | 1.5±0.4 | 1.3±0.4 |
| Blood pressure – mm Hg * | ||||
| Systolic | 135.1±19.3 | 134.9±19.5 | 134.8±19.9 | 132.7±20.1 |
| Diastolic | 81.1±11.9 | 82.1±11.2 | 79.0±11.4 | 82.2±11.1 |
| Body-mass index – weight(kg) / (height(m))2 * | 26.0±4.4 | 26.5±4.6 | 26.8±4.7 | 26.8±4.7 |
| Current smoker – no. (%) | 1425 (27.9) | 1606 (24.5) | 1928 (26.3) | 1611 (31.4) |
| Antihypertensive medication – no. (%) | 443 (8.7) | 761 (11.6) | 979 (13.4) | 953 (18.6) |
| Diabetes mellitus – no. (%) | 172 (3.4) | 323 (4.9) | 358 (4.9) | 291 (5.7) |
| Family history of cardiovascular disease – no. (%) | 1376 (27.0) | 2350 (37.8) | 2426 (33.1) | NA |
| Incident cases – no. (%) | ||||
| Cardiovascular disease | 501 (9.8) | 499 (7.6) | 291 (4.0) | 261 (5.1) |
| Coronary heart disease | 343 (6.7) | 344 (5.2) | 209 (2.9) | 197 (3.8) |
| Acute coronary syndrome | 235 (4.6) | 229 (3.5) | 148 (2.0) | 119 (2.3) |
mean±SD.
Abbreviations: N, number of individuals; LDL, low-density lipoprotein; HDL, high-density lipoprotein; NA, information not available
Association Results
When tested individually, five loci were associated with all cardiovascular endpoints: rs6725887 (reported locus: WDR12), rs12526453 (PHACTR1), rs4977574 (CDKN2A/B, ANRIL), rs1746048 (CXCL12) and rs3825807 (ADAMTS7). In total, 13 loci were significantly associated with at least one of the end points. (Supplementary Table II).
The genetic risk score was associated with all cardiovascular endpoints (Table 2). Subjects in the highest 10% of 28-SNP GRS had 2.07-fold (95% CI 1.68–2.56, P=9.8×10−14) increased risk for coronary heart disease, when compared with the subjects in the middle 20%. When GRS was constructed by using only 13 SNPs that have been identified in the first phase of genome-wide studies 1, the corresponding risk for 13-SNP GRS was 1.55 (95% CI 1.26–1.91, P= 4.7×10−11).
Table 2. Risk for cardiovascular endpoints by genetic risk score.
| Top versus middle 20% | |||||||
|---|---|---|---|---|---|---|---|
| Trait | HR (95 % CI)* | P-value | Top 20% of GRS (95 % CI) | Top 10% of GRS (95 % CI) | Top 5% of GRS (95 % CI) | N events | N |
| CHD | 1.27 ( 1.20 , 1.35 ) | 1.2×10−14 | 1.71 ( 1.42 , 2.06 ) | 2.07 ( 1.68 , 2.56 ) | 2.12 ( 1.62 , 2.77 ) | 1093 | 24124 |
| ACS | 1.27 ( 1.18 , 1.37 ) | 3.1×10−10 | 1.57 ( 1.25 , 1.97 ) | 1.84 ( 1.42 , 2.40 ) | 2.00 ( 1.43 , 2.79 ) | 731 | 24124 |
| CVD | 1.18 ( 1.12 , 1.24 ) | 3.2×10−10 | 1.59 ( 1.36 , 1.86 ) | 1.87 ( 1.56 , 2.24 ) | 1.72 ( 1.35 , 2.18 ) | 1552 | 24124 |
per SD of GRS. Cox regression models were adjusted for sex, total cholesterol, high-density lipoprotein (HDL) cholesterol, body-mass index, systolic blood pressure, antihypertensive treatment, smoking and type 2 diabetes; age was used as the timescale.
Abbreviations: CHD, coronary heart disease; ACS, acute coronary syndrome; CVD, cardiovascular disease; GRS, genetic risk score; HR, Hazard ratio; CI, confidence interval; N, number of individuals
When dividing the risk score into deciles, the highest GRS group was clearly distinguishable from the other groups, especially in CHD and CVD events (Figure 1, Supplementary Figure I). The deviation from the linear risk function was observed for the highest decile in CHD (additional HR over linear risk = 1.38, 95% CI 1.12–1.72, P=0.003) and CVD (HR=1.39, 95% CI 1.15–1.67, P=0.0006), but not for ACS (HR=1.16, 95% CI 0.89–1.51, P=0.27). The non-linearity does not seem to be driven by the weights in the GRS as similar non-linear risk function was observed also for unweighted GRS (Supplementary Figure II).
Figure 1.
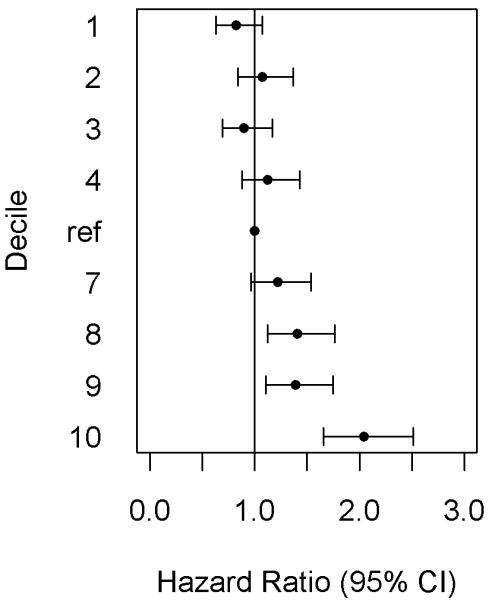
Genetic risk score deciles and risk for coronary heart disease.
The middle deciles (40%–60%) were used as a reference group.
In FINRISK studies, family history of cardiovascular disease was an independent risk factor for all events. Adjusting for the GRS slightly diminished the effects of family history: The estimated risk decreased from 1.46 (95% CI 1.27–1.67) to 1.43 (95% CI 1.25–1.64) for CHD, from 1.37 (95% CI 1.22–1.53) to 1.35 (95% CI 1.21–1.51) for CVD and from 1.45 (95% CI 1.24–1.71) to 1.43 (95% CI 1.21–1.68) for ACS, for the models with and without the GRS, respectively. In comparison, the GRS effects (per SD of GRS) changed from 1.29 (95% CI 1.20–1.38) to 1.28 (95% CI 1.19–1.37) for CHD, from 1.19 (95% CI 1.12–1.26) to 1.18 (95% CI 1.12–1.25) for CVD and from 1.30 (95% CI 1.19–1.40) to 1.29 (95% CI 1.19–1.40) for ACS, when family history was included into the model.
Risk Discrimination and Reclassification
The model including traditional risk factors had the C-index of 0.849 for CHD, 0.835 for CVD and 0.853 for ACS. Adding family history of cardiovascular disease into the models improved C-index by 0.2% (P=0.03 for both CHD and CVD; see also Figure 2). The GRS further improved risk discrimination of all end points over and above traditional risk factors and family history by 0.3–0.5% (P=0.0002 for CHD, P=0.0004 for CVD, P=0.001 for ACS) (Figure 2).
Figure 2.

Changes in C-index in FINRISK cohorts when adding 1) family history of cardiovascular disease and 2) the genetic risk score to the model.
The reference model with the traditional risk factors had the C index of 0.849 for CHD, 0.835 for CVD and 0.853 for ACS
There were 576 incident CHD cases in the combined 14-year follow-up of FINRISK 1992 and 1997. The family history of CVD did not improve the reclassification (NRI=0.1%, P=0.40). Adding the GRS into the model with traditional risk factors and family history resulted in overall NRI=5% (P=0.01). We also observed a significant improvement in reclassification of individuals at the intermediate risk category (clinical NRI=27%, P=1.1×10−8). Overall, 52 CHD cases (27%) and 206 non-cases (20%) in the intermediate risk group were correctly reclassified, when the GRS was added into the model (Table 3). We obtained essentially similar results for NRI in our sensitivity analysis with 2% higher category thresholds (Supplementary Table III). Also, IDI (Value=0.007, P=4.2×10−5) indicated statistically significant improvement in prediction, when genetic information was added to the model. Explained relative risk was 0.43 (se=0.02) for the model without the GRS and 0.45 (se=0.02) with the GRS. Calibration was good for all end points (Hosmer-Lemeshow test, 0.67>P>0.12).
Table 3. Reclassification of individuals in four risk categories after addition of genetic risk score (GRS) to a model with traditional risk factors and family history*.
| Model without GRS | Model with GRS | ||||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| 0-5% | 5-10% | 10-20% | >20% | NRI | Clinical NRI | ||
|
| |||||||
| 0-5% | Events | 79 | 15 | 0 | 0 |
Events: 0.04 (P=0.03) |
Events: 0.15 (P=4.6×10−4) |
|
| |||||||
| Nonevents | 7820 | 199 | 0 | 0 | |||
|
|
|||||||
| All | 7899 | 214 | 0 | 0 |
Nonevents: 0.01 (P-0.002) |
Nonevents: 0.11 (P=3.3×10−12) |
|
|
|
|||||||
| 5-10% | Events | 16 | 94 | 21 | 0 |
All: 0.05 (P=0.01) |
|
|
| |||||||
| Nonevents | 249 | 1080 | 173 | 0 | |||
|
|
|||||||
| All | 265 | 1174 | 194 | 0 |
All: 0.27 (P=1.1×10−8) |
||
|
|
|||||||
| 10-20% | Events | 0 | 22 | 122 | 52 | ||
|
|
|||||||
| Nonevents | 0 | 206 | 745 | 87 | |||
|
|
|||||||
| All | 0 | 228 | 867 | 139 | |||
|
|
|||||||
| >20% | Events | 0 | 0 | 22 | 186 | ||
|
|
|||||||
| Nonevents | 0 | 0 | 105 | 377 | |||
|
|
|||||||
| All | 0 | 0 | 127 | 563 | |||
Traditional risk factors include sex, total cholesterol, high-density lipiprotein (HDL) cholesterol, body-mass index, systolic blood pressure, blood pressure treatment, smoking and type 2 diabetes; age was used as the timescale in the Cox proportional hazards model. Abbreviations: NRI, net reclassification improvement
We assessed the potential over-fitting of the models by using the FINRISK 2002 as a training set and the joint FINRISK 1992 and 1997 data as a test dataset. To keep the training and test datasets comparable, we excluded Health 2000 from the analysis, since it lacks the information on family history. We estimated the effects for two models (with and without the GRS) in the training set data and used the estimated effect sizes from the training models to predict the 14-year absolute risk in the test data. The baseline hazard was estimated from the test data. This approach resulted in essentially similar results as a method where effect sizes were estimated directly from the test dataset (Supplementary Table IV).
Traditional risk factor screening of 100 000 European individuals would classify 64 373 subjects into <10%, 18 223 into 10–20% and 17 404 into ≥20% risk category (Figure 3). Based on the current guidelines 10, 11, only individuals in the highest risk group are eligible for lipid medication. We also classified those subjects with baseline diabetes or lipid treatment directly to the high risk category. Thus, subjects at the intermediate risk category (10–20%) were assumed not to receive statin treatment. Additional GRS screening of these subjects would reclassify 3475 (19%) subjects into the low and 2144 (12%) into the high risk category. Of the subjects reclassified into the high risk category, 676 were expected to experience CHD event within 14 years. Assuming that statins reduce the risk by 20%, additional GRS screening could prevent 135 (676×0.2) CHD cases over 14 years. As a comparison, if statins would be randomly allocated for the same number of subjects (N=2144) in the intermediate risk group, the expected number of prevented cases would be 54 (0.2×272, the expected number of cases). Thus, the GRS screening would prevent 2.5 times more events than the random allocation of statins to a comparable number of individuals predicted with intermediate risk in the absence of the GRS.
Figure 3.

Two-stage risk screening of coronary heart disease in a standard population of 100,000 subjects.
* Based on guidelines 10, 11, the subjects at intermediate risk group (10–20%) are assumed to not receive statin treatment. Statins are currently allocated for the subjects at ≥20% risk group. In addition, subjects with baseline lipid treatment and/or diabetes were assumed treated.
DISCUSSION
We studied four prospective Finnish cohorts with genetic markers from 28 loci that have been associated with coronary heart disease or myocardial infarction in previous studies 2, 3, 12, 13. The genetic risk score based on these variants was strongly associated with cardiovascular events and improved risk discrimination for all end points (C-index change=0.3–0.5%, all P-values≤0.001). GRS also improved risk reclassification of CHD in a joint FINRISK 1992 and FINRISK 1997 analysis (NRI=5%, P=0.01, clinical NRI=27%, P=1.1×10−8). Also IDI and explained relative risk indicated improved prediction. In our clinical modeling of 100 000 individuals, targeted GRS screening of clinically relevant risk group (10–20%) would reclassify 2144 (12%) subjects in the intermediate to high risk category. Statin allocation for reclassified individuals could prevent 135 CHD cases over 14 years.
Our results allow us to draw the following conclusions, which may be of clinical interest when evaluating a healthy individual’s risk for CHD. First, the genetic risk score is associated with incident coronary heart disease with the risk over two-fold for an individual at the top 10% of GRS compared to an average subject. The risk for these individuals is higher than expected by a linear function (P=0.003) and may be underestimated by predictions based only on traditional risk factors. Second, the GRS improved risk prediction of CHD over and above the conventional risk factors and family history, when evaluated with either discrimination or reclassification measures. Improved risk classification led to more accurate risk categorization for individuals at intermediate risk group, which means that a substantial proportion of CHD cases was reclassified upwards and non-cases downwards in the risk scale (0–5%, 5–10%, 10–20%, >20%). As these risk categories have been developed to guide treatment decisions, improved risk reclassification may have public health benefits. To address this, we estimated the effect of reclassification in a population level. Our data suggest that targeted GRS screening in addition to traditional risk factor screening would prevent one additional CHD event over the period of 14 years for every 135 (18 223/135) people screened.
Strengths of our study include a large prospective dataset and accurately defined event definitions, which have been drawn from the validated population registries 14-17. The main end point of our study was CHD (N=1093), rather than more heterogeneous CVD, which has been used in some other genetic risk prediction studies that have failed to show an incremental value of the GRS 5, 7. However, our estimates are comparable to the recent study 6 that analyzed 742 CHD events and constructed the GRS with the comparable SNP set to our study. The authors observed improvements in reclassification (NRI=2.8%, P=0.031), but not in discrimination. The better statistical power due to the larger number of SNPs or CHD events in our study might partially explain why we observed improvement in both reclassification and discrimination. Also, genetic diversity is lower and the extent of linkage disequilibrium is higher in Finland than in other European populations, which might facilitate the study of genetic effects for complex diseases.
Our large population cohort is well suitable for clinical modeling, a concept that has been applied in two recent risk prediction studies. The Emerging Risk Factors Collaboration 9 studied the added utility of lipid-markers in cardiovascular risk prediction. Additional screening based on lipoprotein(a) resulted reclassification of 555 subjects from intermediate to high risk category, and potential prevention of 17 CVD events over 10 years. The other study by the same collaboration 8 resulted approximately 30 prevented CVD cases over 10 years by additional CRP or fibrinogen screening. With restricted 10-year follow-up comparable to these studies, we estimate that additional genetic screening based on 28 SNPs could prevent 61 out of all 6355 CHD cases over 10 years. Thus, the added utility of 28-SNP GRS in risk prediction is superior compared to these other novel cardiovascular risk markers.
Our results should also be interpreted in the context of potential limitations of our study. Although our 28 SNP marker panel consists of lead SNPs from associated loci, we are only catching a fraction of all genetic risk variation for CHD. In our reclassification analyses, we combined two study cohorts and estimated 14-year CHD risk. However, established risk categories are usually applied for a 10-year time frame, and may not be directly applicable to our extended follow-up period. We, however, have addressed this issue by performing a sensitivity analysis with 2% higher risk thresholds. Also, we assumed that all participants eligible for statin treatment would receive them, which might overestimate the benefits of two-stage risk screening. In practice, compliance to treatment might not be complete. Finally, this study was conducted using a Finnish population sample, and the generalizability of these results to other populations, especially to those of non-European ethnicity, needs to be confirmed.
In conclusion, GRS improves CHD risk discrimination and reclassification over and above traditional risk factors and family history. Additional GRS screening of individuals at intermediate cardiovascular risk could help to prevent future cases through more accurate statin allocation. The clinical, economical and practical utility of the genetic testing needs to be further tested.
MATERIAL AND METHODS
Study Populations
FINRISK surveys have been conducted every 5 years since 1972 to monitor the risk of chronic diseases. For each survey, a stratified random sample was selected from the 25-74 year old inhabitants in different regions in Finland. The overlap between the samples is due to a small number of individuals being randomly chosen to consecutive FINRISK surveys. In surveys 1992–2007, 98% of the non-prevalent observations are unique. The survey included a questionnaire and a clinical examination, where a blood sample was drawn. The study protocol has been described elsewhere 1. FINRISK surveys 1992, 1997 and 2002 were included in the current analysis.
Health 2000 was based on a stratified two-stage cluster sampling from the National Population Information system to represent the total Finnish population aged 30 years and over 2. Persons aged ≥ 80 years were oversampled with a sampling weight of 2. The survey included an interview about medical history, health-related lifestyle habits, and a clinical examination at which a blood sample was drawn. A detailed methodology report is available online 3.
During the follow-up, hospitalization and mortality data were obtained from the Finnish National Hospital Discharge Register and the Finnish National Causes-of-Death Register. These registers cover all cardiovascular events that have led either to hospitalization or death in Finland. Cardiovascular diagnoses in these registers have been validated 4-7. CHD was defined as myocardial infarction, unstable angina pectoris, coronary revascularization (coronary artery bypass graft or percutaneous transluminal coronary angioplasty), or death due to CHD. CVD included CHD and ischemic stroke events. ACS was defined as MI, unstable angina or death due to CHD. The follow-up ended on Dec 31, 2010 in FINRISK and on Dec 31, 2008 in Health 2000.
Study protocols have been approved by the ethics committee of the National Institute for Health and Welfare, Finland, and/or the ethics committee of the Helsinki and Uusimaa Hospital District. All participants provided written informed consent.
SNP Selection and Genotyping
Out of 31 loci, which have been associated with myocardial infarction or coronary heart disease in genome-wide association studies 8-11, we included 28 SNPs in the study (Supplementary Table I). Three SNPs were excluded due to the failures in genotype assays or unreliable genotype calling. DNA samples were genotyped with the Sequenom MassARRAY System (Sequenom, San Diego, California), using iPLEX Gold chemistry and standard protocol. Genotyping was done at the Institute for Molecular Medicine Finland FIMM, and at the Wellcome Trust Sanger Institute, UK. All SNPs were in Hardy-Weinberg equilibrium and uncorrelated (r2<0.4), and had the genotype call rate > 98% and sample call rate > 95%. We calculated the GRS as a weighted mean by using the reported effect sizes from the reference studies as weights for the risk allele counts, and divided the sum by the number of the SNPs. Missing genotype data for each SNP was imputed with the average coded allele frequency of the study cohort.
Statistical Methods
CHD was the main cardiovascular end point in our analyses. We excluded individuals who were older than 75 years or had prevalent CVD at baseline. Participants reaching the age of 80 years during the follow-up were censored at their 80th birthday. Associations between the GRS and cardiovascular events were estimated with Cox proportional hazards models adjusted for the traditional risk factors at baseline: sex, total cholesterol, high-density lipoprotein (HDL) cholesterol, body mass index (BMI), systolic blood pressure, blood pressure treatment, current smoking status and diabetes mellitus. Age was used as the time scale in Cox models.
To further quantify the genetic effects for the subjects with different genetic risk load, we divided the GRS into deciles, and estimated the risk for each group by using the middle 20% of individuals as a reference. To estimate differences in risk between the subjects in the highest and middle values of the GRS, we compared the extreme 20%, 10% and 5% ends of the GRS with the middle 20% reference group. Deviation from the linear risk function was tested by fitting the joint model with both continuous GRS and an indicator variable, where the subjects in the highest 10% of the GRS were assigned as 1 and others as 0. We also studied the effects of unweighted GRS by calculating the number of risk alleles for each subject, and tested the association between cardiovascular end points and unweighted GRS using Cox models.
The information on family history of CVD was available in FINRISK 1992, FINRISK 1997 and FINRISK 2002 cohorts (N=19 001). We estimated the effect of family history on CHD, CVD and ACS using Cox models adjusted for traditional risk factors. We further adjusted the models for the GRS to examine how much the effects of these variants explain the familial risk. As a comparison, we studied the genetic effects with and without the adjustment for family history.
We found no evidence on heterogeneity in effect estimates between studies, and thus fixed effects meta-analysis was used to combine the results from each cohort. The validity of proportional hazards assumption was tested with scaled Schoenfeld residuals 12.
To evaluate the improvement in risk discrimination by using the genetic information and family history, we compared C-indices 13 for the models with and without the GRS and family history indicator in FINRISK cohorts. The change in C-index was estimated in each cohort separately and then combined across studies as proposed previously 14.
We then studied risk reclassification by using a restricted 14-year follow-up of FINRISK 1992 and 1997 cohorts. We modeled risk reclassification jointly in these two datasets and adjusted the analysis with the cohort indicator and traditional risk factors, including family history. Net reclassification improvement (NRI) was calculated from prospective data 15 using four risk categories: 0–5%, 5–10%, 10–20% and >20%. Clinical NRI was calculated for the subjects, who were classified to the intermediate risk group (10–20%) by the conventional model (model without the genetic data) 16. Since these risk categories are usually applied for 10-year time period, we performed additional sensitivity analysis by rising all thresholds by 2%. We also calculated integrated discrimination improvement (IDI) 17, explained relative risk 18 and evaluated model calibration with Hosmer-Lemeshow goodness-of-fit test 19.
Following the concept of two-stage risk screening used in two recent studies 14, 20, we estimated the clinical benefit of the GRS in a cardiovascular risk screening in a standard European population of 100 000 subjects. We assumed that all participants were first classified into cardiovascular risk categories based on traditional risk factors, and then additional GRS screening was targeted to those at the intermediate risk category (predicted risk 10–20%). The subjects at the intermediate risk were considered as clinically relevant subgroup based on the following assumptions; 1) statin medication is allocated to the subjects at the high risk category (≥20%) and the subjects with diabetes 21, 22, and 2) statins reduce cardiovascular risk by 20% in subjects without prevalent CVD 23. Reclassification was calculated separately for males and females and in four age groups (40–50, 50–60, 60–70, ≥ 70). Assuming that age- and sex-specific incidences of CHD in the European standard population are comparable to the current study, we estimated incidence rates from the FINRISK 1992 and 1997. We weighed reclassification tables with estimated incidence rates multiplied by the group-specific counts based on standard European population.
The statistical package R (version 2.12.2) was used for all analyses. We considered two-sided P<0.05 to be statistically significant.
Supplementary Material
SIGNIFICANCE.
The majority of the cardiovascular events occur within a population who are not classified as ‘high risk’ on the basis of the traditional risk factors. This has motivated the search for new potential risk markers. Genome-wide association studies have identified several common SNPs associated with CHD in case-control datasets. In this study, we show that the genetic risk score of these variants improves the risk discrimination and reclassification of CHD over and above traditional risk factors and family history in a prospective study setting. We applied the reclassification results into standard European population of 100 000 individuals, and showed that targeted genetic screening of individuals at intermediate risk (10–20%) could prevent one additional CHD event over the period of 14 years for every 135 (18 223/135) people screened. The clinical, economical and practical utility of genetic screening of individuals in the intermediate risk for CHD needs to be further tested.
Acknowledgments
SOURCES OF FUNDING
Wellcome Trust Sanger Institute supported the genotyping. S.R. was supported by the Academy of Finland Center of Excellence in Complex Disease Genetics (#213506 and #129680), Academy of Finland (#251217), the Finnish foundation for Cardiovascular Research and the Sigrid Juselius Foundation. V.S. was supported by grants #139635 and #129494 from the Academy of Finland and the Finnish Foundation for Cardiovascular Research. Authors declare that they have no competing interest. E.T. had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Abbreviations
- SNP
single-nucleotide polymorphism
- GRS
genetic risk score
- CHD
coronary heart disease
- CVD
cardiovascular disease
- ACS
acute coronary syndrome
Footnotes
DISCLOSURES
None.
Subject codes: [8] Epidemiology, [89] Genetics of cardiovascular disease, [135] Risk Factors
REFERENCES
- 1.Musunuru K, Kathiresan S. Genetics of coronary artery disease. Annu Rev Genomics Hum Genet. 2011;11:91–108. doi: 10.1146/annurev-genom-082509-141637. [DOI] [PubMed] [Google Scholar]
- 2.Schunkert H, Konig IR, Kathiresan S, et al. Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nat Genet. 2011;43:333–338. doi: 10.1038/ng.784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Coronary Artery Disease (C4D) Genetics Consortium A genome-wide association study in Europeans and South Asians identifies five new loci for coronary artery disease. Nat Genet. 2011;43:339–344. doi: 10.1038/ng.782. [DOI] [PubMed] [Google Scholar]
- 4.Ripatti S, Tikkanen E, Orho-Melander M, et al. A multilocus genetic risk score for coronary heart disease: case-control and prospective cohort analyses. The Lancet. 2010;376:1393–1400. doi: 10.1016/S0140-6736(10)61267-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thanassoulis G, Peloso GM, Pencina MJ, Hoffmann U, Fox CS, Cupples LA, Levy D, D’Agostino RB, Hwang SJ, O’Donnell CJ. A genetic risk score is associated with incident cardiovascular disease and coronary artery calcium: the Framingham Heart Study. Circ Cardiovasc Genet. 2012;5:113–121. doi: 10.1161/CIRCGENETICS.111.961342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vaarhorst AA, Lu Y, Heijmans BT, et al. Literature-based genetic risk scores for coronary heart disease: the Cardiovascular Registry Maastricht (CAREMA) prospective cohort study. Circ Cardiovasc Genet. 2012;5:202–209. doi: 10.1161/CIRCGENETICS.111.960708. [DOI] [PubMed] [Google Scholar]
- 7.Paynter NP, Chasman DI, Pare G, Buring JE, Cook NR, Miletich JP, Ridker PM. Association between a literature-based genetic risk score and cardiovascular events in women. JAMA. 2010;303:631–637. doi: 10.1001/jama.2010.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kaptoge S, Di Angelantonio E, Pennells L, et al. C-reactive protein, fibrinogen, and cardiovascular disease prediction. N Engl J Med. 2012;367:1310–1320. doi: 10.1056/NEJMoa1107477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Di Angelantonio E, Gao P, Pennells L, et al. Lipid-related markers and cardiovascular disease prediction. JAMA. 2012;307:2499–2506. doi: 10.1001/jama.2012.6571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. [Accessed August 7, 2012];National Institute for Health and Clinical Excellence. Statins for the prevention of cardiovascular events. http://www.nice.org.uk/nicemedia/pdf/TA094guidance.pdf.
- 11.Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults Executive Summary of The Third Report of The National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, And Treatment of High Blood Cholesterol In Adults (Adult Treatment Panel III) JAMA. 2001;285:2486–2497. doi: 10.1001/jama.285.19.2486. [DOI] [PubMed] [Google Scholar]
- 12.Kathiresan S, Voight BF, Purcell S, et al. Genome-wide association of early-onset myocardial infarction with single nucleotide polymorphisms and copy number variants. Nat Genet. 2009;41:334–341. doi: 10.1038/ng.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Erdmann J, Grosshennig A, Braund PS, et al. New susceptibility locus for coronary artery disease on chromosome 3q22.3. Nat Genet. 2009;41:280–282. doi: 10.1038/ng.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mähönen M, Jula A, Harald K, Antikainen R, Tuomilehto J, Zeller T, Blankenberg S, Salomaa V. The validity of heart failure diagnoses obtained from administrative registers. Eur J Prev Cardiol. 2013;20:254–259. doi: 10.1177/2047487312438979. [DOI] [PubMed] [Google Scholar]
- 15.Pajunen P, Koukkunen H, Ketonen M, et al. The validity of the Finnish Hospital Discharge Register and Causes of Death Register data on coronary heart disease. Eur J Cardiovasc Prev Rehabil. 2005;12:132–137. doi: 10.1097/00149831-200504000-00007. [DOI] [PubMed] [Google Scholar]
- 16.Tolonen H, Salomaa V, Torppa J, Sivenius J, Immonen-Raiha P, Lehtonen A. The validation of the Finnish Hospital Discharge Register and Causes of Death Register data on stroke diagnoses. Eur J Cardiovasc Prev Rehabil. 2007;14:380–385. doi: 10.1097/01.hjr.0000239466.26132.f2. [DOI] [PubMed] [Google Scholar]
- 17.Sund R. Quality of the Finnish Hospital Discharge Register: A systematic review. Scand J Public Health. 2012;40:505–515. doi: 10.1177/1403494812456637. [DOI] [PubMed] [Google Scholar]
MATERIAL AND METHODS REFERENCES
- 1.Vartiainen E, Laatikainen T, Peltonen M, Juolevi A, Mannisto S, Sundvall J, Jousilahti P, Salomaa V, Valsta L, Puska P. Thirty-five-year trends in cardiovascular risk factors in Finland. Int J Epidemiol. 2010;39:504–518. doi: 10.1093/ije/dyp330. [DOI] [PubMed] [Google Scholar]
- 2.Kattainen A, Salomaa V, Harkanen T, Jula A, Kaaja R, Kesaniemi YA, Kahonen M, Moilanen L, Nieminen MS, Aromaa A, Reunanen A. Coronary heart disease: from a disease of middle-aged men in the late 1970s to a disease of elderly women in the 2000s. Eur Heart J. 2006;27:296–301. doi: 10.1093/eurheartj/ehi630. [DOI] [PubMed] [Google Scholar]
- 3.National Public Health Institute Methodology report. Health 2000 survey. http://www.terveys2000.fi/doc/methodologyrep.pdf.
- 4.Mähönen M, Jula A, Harald K, Antikainen R, Tuomilehto J, Zeller T, Blankenberg S, Salomaa V. The validity of heart failure diagnoses obtained from administrative registers. Eur J Prev Cardiol. 2013;20:254–259. doi: 10.1177/2047487312438979. [DOI] [PubMed] [Google Scholar]
- 5.Pajunen P, Koukkunen H, Ketonen M, et al. The validity of the Finnish Hospital Discharge Register and Causes of Death Register data on coronary heart disease. Eur J Cardiovasc Prev Rehabil. 2005;12:132–137. doi: 10.1097/00149831-200504000-00007. [DOI] [PubMed] [Google Scholar]
- 6.Tolonen H, Salomaa V, Torppa J, Sivenius J, Immonen-Raiha P, Lehtonen A. The validation of the Finnish Hospital Discharge Register and Causes of Death Register data on stroke diagnoses. Eur J Cardiovasc Prev Rehabil. 2007;14:380–385. doi: 10.1097/01.hjr.0000239466.26132.f2. [DOI] [PubMed] [Google Scholar]
- 7.Sund R. Quality of the Finnish Hospital Discharge Register: A systematic review. Scand J Public Health. 2012;40:505–515. doi: 10.1177/1403494812456637. [DOI] [PubMed] [Google Scholar]
- 8.Kathiresan S, Voight BF, Purcell S, et al. Genome-wide association of early-onset myocardial infarction with single nucleotide polymorphisms and copy number variants. Nat Genet. 2009;41:334–341. doi: 10.1038/ng.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Erdmann J, Grosshennig A, Braund PS, et al. New susceptibility locus for coronary artery disease on chromosome 3q22.3. Nat Genet. 2009;41:280–282. doi: 10.1038/ng.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schunkert H, Konig IR, Kathiresan S, et al. Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nat Genet. 2011;43:333–338. doi: 10.1038/ng.784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Coronary Artery Disease (C4D) Genetics Consortium A genome-wide association study in Europeans and South Asians identifies five new loci for coronary artery disease. Nat Genet. 2011;43:339–344. doi: 10.1038/ng.782. [DOI] [PubMed] [Google Scholar]
- 12.Schoenfeld D. Residuals for the proportional hazards regression model. Biometrika. 1982;69:239–241. [Google Scholar]
- 13.Antolini L, Nam B, D’Agostico R. Inference on correlated discrimination measures in survival analysis: A nonparametric approach. Communications in statistics. Theory and methods. 2004;33:2117–2135. [Google Scholar]
- 14.Di Angelantonio E, Gao P, Pennells L, et al. Lipid-related markers and cardiovascular disease prediction. JAMA. 2012;307:2499–2506. doi: 10.1001/jama.2012.6571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pencina MJ, D’Agostino RB, Sr, Steyerberg EW. Extensions of net reclassification improvement calculations to measure usefulness of new biomarkers. Stat Med. 2011;15:11–21. doi: 10.1002/sim.4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cook NR. Comments on ‘Evaluating the added predictive ability of a new marker: From area under the ROC curve to reclassification and beyond’ by M. J. Pencina et al., Statistics in Medicine (DOI: 10.1002/sim.2929) Stat Med. 2008;27:191–195. doi: 10.1002/sim.2987. [DOI] [PubMed] [Google Scholar]
- 17.Pencina MJ, D’Agostino RB, Sr., D’Agostino RB, Jr., Vasan RS. Evaluating the added predictive ability of a new marker: from area under the ROC curve to reclassification and beyond. Stat Med. 2008;27:157–172. doi: 10.1002/sim.2929. [DOI] [PubMed] [Google Scholar]
- 18.Heller G. A measure of explained risk in the proportional hazards model. Biostatistics. 2011;13:315–325. doi: 10.1093/biostatistics/kxr047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hosmer DW, Lemeshow S. Applied Survival Analysis: Regression Modelling of Time to Event Data. Wiley; New York: 1999. [Google Scholar]
- 20.Kaptoge S, Di Angelantonio E, Pennells L, et al. C-reactive protein, fibrinogen, and cardiovascular disease prediction. N Engl J Med. 2012;367:1310–1320. doi: 10.1056/NEJMoa1107477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. [Accessed August 7, 2012];National Institute for Health and Clinical Excellence. Statins for the prevention of cardiovascular events. http://www.nice.org.uk/nicemedia/pdf/TA094guidance.pdf.
- 22.Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults Executive Summary of The Third Report of The National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, And Treatment of High Blood Cholesterol In Adults (Adult Treatment Panel III) JAMA. 2001;285:2486–2497. doi: 10.1001/jama.285.19.2486. [DOI] [PubMed] [Google Scholar]
- 23.Baigent C, Keech A, Kearney PM, Blackwell L, Buck G, Pollicino C, Kirby A, Sourjina T, Peto R, Collins R, Simes R. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet. 2005;366:1267–1278. doi: 10.1016/S0140-6736(05)67394-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.