

Abstract
Genetic factors may be learnt from families with gene mutations that render nerve-injury susceptibility even to ordinary physical activities. A typical example is hereditary neuropathy with liability to pressure palsies (HNPP). HNPP is caused by a heterozygous deletion of PMP22 gene. PMP22 deficiency disrupts myelin junctions (such as tight junction and adherens junctions), leading to abnormally increased myelin permeability that explains the nerve susceptibility to injury. This finding should motivate investigators to identify additional genetic factors contributing to nerve vulnerability of injury.
Keywords: nerve injury, peripheral myelin protein-22, PMP22, Charcot-Marie-Tooth disease, myelin, tight junction, adherens junction, action potential propagation, myelin permeability
Introduction
Our understanding on genetic factors affecting nerve injury and regeneration is primarily derived from numerous mouse models. Individual genes are inactivated in the mouse genome. Alterations of nerve regeneration are then observed in these mice (Osterloh et al., 2012; Wilhelm et al., 2012). Although critical information has been learnt from these models, it is often difficult to know how these findings can be translated into humans. The mechanical force during each injury cannot be the same, which makes any controlled study formidable among humans with nerve injures.
Nerves in humans with PMP22 deficiency exhibit susceptibility to mechanical pressure
One way to circumvent this obstacle is to ask questions differently. Are there any individuals or families with specific genetic mutations that would render neurological deficits when mechanical stress on these individuals is no more than ordinary physical activities? These mechanical stresses result in no symptoms in normal subjects, but are sufficient to cause dysfunction of the nerves with the mutation. Hereditary neuropathy with liability to pressure palsies (HNPP) says “yes” to this question. As its name denotes, this autosomal dominant inherited disorder typically presents with focal sensory loss and/or muscle weakness when the related peripheral nerves are challenged by mechanical stress. For instance, a patient with HNPP sits with one leg crossed on the other leg, which imposes mechanical pressure on the peroneal nerve at the fibular head. A half hour of this benign pressure is often sufficient to induce a foot drop on the crossed leg that may last hours to months in patients with HNPP (Earl et al., 1964; Li et al., 2004) but would not produce any symptoms in normal subjects. Strenuous physical activities in HNPP patients, such as running 10 miles with a 50lb backpack, may lead to severe arm paralysis and protracted recovery (Horowitz et al., 2004). This phenotype clearly demonstrates nerve susceptibility to mechanical stress.
Gene mapping has revealed that patients with HNPP are associated with a heterozygous deletion of chromosome 17p12 (c17p12) (Chance et al., 1993). The c17p12 contains 9 genes, including peripheral myelin protein-22 (PMP22). Humans with a heterozygous truncation mutation of PMP22 manifest an HNPP phenotype identical to that in patients with the heterozygous deletion of c17p12, supporting a causal role of loss of PMP22 function but not other genes in c17p12 (Nicholson et al., 1994; Li et al., 2007). Mice with heterozygous knockout of PMP22 gene recapitulate the pathology of humans with HNPP (Adlkofer et al., 1995). Application of mechanical compression on PMP22+/– mouse nerves induced conduction block (failure of action potential propagation) more rapidly than that in PMP22+/+ mouse nerves. Recovery after the compression on PMP22+/– nerves was very prolonged. This finding is well in line with the focal sensory loss and muscle weakness in HNPP patients when their nerves are exposed to mild mechanical stress (Li et al., 2002; Bai et al., 2010). Therefore, these mice have become an authentic model of HNPP.
Junction disruption and abnormally increased permeability in PMP22-deficient myelin
Utilizing the PMP22+/– mouse model, molecular mechanism underlying the impaired action potential propagation in HNPP has been investigated lately (Guo et al., 2014). PMP22 is a tetra-span membrane protein. It is primarily localized in adult myelinating Schwann cells, while its expression is diffuse in the developing nervous system (Parmantier et al., 1995, 1997; Li et al., 2013). Interestingly, demyelination is not found until the late stage of the disease (Bai et al., 2010). Although demyelination is widely regarded as one of the most important mechanisms which alter nerve conduction, effective nerve conduction is also thought to require a proper myelin seal through myelin junctions (such as tight junctions, adherens junctions). These junctions seal the spaces between adjacent myelin lamellae as well as spaces between the myelin and axolemma (Hartline and Colman, 2007). We found that deficiency of PMP22 dislocates junction protein complexes in myelin (Figure 1). This change yields excessively permeable myelin that allows an entry of dextran molecules up to a size of 70 kDa (Guo et al., 2014).
Figure 1.
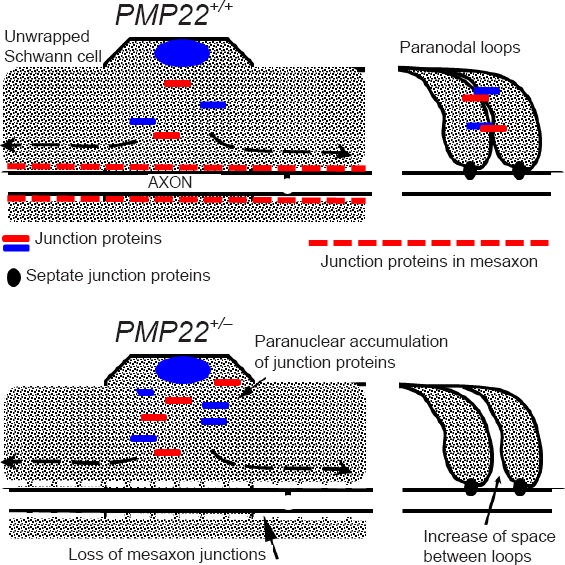
Myelin junction disruption (modified from Figure 6H–I in Guo et al., Annals of Neurology 2014).
Upper panel: Myelin junctions in a PMP22+/+ nerve fiber are depicted in paranodes and mesaxons. Schmidt-Lanterman incisures in inter-nodes are omitted since junctions in the incisures have changes similar to those in paranodes and mesaxons. These junctions prevent axonal current from leaking out. Lower panel: A PMP22+/– nerve fiber shows disruption or loss of junction protein complexes (tight junctions, ad-herens junctions) in paranodes and/or mesaxons. These junction pro-teins may be found in aberrant locations, including pernuclear regions of myelinating Schwann cells. Abnormal assembly of these junctions (including JAM-C transmembrane adhesion) also loosens adhesion between paranodal myelin lamellae (arrow on the right). In contrast, septate junctions in PMP22+/– nerves are still preserved. These changes increase myelin permeability that shunts current out of nerve fiber in the absence of demyelination, called “functional demyelination”.
The severity of abnormally increased myelin permeability was found to vary in different nerve fibers (Guo et al., 2014), and would produce two different populations of myelinated nerve fibers. Those in the first group have severely “leaky” myelin that would shunt current out of nerve fibers, leading to failure of action potential propagation in the absence of demyelination. We call this “functional demyelination” (Figure 1). Those in the second group have a mildly increased permeability of myelin, which still allows action potential to propagate, but would compromise the safety factor of action potential propagation. The nodes of Ranvier in myelinated nerve fibers typically generate depolarizing currents five times higher than the minimum required for the induction of an action potential. This surplus is called the safety factor (Kaji et al., 2000). This partially compromised safety factor would put the PMP22-deficient nerve fiber at risk to conduction failure if the fiber is challenged by additional external factors, such as mechanical stress. Taken together, studies in HNPP reveal a new concept that specific human genetic factor, such as PMP22, may critically affect human nerve susceptibility to injures and recovery after the trauma. One mechanism to achieve this biological effect is through PMP22's regulation of myelin junction formation and stability.
There are three types of junctions in myelin, including tight junctions, adherens junctions, and septate junctions. The first two are often autotypic junctions between myelin laminae of the same cell. The third one is situated between the most inner lamina of Schwann cells and axolemma (Hartline and Colman, 2007). Desmosomes were initially reported in myelin, but were later proven to be adherens junction (Fannon et al., 1995).
Under freeze-fracture electron microscopy, tight junctions appear as micro-strands extruding out of the membrane (Tetzlaff, 1978). In the peripheral nerve myelin, these junctions are localized in non-compact myelin such as paranodal loops, Schmidt-Lanterman incisures, and inner/outer mesaxons (Poliak et al., 2002). The strands are formed by polymerization of claudins, a family of tetraspan membrane proteins. C-terminals of claudins interact with a group of cytoplasmic proteins containing PDZ-domains such as ZO1 or ZO2 (Itoh et al., 1999). On the other hand, these PDZ-containing proteins also interact with actins and link the tight junction strands to the cytoskeleton for junction stabilization (Hartsock and Nelson, 2008).
A similar molecular organization is employed in adherens junctions. E-cadherins have a large glycosylated extracellular domain, a single transmembrane domain and a cytoplasmic tail at the c-terminal that interacts with catenins (α-catenin, β-catenin and p120 catenin). α-Catenins interact with actin filaments (Hartsock and Nelson, 2008).
There is another family of proteins, called JAM (JAM-A, JAM-B and JAM-C) that expresses nearby junctions. JAM contains a large extracellular Ig-domain, a transmembrane domain, and a cytoplasmic c-terminal. Interactions between the Ig-domain of JAM from two opposing membranes may form homotypic dimers. The dimers act like a“zipper” for transmembrane adhesion (Bazzoni et al., 2000a), sealing the opposing membranes juxtaposed to tight/adherens junctions, and further strengthening the seal of myelin inter-membrane space (Bazzoni et al., 2000b; Ebnet et al., 2000). JAM-C is the predominate form of JAMs in myelin. Ablation of Jam-c in mice results in HNPP-like pathology and alters nerve conduction (Scheiermann et al., 2007).
Finally, septate junctions are localized between paranodal myelin loops and axolemma. The protein constituents of septate junction include neurofascin-155 (Nf155) located at the tips of paranodal myelin loops and the Nf155-interacting partners (Caspr and contactin) on the axolemma. Under freeze fracture EM, septate junctions (also called transverse bands) are ridges spiraling along the paranodal axolemma which bridge the tips of paranodal myelin loops and axolemma (Rosenbluth, 2009). Removal of any septate junction protein detaches paranodal myelin loops from the axolemma and impairs action potential propagation (Bhat et al., 2001; Boyle et al., 2001; Sherman et al., 2005). However, the septate junctions are still preserved in HNPP mouse model.
Our observation in PMP22+/– mice shows abnormal formation and maintenance of these tight/adherence junctions in PMP22 deficiency. This finding not only offers a novel mechanism to explain nerve conduction defect in the disease (Guo et al., 2014), but also has additional physiological implications. After the developmental stage, when the myelinated nerve fiber has matured, its diameter, internodal length and myelin thickness remain stable. Our study provides an alternative mechanism that may fine-tune conduction by tightening or loosening the myelin junctions.
Lessons may be learnt from additional genetic mutations
Acquired or sporadic diseases can be studied by utilizing genetic models. A typical example is those studies in amyotrophic lateral sclerosis (ALS). A variety of rodent genetic models, such as the SOD1 and TDP43 transgenic mice, have been used to study the pathogenic mechanisms of the disease. While ALS patients with mutations in SOD1 or TDP43 are rare, investigations using these transgenic mice have made remarkable contributions to our understanding in the pathogenesis of ALS in general (Swarup and Julien, 2010). We believe that a similar advance in nerve injures could be achieved through the use of genetic models. HNPP and its mouse model are one step closer toward this goal. This strategy should motivate investigators to seek more families that might reveal additional genetic factors relevant to nerve injures.
SH3TC2 (Src homology 3 domain and tetratricopeptide repeats) may become the next candidate gene. Autosomal recessive mutations in SH3TC2 have been associated with an inherited peripheral nerve disease, called Charcot-Marie-Tooth disease type-4C (CMT4C). Patients with CMT4C usually present with an early onset neuropathy with severe axonal loss and dysmyelination (Kessali et al., 1997). However, humans with heterozygous mutations in SH3TC2 may present with carpal tunnel syndrome only (Lupski et al., 2010), implicating a nerve susceptibility to mechanical pressure. SH3TC2 is exclusively expressed in Schwann cells in peripheral nerves. It is tethered to cellular membrane via its myristic acid anchor and is involved in regulation of endosome recycling through its interaction with Rab11 (Stendel et al., 2010). How these molecular functions relate to nerve resistance to mechanical stress is still unknown.
Finally, nerve entrapments (focal compression) are common neurological conditions in humans; including median nerve entrapment at the wrist (carpal tunnel syndrome), ulnar nerve across the elbow, and peroneal nerve across the fibular head. Genetic factors contributing to these conditions are largely unknown. Dissecting out these factors should provide valuable insights into our understanding in nerve injures.
Footnotes
Funding: This research is, in part, supported by grants from NINDS R01NS066927 and Department of Veterans Affairs R&D funds.
References
- 1.Adlkofer K, Martini R, Aguzzi A, Zielasek J, Toyka KV, Suter U. Hypermyelination and demyelinating peripheral neuropathy in Pmp22-deficient mice. Nat Genet. 1995;11:274–280. doi: 10.1038/ng1195-274. [DOI] [PubMed] [Google Scholar]
- 2.Bai Y, Zhang X, Katona I, Saporta MA, Shy ME, O’Malley HA, Isom LL, Suter U, Li J. Conduction block in PMP22 deficiency. J Neurosci. 2010;30:600–608. doi: 10.1523/JNEUROSCI.4264-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bazzoni G, Martinez-Estrada OM, Mueller F, Nelboeck P, Schmid G, Bartfai T, Dejana E, Brockhaus M. Homophilic interaction of junctional adhesion molecule. J Biol Chem. 2000a;275:30970–30976. doi: 10.1074/jbc.M003946200. [DOI] [PubMed] [Google Scholar]
- 4.Bazzoni G, Martinez-Estrada OM, Orsenigo F, Cordenonsi M, Citi S, Dejana E. Interaction of junctional adhesion molecule with the tight junction components ZO-1, cingulin, and occludin. J Biol Chem. 2000b;275:20520–20526. doi: 10.1074/jbc.M905251199. [DOI] [PubMed] [Google Scholar]
- 5.Bhat MA, Rios JC, Lu Y, Garcia-Fresco GP, Ching W, St MM, Li J, Einheber S, Chesler M, Rosenbluth J, Salzer JL, Bellen HJ. Axon-glia interactions and the domain organization of myelinated axons requires neurexin IV/Caspr/Paranodin. Neuron. 2001;30:369–383. doi: 10.1016/s0896-6273(01)00294-x. [DOI] [PubMed] [Google Scholar]
- 6.Boyle ME, Berglund EO, Murai KK, Weber L, Peles E, Ranscht B. Contactin orchestrates assembly of the septate-like junctions at the paranode in myelinated peripheral nerve. Neuron. 2001;30:385–397. doi: 10.1016/s0896-6273(01)00296-3. [DOI] [PubMed] [Google Scholar]
- 7.Chance PF, Alderson MK, Leppig KA, Lensch MW, Matsunami N, Smith B, Swanson PD, Odelberg SJ, Disteche CM, Bird TD. DNA deletion associated with hereditary neuropathy with liability to pressure palsies. Cell. 1993;72:143–151. doi: 10.1016/0092-8674(93)90058-x. [DOI] [PubMed] [Google Scholar]
- 8.Earl CJ, Fullerton PM, Wakefield GS, Schutta HS. Hereditary Neuropathy with liability to pressure palsies; a clinical and electrophysiological studies in four families. Q J Med. 1964;33:481–498. [PubMed] [Google Scholar]
- 9.Ebnet K, Schulz CU, Meyer zu Brickwedde MK, Pendl GG, Vestweber D. Junctional adhesion molecule interacts with the PDZ domain-containing proteins AF-6 and ZO-1. J Biol Chem. 2000;275:27979–27988. doi: 10.1074/jbc.M002363200. [DOI] [PubMed] [Google Scholar]
- 10.Fannon AM, Sherman DL, Ilyina-Gragerova G, Brophy PJ, Friedrich VL, Jr, Colman DR. Novel E-cadherin-mediated adhesion in peripheral nerve: Schwann cell architecture is stabilized by autotypic adherens junctions. J Cell Biol. 1995;129:189–202. doi: 10.1083/jcb.129.1.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guo J, Wang L, Zhang Y, Wu J, Arpag S, Hu B, Imhof BA, Tian X, Carter BD, Suter U, Li J. Abnormal junctions and permeability of myelin in PMP22-deficient nerves. Ann Neurol. 2014;75:255–265. doi: 10.1002/ana.24086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hartline DK, Colman DR. Rapid conduction and the evolution of giant axons and myelinated fibers. Curr Biol. 2007;17:R29–35. doi: 10.1016/j.cub.2006.11.042. [DOI] [PubMed] [Google Scholar]
- 13.Hartsock A, Nelson WJ. Adherens and tight junctions: structure function and connections to the actin cytoskeleton. Biochim Biophys Acta. 2008;1778:660–669. doi: 10.1016/j.bbamem.2007.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Horowitz SH, Spollen LE, Yu W. Hereditary neuropathy with liability to pressure palsy: fulminant development with axonal loss during military training. J Neurol Neurosurg Psychiatry. 2004;75:1629–1631. doi: 10.1136/jnnp.2003.029314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Itoh M, Furuse M, Morita K, Kubota K, Saitou M, Tsukita S. Direct binding of three tight junction-associated MAGUKs, ZO-1, ZO-2, and ZO-3, with the COOH termini of claudins. J Cell Biol. 1999;147:1351–1363. doi: 10.1083/jcb.147.6.1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kaji R, Bostock H, Kohara N, Murase N, Kimura J, Shibasaki H. Activity-dependent conduction block in multifocal motor neuropathy. Brain. 2000;123:1602–1611. doi: 10.1093/brain/123.8.1602. [DOI] [PubMed] [Google Scholar]
- 17.Kessali M, Zemmouri R, Guilbot A, Maisonobe T, Brice A, LeGuern E, Grid D. A clinical, electrophysiologic, neuropathologic, and genetic study of two large Algerian families with an autosomal recessive demyelinating form of Charcot-Marie-Tooth disease. Neurology. 1997;48:867–873. doi: 10.1212/wnl.48.4.867. [DOI] [PubMed] [Google Scholar]
- 18.Li J, Krajewski K, Shy ME, Lewis RA. Hereditary neuropathy with liability to pressure palsy: the electrophysiology fits the name. Neurology. 2002;58:1769–1773. doi: 10.1212/wnl.58.12.1769. [DOI] [PubMed] [Google Scholar]
- 19.Li J, Krajewski K, Lewis RA, Shy ME. Loss-of-function phenotype of hereditary neuropathy with liability to pressure palsies. Muscle Nerve. 2004;29:205–210. doi: 10.1002/mus.10521. [DOI] [PubMed] [Google Scholar]
- 20.Li J, Ghandour K, Radovanovic D, Shy RR, Krajewski KM, Shy ME, Nicholson GA. Stoichiometric alteration of PMP22 protein determines the phenotype of HNPP. Arch Neurol. 2007;64:974–978. doi: 10.1001/archneur.64.7.974. [DOI] [PubMed] [Google Scholar]
- 21.Li J, Parker B, Martyn C, Natarajan C, Guo J. The PMP22 Gene and Its Related Diseases. Mol Neurobiol. 2013;47:673–698. doi: 10.1007/s12035-012-8370-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lupski JR, Reid JG, Gonzaga-Jauregui C, Rio Deiros D, Chen DC, Nazareth L, Bainbridge M, Dinh H, Jing C, Wheeler DA, McGuire AL, Zhang F, Stankiewicz P, Halperin JJ, Yang C, Gehman C, Guo D, Irikat RK, Tom W, Fantin NJ, et al. Whole-genome sequencing in a patient with Charcot-Marie-Tooth neuropathy. N Engl J Med. 2010;362:1181–1191. doi: 10.1056/NEJMoa0908094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nicholson GA, Valentijn LJ, Cherryson AK, Kennerson ML, Bragg TL, DeKroon RM, Ross DA, Pollard JD, McLeod JG, Bolhuis PA. A frame shift mutation in the PMP22 gene in hereditary neuropathy with liability to pressure palsies. Nat Genet. 1994;6:263–266. doi: 10.1038/ng0394-263. [DOI] [PubMed] [Google Scholar]
- 24.Osterloh JM, Yang J, Rooney TM, Fox AN, Adalbert R, Powell EH, Sheehan AE, Avery MA, Hackett R, Logan MA, MacDonald JM, Ziegenfuss JS, Milde S, Hou YJ, Nathan C, Ding A, Brown RH, Jr, Conforti L, Coleman M, Tessier-Lavigne M, et al. dSarm/Sarm1 is required for activation of an injury-induced axon death pathway. Science. 2012;337:481–484. doi: 10.1126/science.1223899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Parmantier E, Cabon F, Braun C, D’Urso D, Muller HW, Zalc B. Peripheral myelin protein-22 is expressed in rat and mouse brain and spinal cord motoneurons. Eur J Neurosci. 1995;7:1080–1088. doi: 10.1111/j.1460-9568.1995.tb01095.x. [DOI] [PubMed] [Google Scholar]
- 26.Parmantier E, Braun C, Thomas JL, Peyron F, Martinez S, Zalc B. PMP-22 expression in the central nervous system of the embryonic mouse defines potential transverse segments and longitudinal columns. J Comp Neurol. 1997;378:159–172. [PubMed] [Google Scholar]
- 27.Poliak S, Matlis S, Ullmer C, Scherer SS, Peles E. Distinct claudins and associated PDZ proteins form different autotypic tight junctions in myelinating Schwann cells. J Cell Biol. 2002;159:361–372. doi: 10.1083/jcb.200207050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rosenbluth J. Multiple functions of the paranodal junction of myelinated nerve fibers. J Neurosci Res. 2009;87:3250–3258. doi: 10.1002/jnr.22013. [DOI] [PubMed] [Google Scholar]
- 29.Scheiermann C, Meda P, Aurrand-Lions M, Madani R, Yiangou Y, Coffey P, Salt TE, Ducrest-Gay D, Caille D, Howell O, Reynolds R, Lobrinus A, Adams RH, Yu AS, Anand P, Imhof BA, Nourshargh S. Expression and function of junctional adhesion molecule-C in myelinated peripheral nerves. Science. 2007;318:1472–1475. doi: 10.1126/science.1149276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sherman DL, Tait S, Melrose S, Johnson R, Zonta B, Court FA, Macklin WB, Meek S, Smith AJ, Cottrell DF, Brophy PJ. Neurofascins are required to establish axonal domains for saltatory conduction. Neuron. 2005;48:737–742. doi: 10.1016/j.neuron.2005.10.019. [DOI] [PubMed] [Google Scholar]
- 31.Stendel C, Roos A, Kleine H, Arnaud E, Ozcelik M, Sidiropoulos PN, Zenker J, Schupfer F, Lehmann U, Sobota RM, Litchfield DW, Luscher B, Chrast R, Suter U, Senderek J. SH3TC2, a protein mutant in Charcot-Marie-Tooth neuropathy, links peripheral nerve myelination to endosomal recycling. Brain. 2010;133:2462–2474. doi: 10.1093/brain/awq168. [DOI] [PubMed] [Google Scholar]
- 32.Swarup V, Julien JP. ALS pathogenesis: Recent insights from genetics and mouse models. Prog Neuropsychopharmacol Biol Psychiatry. 2010;35:363–369. doi: 10.1016/j.pnpbp.2010.08.006. [DOI] [PubMed] [Google Scholar]
- 33.Tetzlaff W. The development of a zonula occludens in peripheral myelin of the chick embryo. A freeze-fracture study. Cell Tissue Res. 1978;189:187–201. doi: 10.1007/BF00209269. [DOI] [PubMed] [Google Scholar]
- 34.Wilhelm JC, Xu M, Cucoranu D, Chmielewski S, Holmes T, Lau KS, Bassell GJ, English AW. Cooperative roles of BDNF expression in neurons and Schwann cells are modulated by exercise to facilitate nerve regeneration. J Neurosci. 2012;32:5002–5009. doi: 10.1523/JNEUROSCI.1411-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang Y, Bekku Y, Dzhashiashvili Y, Armenti S, Meng X, Sasaki Y, Milbrandt J, Salzer JL. Assembly and maintenance of nodes of ranvier rely on distinct sources of proteins and targeting mechanisms. Neuron. 2012;73:92–107. doi: 10.1016/j.neuron.2011.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]