

Abstract
Dentinogenesis imperfecta (DI) is an inherited disorder affecting dentin. Defective dentin formation results in discolored teeth that are prone to attrition and fracture. Mutation in dentin sialophosphoprotein (DSPP) has been found to cause the dentin disorders DI - I and II (shields II and III). Early diagnosis and treatment of DI is recommended as it may prevent or intercept deterioration of the teeth and occlusion and improve esthetics. Here, we report a case with characteristic clinical, radiological and histological features of DI-I. The etiology and classification followed in literature is confusing since dentinoenamel junction (DEJ) in DI seems to be structurally and functionally normal and DI is clearly a disorder distinct from osteogenesis imperfecta (OI), but we still relate etiology of DI to DEJ and follow Shields classification. Therefore, we have briefly reviewed etiology and nomenclature system of DI.
Keywords: Dentin, odontoblasts, reciprocal induction
INTRODUCTION
Dentinogenesis imperfecta (DI) is described as a localized form of mesodermal dysplasia observed in histodifferentiation and corresponds to a congenital hereditary change involving deciduous and permanent teeth. DI was first reported by Tolbot as an autosomal dominant trait. Roberts and Schonr were the first to use the term DI, describing it as a condition similar to that found in OI.[1]
The disease is inherited in a simple autosomal dominant mode with high penetration and a low mutation rate.[2] Teeth have typical amber like translucency against reflected light varying from grayish purple to purple brown or yellowish brown. Radiographic features of DI include bulbous crowns with constricted short roots, progressive obliteration of the pulp chambers. Histologically, dentinal tubules are irregular and bigger in diameter and areas of uncalcified matrix are seen. The purpose of this article is to stress on the need to rethink the nomenclature system of DI.
CASE REPORT
An 18-year-old female patient reported with a decreased lower facial height. According to her parents, there was a discoloration of primary teeth and chipping of enamel. She was born from a second degree consanguineous marriage. None of the family (siblings and parents) members have similar complaint.
There was decrease in lower facial height due to severe attrition to the level of gingiva. Intraorally, generalized brownish discoloration of teeth with loss of enamel was seen [Figure 1]. Dental caries cannot develop in these cases owing to the absence of dentinal tubules and inability of caries to develop on a surface where enamel is rapidly being lost due to abrasion and fracture. The case presented here confirms this with the absence of carious lesions.
Figure 1.
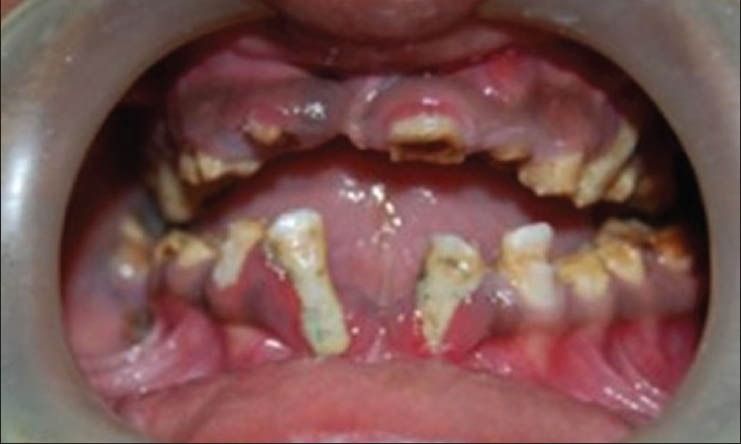
Intraoral view with brownish discoloration and severe attrition
Panoramic study showed narrow pulp chamber with severe coronal attrition and multiple periapical radiolucencies [Figure 2]. Unstained ground section with photomicrography showed irregular dentinal tubules with increased hypomineralized interglobular dentin and obliterated pulp chamber [Figures 3–5].
Figure 2.
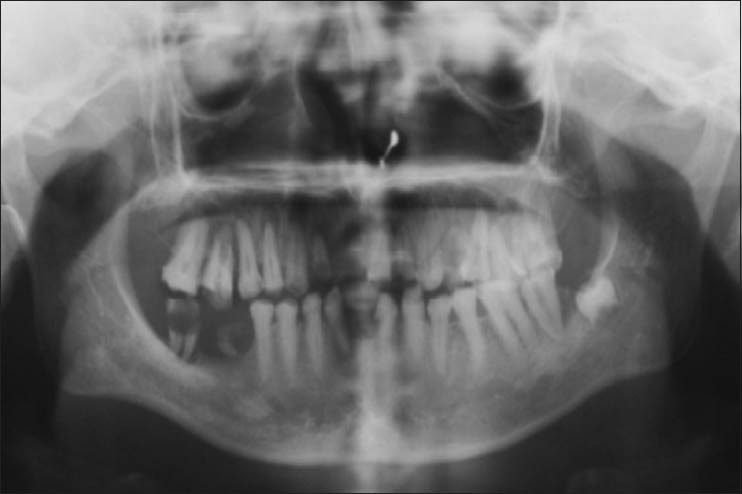
Panoramic Radiograph showing obliterated pulp chambers
Figure 3.

Photomicrograph showing irregular dentinal tubules
Figure 5.

Photomicrograph showing obliterated pulp chamber
Figure 4.

Photomicrograph showing increased interglobular dentin
DISCUSSION
DI is a localized mesodermal dysplasia affecting both primary and permanent dentition.[2] Inheritance of dentin defects is typically autosomal dominant, although autosomal recessive and X-linked cases of dentin defects associated with syndromes are reported. Dentin defects occur as a feature in a number of syndromes, including osteogenesis imperfecta (OI), Ehlers-Danlos syndrome (EDS), tumoral calcinosis and hypophosphatemic rickets.[3]
DI is an inherited disorder affecting dentin. Mutation in dentin sialophosphoprotein (DSPP) is the cause for this defect. The DSPP gene is located at 4q21.3 in a cluster of dentin and bone matrix genes. DSPP encodes both dentin sialoprotein (DSP) and dentin phosphoprotein (DPP) as one precursor protein that is cleaved before secretion. DSP and DPP have different roles in dentinogenesis. DPP serves as a nucleator of mineralization and induces apatite formation.[4]
Shields et al. proposed three types of DI: DI type 1 is associated with OI. DI type 2 has essentially the same clinical radiographic and histological features as DI type 1 but without OI; DI type 3 is rare and is only found in the triracial Brandywine population of Maryland. These systems are well accepted but not completely satisfactory. The best nomenclature system was suggested by Levin [Table 1]. Extensive pedigrees of individuals with DI have been studied and none have exhibited other changes suggestive of OI. Therefore, DI is clearly a disorder distinct from OI.[7] So far, no definitive relation between the type of OI and the dental involvement can be established. Familial occurrence of DI with OI cannot be comprehensively explained by mutations in type I collagen genes.[9]
Table 1.
Various classifications of DI

There is no substitute in the present classification for the category designated as DI-I of Shields classification. Therefore, in present classification, there are only two types: Type I DI without OI and Type II – Brandywine Type with shell tooth. The diagnosis of DI should be reserved for defective dentin formation with opalescent teeth in the absence of systemic disease. Appropriately, dentin defects associated with the systemic bone disease are termed OI with opalescent teeth.
The color of teeth varies from brown to blue, sometimes described as amber or gray, with an opalescent sheen. The enamel may show hypoplastic or hypocalcified defects in about one-third of the patients and tends to crack away from the defective dentin in an affected patient. The exposed dentin may undergo severe and rapid attrition.[2] Originally, it was believed that a defective dentinoenamel junction (DEJ) was resulting in chipping of enamel, but scanning electron microscopic studies have disclosed a normal junction.[10] The enamel does not actually fall off the DEJ, as some believe; the DEJ in DI seems to be structurally and functionally normal [Figure 6].
Figure 6.

DEJ is normal, the mantle dentin is slightly abnormal and the secondary dentin is significantly abnormal
It needs to be appreciated that odontoblasts differentiate in the pre-existing ground substance of the dental papilla and that into this ground substance the first dentin collagen is deposited.[11] Odontoblast differentiation follows three steps: (a) Induction, (b) Competence and (c) Terminal differentiation.[12] In the first formed dentin (mantle dentin), some of the matrix components are contributed by dental pulp cells beneath the odontoblasts. The odontoblasts are still undergoing the late stage of differentiation as the first layer of dentin matrix is being deposited.[13] The need for an alternate mechanism of mineralization during mantle dentin formation may relate to the fact that odontoblasts are still completing their terminal differentiation at this stage and may not be able to fully exhibit the odontoblast phenotype in terms of expression of dentin-specific matrix components.[12] The result is a near normal DEJ because of the less affected mantle dentin. Imperfect primary dentin in DI is a result of matrix components that are completely formed by defective odontoblasts. They secrete an abnormal collagen, which is undermineralized and fails to form odontoblastic tubules.
Some amount of enamel wears off excessively because of hypoplastic hypocalcified areas within the enamel rods. This is presumably due to the fact that the cells of the internal dental epithelium were needed to induce odontoblasts and their product, dentin, becomes the initiator for further differentiation of the internal dental epithelial cells in a process of reciprocal induction.[11] The abnormality in dentin formation results secondarily in enamel that is also microscopically somewhat defective.[14] The exposed dentin, which is soft, undergoes quick attrition to such an extent that the dentin becomes smooth and continuous with the gingival tissue.
Radiographically, the teeth have bulbous crowns with constricted short roots. Initially, pulp chambers may be abnormally wide and resemble “shell teeth,” but they will progressively obliterate. Histologically, the enamel, although normal in structure, tends to crack. The DEJ is not scalloped. The enamel appears defective with subtle hypocalcification defects in the enamel rods just above the DEJ. The DEJ appears flattened although it appears qualitatively normal. In most cases, the structure of the mantle dentin is normal, whereas the dentinal tubules of the circumferential dentin are coarse and branched and the total number of tubules is reduced. The presence of an atubular area in the dentin with reduced mineralization and a reduced number of odontoblasts are consistent findings. Pulpal inclusions and much interglobular dentin are also frequent.[2] Odontoblasts entrapment may be seen within the dentinal matrix. Large areas of unmineralized dentin and irregular border between the unmineralized and mineralized dentin is seen.
Treatment of DI has several objectives: To maintain dental health and preserve vitality, form and size of the dentition; to provide the patient with an esthetic appearance at an early age in order to prevent psychological problems; to provide the patient with a functional dentition; to prevent loss of vertical dimension; to avoid interfering with the eruption of the remaining permanent teeth and to allow normal growth of the facial bones and temporomandibular joint.
Treatment of mixed and permanent dentition is challenging and frequently demands a multidisciplinary approach. Collaboration of the pediatric dentist with a prosthodontist and an orthodontist is often imperative.
Footnotes
Source of Support: Nil.
Conflict of Interest: None declared.
REFERENCES
- 1.Mayordomo FG, Estrela F, de Aldecoa EA. Dentinogenesis imperfecta: A case report. Quintessence Int. 1992;23:795–802. [PubMed] [Google Scholar]
- 2.Sapir S, Shapira J. Dentinogenesis imperfecta: An early treatment strategy. Pediatr Dent. 2001;23:232–7. [PubMed] [Google Scholar]
- 3.Hart PS, Hart TC. Disorders of human dentin. Cells Tissues Organs. 2007;186:70–7. doi: 10.1159/000102682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Holappa H, Nieminen P, Tolva L, Lukinmaa PL, Alaluusua S. Splicing site mutations in dentin sialophosphoprotein causing dentinogenesis imperfecta type II. Eur J Oral Sci. 2006;114:381–4. doi: 10.1111/j.1600-0722.2006.00391.x. [DOI] [PubMed] [Google Scholar]
- 5.Shields ED, Bixler D, el-Kafrawy AM. A proposed classification for heritable human dentin defects with a description of a new entry. Arch Oral Boil. 1973;18:543–53. doi: 10.1016/0003-9969(73)90075-7. [DOI] [PubMed] [Google Scholar]
- 6.Witkop CJ., Jr Hereditary defects of dentin. Dent Clin North Am. 1975;19:25–45. [PubMed] [Google Scholar]
- 7.Neville BW, Damm DD, Bauquot JE, Allen C. 2nd ed. Amsterdam: Elsevier; 2005. Oral and Maxillofacial Pathology; pp. 94–6. [Google Scholar]
- 8.Shafer WG, Hine MK, Levy BM. 5th ed. Amsterdam: Elsevier; 2006. Text Book of Oral Pathology; pp. 75–7. [Google Scholar]
- 9.Ranta H, Lukinmaa PL, Waltimo J. Heritable dentin defects: Nosology, pathology, and treatment. Am J Med Genet. 1993;45:193–200. doi: 10.1002/ajmg.1320450209. [DOI] [PubMed] [Google Scholar]
- 10.Eversole LR. 4th ed. US: PMPH; 2001. Clinical outline of oral pathology: Diagnosis and treatment; p. 363. [Google Scholar]
- 11.Nanci A. 7th ed. Maryland Heights: Mosby; 2008. Ten Cate's Oral histology: Development, structure, and function; pp. 191–238. [Google Scholar]
- 12.Hargreaves KM, Goodis HE. 1st ed. UK: Quintessence publication; 2002. Seltzer and Bender's Dental Pulp; pp. 31–46. [Google Scholar]
- 13.Berkovitz BK, Holland GR, Moxham BJ. 3rd ed. Maryland Heights: Mosby; 2002. Oral Anatomy, Histology and Embryology; p. 322. [Google Scholar]
- 14.Marx RE, Stern D. 1st ed. UK: Quintessence; 2003. Oral and Maxillofacial Pathology; pp. 232–4. [Google Scholar]