

Abstract
We have shown that loss of ELF, a stem cell adaptor protein, disrupts TGF-β signaling through Smad3 and Smad4 localization. Notably elf+/−/smad4+/− mice develop gastric cancer presenting this as an important model for analyzing molecular event in gastric carcinogenesis. To gain further insight into the functional role of ELF in gastric cancer suppression, we carried out a detailed characterization of cell cycle events leading to gastric tumorigenesis. elf−/− cells and elf+/−/smad4+/− mice demonstrate a marked alteration of cell cycle regulators, such as Cdk4, K-Ras, and p21. Levels of Cdk4 increased compared to normal controls, suggesting loss of ELF results in functional abnormalities in cell cycle regulation. We further demonstrate that the elf−/− MEFs show a disruption of G1/S cell cycle transition and a significant reduction in senescence. Thus, in response to ELF deficiency, the abnormalities of G1/S checkpoint and senescence contribute their increment of susceptibility to malignant transformation.
Keywords: ELF, Gastric cancer, Smad4, Cell cycle, Senescence
Gastric carcinoma is a leading cause of cancer-related deaths worldwide. With approximately 755,000 new cases diagnosed each year, it ranks second in incidence only to lung cancer. Most gastric cancers are diagnosed at an advanced stage, contributing to a dismal 5-year survival rate of under 20% [1–3]. The factors that govern progression from gastric epithelial cell hyperplasia through dysplasia to in situ carcinoma and invasive disease remain poorly understood and are thought to involve multiple pathways.
Helicobacter pylori is one of the most prominent reasons in gastric oncogenic activation by the cytotoxic-associated antigen A (cag A), which is known to bind signaling molecules such as erb2, c-met, and zo-1. Infection with H. pylori perturbs several signaling pathways thus promoting gastric carcinogenesis [4]. Germ line mutations in E-cadherin has been one of the first few genes clearly implicated in diffuse type of gastric cancer. Subsequently oncogenic activation of β-catenin (17–27%) in the differentiated type, K-Ras (up to 18%) in diffuse and differentiated types, as well as amplification of c-erbB2 or c-met in approximately 10% of both cancer types have been identified. Among tumor suppressors, APC mutations although frequent in gastric adenoma are rarely found in gastric cancers. Similarly, mutations in p16 and p53 are seen in up to 20% of cases. Microsatellite instability (MSI) is present in up to 33% of gastric tumors [5], and is in turn associated with more frequent frameshift mutations of TGF RII, BAX, and hMSH3 genes [6]. Other involved molecules are CD-44, c-erb2, tie-1, c-met, and more recently, Smad4 [7].
Recent genetic studies using mouse knockouts have demonstrated a strong role for the TGF-β signaling pathway in gastric carcinogenesis. The TGF-β family encompasses a number of ubiquitous growth and differentiation factors. These proteins control cell fate by regulating differentiation, proliferation, migration, and apoptosis. Their signaling mechanism is mediated through type I and II receptors, transmembrane serine/threonine kinases, and transmitted to Smads, specific intracellular mediators. Activation of Smads results in nuclear translocation and activation of gene expression [8–10]. Vertebrates possess at least nine Smad proteins falling into three functional classes: (i) Receptor activated Smads (R-Smads): Smad1, Smad2, Smad3, Smad5, and Smad8; (ii) Co-mediator Smads: Smad4 and Smad10; and (iii) Inhibitory Smads: Smad6 and Smad7. Smad2 and Smad4 are known tumor suppressors in humans, and inactivation of Smad4 is a common aberration noted in human gastrointestinal tumors [11–13]. The pleiotrophic effects of Smad proteins and their interactions with various transcriptional factors, adaptors, and ubiquitinators. Among these, Runx transcriptional activators play a prominent role in suppressing gastric cancer. Runx3-null mice exhibit gastric hyperplasia, and approximately half of human cancers lack Runx expression due to hemizygous deletion and DNA methylation of the Runx promoter [14,15].
In addition, the activities of Smads can be modulated by adaptors such as embryonic liver fodrin (ELF), filamin or Smad anchor for receptor activation (SARA), as well as functional interactions with multiple signal transduction pathways [16,17]. These adaptor proteins play a critical role in localizing Smad and facilitating the functions of TGF-β. ELF is a key protein involved in endodermal stem/progenitor cell commitment to foregut lineage [17]. It is also crucial for protein sorting, cell adhesion, and the development of a polarized differentiated epithelial cell. In prior work, we have demonstrated that TGF-β triggers phosphorylation and association of ELF, a β-spectrin, with Smad3 and Smad4 resulting in nuclear translocation [16]. elf+/− mice display a phenotype similar to mice with compound haploinsufficiency at smad2 and smad3 loci. Disruption of elf gene expression may be an important modulator of TGF-β inactivation. An examination of elf+/− and elf+/−/smad4+/− mice for tumor development revealed that 40% of elf+/− mice developed tumors of varying etiology. This tumor incidence was comparable to that seen in smad4+/− mice (45%). Interestingly, 90% of elf+/−/smad4+/− double heterozygous mutants developed tumors, suggesting a cooperative interaction between ELF and Smad4 leading to enhanced tumorigenesis [18,19].
Inhibition of cell proliferation is central to the TGF-β response in epithelial, endothelial, hematopoietic, neural, and certain types of mesenchymal cells. Escape from this response is a hallmark of many cancer cells. TGF-β can induce anti-proliferative gene responses at any point during the division cycle. These responses are effective at inhibiting cell cycle progression primarily during G1. Once a cell becomes committed to executing DNA replication in late G1, the division cycle will proceed for the most part undeterred by TGF-β until the cell enters G1 again following mitosis, at which point the cell cycle will arrest [20]. The mechanism of TGF-β mediated growth arrest at the G1 phase in epithelial cells includes the inhibition of cyclin D, E, and A mRNA expressions [21–23], a reduction of cyclin-dependent kinase 4 (Cdk4) synthesis, and a down-regulation of Cdk2 kinase activity [24,25]. Additionally, p21cip1, a Cdk inhibitor, has been reported to be a potential cellular mediator in TGF-β induced cell cycle arrest [26–28]. The induction or redistribution of these molecules by TGF-β effectively suppresses the activity of the G1 cyclin/Cdk complex.
Epistatistically increased gastric tumors in elf+/−/smad4+/− mutant mice present an ideal model for further analysis of the genetic cooperation between ELF and Smad4. In this study, we sought to demonstrate changes in cell proliferation and expression of proteins involved in cell cycle regulation, such as Cdk4, Ras, and p21 in elf−/− cells and elf+/−/smad4+/− mice. Interestingly, we found that loss of ELF upregulates the level of these proteins in cultured cells and hyperplastic tissues. We further demonstrate that elf−/− MEFs show impairment of transition at the G1/S cell cycle checkpoint and a significant reduction of senescence. Our results indicate that the loss of ELF, a modulator of the TGF-β pathway, leads to deregulation of cell cycle control and senescence. The presence of a prominent gastric tumor phenotype in the elf+/−/smad4+/− with the significant disruption of key cell cycle regulation indicates a strong tumor suppressor role for ELF and Smad4 in gastric carcinogenesis.
Materials and methods
Mouse maintenance and analysis
Mice were monitored at least twice a week for possible symptoms related to tumor formation. At autopsy, the esophagus and stomach were bisected and flattened during fixation or were expanded with fixative by clamping the adjacent duodenum/esophagus and infusing the lumen with a syringe. They were then cleaned of blood, food, and feces, fixed overnight in 10% buffered formalin, processed, and embedded in paraffin. Animal care was in accordance with institutional guidelines and under approved animal care protocols.
MEFs generation and culture
MEFs were derived from E14.5 embryos generated from intercrosses between elf+/− mice. The comparisons between wild type and mutant cells were performed between littermates. To immortalize the wild type and mutant MEFs, the cells were seeded 1 × 106 cells in a 10-cm dish and left untouched (except for the medium change every 3 day) for at least a month. Individual colonies were picked and sub-cultured sequentially through 24-well, 12-well, 6-well, and 10-cm plates.
G1/S cell cycle analysis
For G1/S checkpoint analysis, MEFs plated 1 day before were synchronized at G0 by incubation for 4 days in starvation medium containing 0.5% fetal bovine serum (FBS). The synchronized cells were released by changing medium to complete medium containing 15% FBS. After harvesting the cells by trypsin treatment, the cells were fixed by 70% ethanol, stained by propidium iodide, and analyzed by using FAC-SCalibur flowcytometer (BD Biosciences). Three embryos from different littermates representing each genotype were used. Data are represented as the average of three experiments. The DNA contents and cell cycle of MEFs were analyzed by CellQuest (BD Biosciences).
Cell senescence analysis
MEFs were plated as 5 × 104 MEF cells per 1-well chamber slide (Becton Dickinson) for 3 day, and then processed for acidic β-galactosidase activity. In brief, cells were washed, fixed 5 min in 1 mM MgCl2, 0.5% glutaraldehyde in PBS, washed, and incubated at 37 °C with freshly prepared senescence staining solution: 1 mg/ml 5-bro-mo-4-chloro-3-indolyl β-D-galactoside/40 mM citric acid/sodium phosphate (pH 6.0)/5 mM potassium ferrocyanide/5 mM potassium ferricyanide/150mM NaCl/2mM MgCl2.
Histology, immunohistochemical staining, and Western blotting
Sections of hyperplastic gastric tissue (5 μm thickness) were immersed in xylene to remove paraffin, then dehydrated in graded alcohol, and rinsed in PBS. Endogenous peroxide was quenched using 3% hydrogen peroxide (Sigma). Non-specific binding sites were blocked using PBS containing 3% goat serum and 3% BSA. The sections were incubated overnight at 4 °C in a humid chamber with primary antibody. The sections were then incubated with peroxidase-conjugated secondary antibody (Jackson Immunoresearch Laboratories). Between reagent changing, tissues were extensively washed by PBS with 0.1% Tween 20. The insoluble peroxidase substrate, DAB (Sigma), was added to cover the entire tissue on the slide, and color development was monitored under the microscope. After rinsing in distilled water for 2 min, counterstaining was performed with modified Harris hematoxylin (Sigma) for 2 min followed by a rinse in distilled water for 3 min. Sections were dehydrated by passage through graded alcohol concentrations and finally xylene. Coverslips were mounted using DPX (Fluka) before observation. Western blot analysis was carried out according to standard procedures using ECL detection (Amersham). The following primary antibodies were used: ELF (against residues 2–14) Cdk4, cyclin D1, K-Ras, p21, Smad4, β-actin (Santa Cruz), Mdm2 (Calbiochem), and p53 (Oncogen Research Products). Horseradish peroxidase-conjugated donkey anti-rabbit or sheep anti-mouse antibodies (Jackson Immuno Research) were used as secondary antibodies.
Statistical analysis
Global χ2 test was used to test the hypothesis that the coefficient of each variable was equal to 0. Sample sets of data were compared to assess the significance. A P value <0.05 was required for statistical significance, and all tests were two-sided. All tests were done with SPSS 10.1 software (SPSS Incorporation).
Results
Overexpression of Cdk4 in elf−/− MEFs
Loss of ELF results in rapid development of gastric cancer within a year in mutant mice. Strong expression of ELF in normal gastric tissues was observed in epithelial cells of the glandular stomach and weak expression in the fore-stomach epithelia. ELF expression was greater in parietal and surface mucous cells than in the chief cells. Strongest biosignals could be found in the stem cell zone (Fig. 1A). ELF inactivation leads to multiple gastrointestinal tumors. We therefore hypothesized that ELF inactivation leads to multiple gastrointestinal tumors and loss of ELF leads to deregulation of cell cycle control in elf+/− and elf+/−/smad4+/− mutant mice. To explore our hypothesis that cell cycle proliferation and deregulation occur in elf+/− and elf+/−/smad4+/− mutant mice, we focused on expression of G1 checkpoint and G1/S transition proteins in the absence of ELF. We compared the expression level of Cdk4 in the early passage of elf−/− MEFs with wild type control. The levels of Cdk4 were increased over twice in elf−/− MEFs (Fig. 1B). This result suggests that loss of ELF causes upregulation of Cdk4 and may induce deregulation of the cell cycle.
Fig. 1.
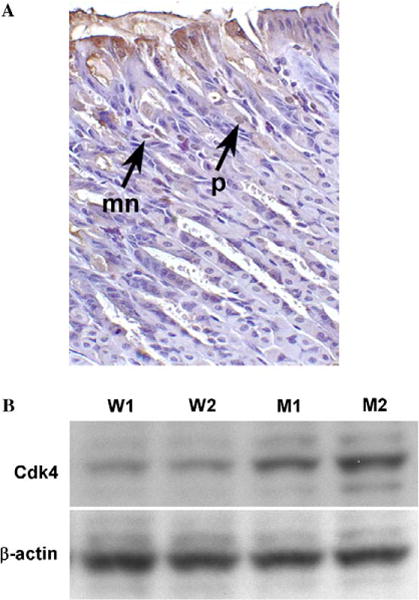
Expression of elf in normal gastric tissue and overexpression of Cdk4 in elf−/− MEFs. (A) Immunohistochemical staining of gastric tissue sections from wild type mice. Images of sections were detected by the primary antibodies against ELF. p and mn represent parietal and mucous cells, respectively. (B) Cdk4 expression patterns of wild type (W1, W2) and elf−/− (M1, M2) MEFs. β-Actin was used as loading control.
Loss of ELF results in abrogation of G1/S cell cycle transition and senescence
Since ELF-deficient MEFs exhibit markedly upregulated pattern of Cdk4 as compared to wild type, we tested whether loss of ELF prevented proper control of G1/S checkpoint proteins. elf−/− and wild type cells were synchronized by starvation and released by changing to complete medium. elf−/− mutant cells showed reduction of G1 population comparing wild type control indicating that loss of ELF promotes faster entry into S-phase (Fig. 2A). To confirm the function of ELF in G1/S checkpoint regulation, we studied the expression of p21, a critical regulator of the G1/S checkpoint in the cultured cell (Fig. 2B). The expression of p21 was only 62% in mutant MEFs as compared to the wild type. These results suggest that loss of ELF induced the abnormal G1/S checkpoint transition contributing for genetic stability and tumor formation.
Fig. 2.
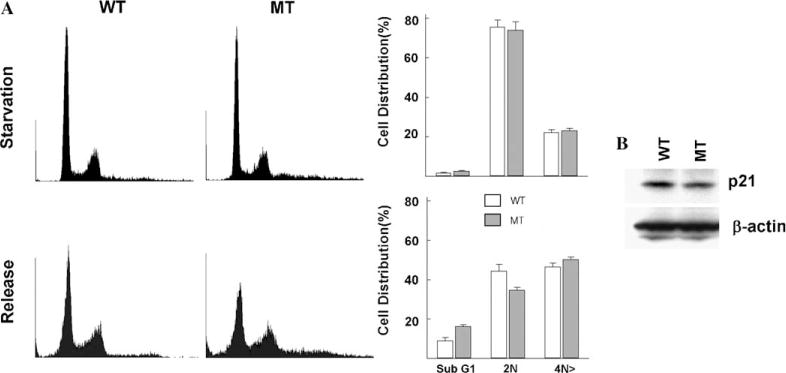
Loss of ELF alters of G1/S arrest of cell cycle. (A) Representative histograms of FACS showing DNA content of synchronized and released wild type (WT) or elf−/− (MT) MEFs. When releasing, cells were also treated with nocodazole (100 ng/ml) to prevent the continuous cycling of cells. Graphs represent percentages of cells distribution in indicated stages of cell cycle in flow cytometry analysis. (B) Western blot analysis of p21 in type (WT) or elf−/− (MT).
We next investigated whether deregulated S-phase entry of mutant cells interferes with their ability to undergo senescence, which prevents the growth of defective cells. When we cultured the wild type and mutant MEFs followed by NIH 3T3 culture protocol, we did not find any significant difference in early passage (P1). However, on progressive passage, elf−/− cells appeared smaller than comparing flattened wild type. To see whether this changing of cell shape is related with the senescence, we performed the X-gal stain in several different passages of MEFs (Fig. 3). At P3 passage, statistically significant reduced X-gal positive population of elf−/− MEFs indicates that loss of ELF prevents proper senescence, leading to the accumulation of defective cells which are more susceptible to tumorigenesis.
Fig. 3.
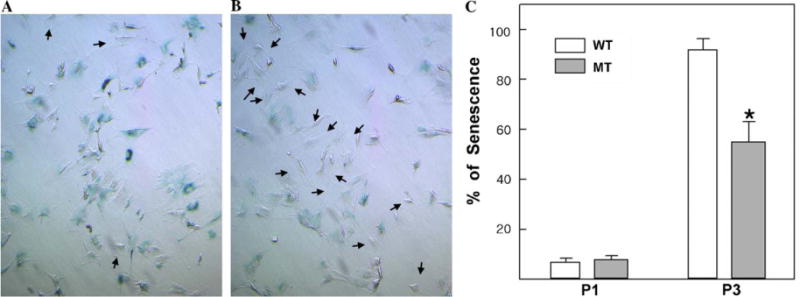
Senescence phenotype in elf−/− MEFs. Photographs of acidic β-galactosidase activity staining of wild type (A) and elf−/− (B) MEFs in passage 3. Arrows indicate the senescence-resistance cells. (C) To provide a quantitative comparison, X-gal stained passage 1 (P1) and passage 3 (P3) of wild type (WT) or elf−/− (MT) MEFs were counted. Statistically significant differences (P> 0.01) are indicated by asterisks.
Loss of ELF leads to an alteration of oncogenic and cell cycle proteins
To provide further insight into the progression to tumorigenesis, we generated immortalized cell lines from the primary MEFs and examined the expression pattern of regulatory protein by Western blot (Fig. 4A). elf mutant cells showed a higher expression of Cdk4 (183%) and Smad4 (570%), indicating abnormal cell cycle arrest and over-compensation of the TGF-β signaling pathway, respectively, during immortalization. In order to find out the relationship between loss of ELF and gastric malignancy, we examined the expressions of ELF, p53, and p21 in human cancer cell lines of gastric origin (Fig. 4B). Among the seven gastric cell lines, loss of ELF was found in two cell lines with stabilized but non-functional p53 which cannot transactivate the expression of p21. These results suggested that loss of ELF may require the inactivation of p53 pathway in the immortalization process to overcome p53-dependent tumor protection system including G1/S checkpoint and senescence.
Fig. 4.
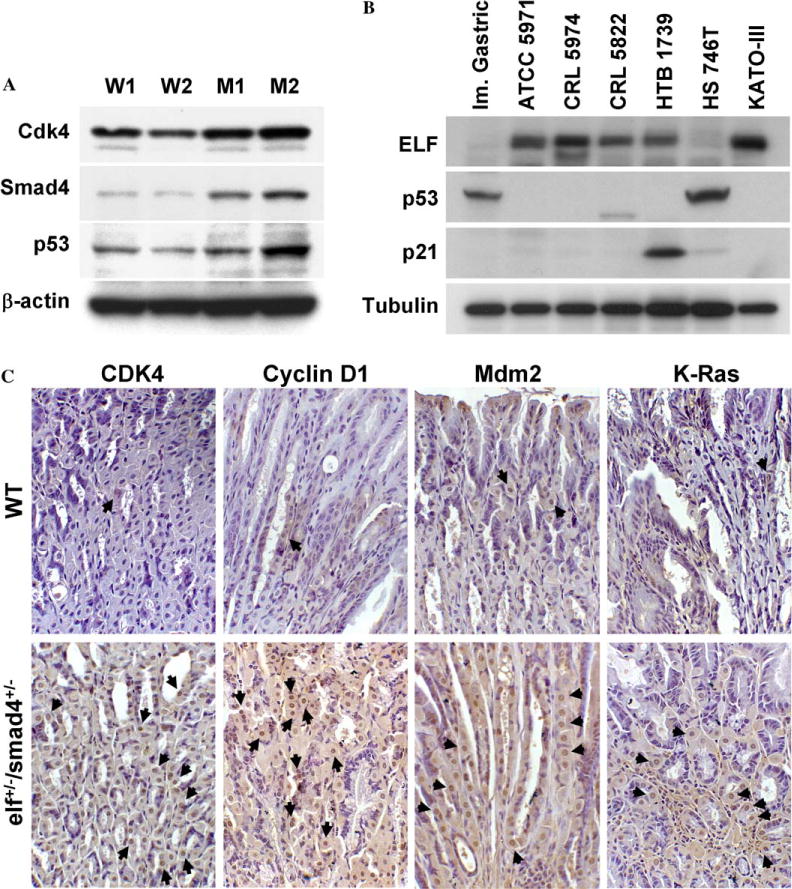
Loss of ELF leads to an alteration of oncogenic and cell cycle proteins in vitro and in vivo. Protein expression patterns in immortalized MEFs (A) and cancer cell lines (B). (C) Loss of elf increase in cyclin D1, Cdk4, Mdmd2, and K-Ras in elf+/−/smad4+/− mutant mice, as compared to wild type controls. Arrows indicate the positive signals.
To explore our hypothesis that cell cycle deregulation occurs due to loss of ELF, we examined the expression of Cdk4 and cyclin D1, G1/S checkpoint proteins, in hyperplastic gastric tissue of elf+/−/smad4+/− mice (Fig. 4C). Immunohistochemical analysis showed an increased expression of Cdk4 and cyclin D1 proteins in the gastric tissue of elf+/−/smad4+/− mice compared to the wild type. These results indicate that escaping from TGF-β growth inhibition results in increased cyclin D1 and activation of Cdk4 kinase activity. Next, to examine whether these tumors contain common genetic alterations or different secondary genetic events, we analyzed the expression of Mdm2 and K-Ras. Increased expression of Mdm2 and K-Ras was seen in elf+/−/smad4+/− gastric tissue compared to wild type controls. In summary, our results suggest that a mutation in the elf locus enables the cells to undergo faster entry into the S-phase of cell cycle, conferring resistance to senescence and apoptotic stimuli and susceptibility to transformation.
Discussion
The TGF-β pathway occupies a central position in the signaling networks that control growth, differentiation, and apoptosis [23]. TGF-β induces inhibition of cell cycle progression during the G1 phase through the control of Cdks. In mammalian cells, tightly regulated cyclins-Cdks act sequentially during the G1/S transition and are required for cell cycle progression through this period. Loss of TGF-β responsiveness results in deregulated cellular growth and is believed to be a crucial step in the development of various tumors, including gastric cancer [29]. We have previously noted a higher rate of gastric proliferation in elf+/−/smad4+/− mutant mice, suggesting that the disruption of TGF-β signaling noted in elf-deficient mice results in inhibition of growth arrest [18]. Our preliminary experiments show that elf+/−/smad4+/− and smad4+/− mice exhibit a tendency to develop tumors as a function of age. Importantly, smad4+/− mice are a well-established model for gastric carcinoma. smad4+/− mice develop fewer tumors at a later time point (45% at 12–14 months) than those of elf+/− smad4+/− animals (90% at 7–10 months). Our present work seeks to further elucidate the contribution of ELF in the enhanced susceptibility of the elf+/−/smad4+/− mice to the development of gastric tumors and determine the nature of secondary events that lead to tumor formation.
The mechanisms whereby TGF-β arrests the cell cycle have been studied mainly in epithelial cells, such as mink lung epithelial cells and human keratinocytes, focusing on the regulation of G1 cyclin-dependent kinases. In mink lung epithelial cells, TGF-β treatment induces inhibition of Cdk4 synthesis and Cdk2 inactivation and subsequent G1 arrest [30]. In HaCaT cells, TGF-β induces growth arrest through the downregulation of cell-cycle regulators, including cyclin E, cyclin A, Cdk2, and Cdk4 [31]. Several cyclin-dependent kinase (Cdk) inhibitors have been implicated in TGF-β induced cell cycle arrest. TGF-β binds to cyclin D/Cdk4, consequently releasing the p27 proteins from Cdk4, which then binds to Cdk2, blocking its activity [32–34]. Thus, sequential action results in inhibition of the cyclin-dependent kinases, which prevent progression of the G1 phase of the cell cycle. The Cdk inhibitor p21 has also been implicated in TGF-β mediated growth inhibition in many different cell types. Expression of p21 is induced by TGF-β through the transcription factors, Sp-1 and Sp-3, thus inhibiting the kinase activity of cyclin E/Cdk2 [33]. However, the fact that there exist multiple cell cycle regulators involved in TGF-β action prompts several questions relating to their actual roles in a certain cell type, whether they act via redundancy or in a coordinate manner, and whether their relative importance is largely common or different depending on cell type.
Our study specifically examines the role of ELF, a modulator of TGF-β signaling, in the dysregulation of cell cycle events leading to gastric cancer. Our results demonstrate a markedly higher expression of Cdk4 in elf−/− cells and elf+/−/smad4+/− mice, implying that TGF-β induced G1 arrest has been disrupted by the loss of ELF. The expression of p21 was also markedly decreased in elf−/− cells even in the presence of TGF-β. This is consistent with previous data in which p21 was demonstrated to be a major downstream target of TGF-β in gastric cancer cell lines [35]. TGF-β treatment of SNU16 gastric cancer cells induces expression of p21 with subsequent inhibition of G1-cyclin-associated Cdks [35]. Inhibition of G1-Cdk enzymatic activities results in under phosphorylation of p130, that binds to transcription factors such as the E2F family. Upregulation of p21 was shown to be sufficient to arrest the cells in G1 phase [35]. Furthermore, inhibition of p21 by anti-sense RNA results in the abrogation of sensitivity to TGF-β induced growth arrest [35]. Thus loss of the p21 gene or failure of p21 induction may contribute to TGF-β resistance in gastric carcinoma cells.
Senescence is a programmed cellular response in normal cells, the induction of which depends on the accumulated number of cell divisions [36]. Unlike cells undergoing apoptosis, senescent cells have a large and flat morphology, express acidic β-galactosidase, and show a permanent cell cycle G1 phase arrest [37]. Senescent cells contain an activated p53 transcription factor [38,39] and elevated levels of p21 [40]. The elevated level of p53 in response to multiple stress signals results in an increase in the transcription of the p21 gene. The p21 protein is a general inhibitor of CDKs, which phosphorylate Rb and allow G1 to S-phase progression [41,42]. In addition, p21 protein also inhibits PCNA-mediated DNA replication, a mechanism by which p21 functions as a growth inhibitor independent of its CDK inhibitor activity [43,44]. Taken together, these results indicate the possibility that the G1/S cell cycle checkpoints are critical controls for senescence. Our results demonstrated that the loss of ELF causes reduced senescence, and decreases the ability to abolish defective cells, leading to an accumulation of mutated cells that are more susceptible to tumorigenesis.
Our data suggest that loss of ELF also results in common genetic alterations or a group of diverse secondary genetic events such as stabilization of p53 and induction of oncogenic Ras. p53 and K-Ras genes are frequently mutated and stabilized in cancer [45,46]. K-Ras mutations are seen up to 28% of gastric carcinomas [47–49]. Our study demonstrates increased expression of p53 and K-Ras in elf−/− cells and elf+/−/smad4+/− gastric tissue, respectively. p21 is a downstream effector of activated K-Ras, and downregulation of p21 in elf mutated tissue may induce the development of tumors.
In summary, we have demonstrated abnormalities in the expression of several cell-cycle proteins in elf−/− cells and the gastric tissues of elf+/−/smad4+/− mutant mice. We also demonstrate that the loss of ELF leads to the abnormality of G1/S checkpoint and senescence. This, in turn, then leads to cancer development in mutant mice. Our data suggest that the disruptions in TGF-β signaling mediated by the loss of ELF result in profound and previously unrecognized interactions with cell-cycle regulation. We are thus provided with intriguing and potentially useful insights into the tumor biology of gastric cancers, and in the future may be able to utilize these cell cycle proteins in the early diagnosis and targeted therapeutics of these potentially lethal cancers.
Acknowledgments
This work was supported by NIH Grants RO1 CA106614A (L.M.), RO1 DK56111 (L.M.), RO1 DK58637 (B.M.), VA Merit Award (L.M.), and R. Robert and Sally D. Funderburg Research Scholar (L.M.).
References
- 1.Guilford P, Hopkins J, Harraway J, McLeod M, McLeod N, Harawira P, Taite H, Scoular R, Miller A, Reeve AE. E-cadherin germline mutations in familial gastric cancer. Nature. 1998;392:402–405. doi: 10.1038/32918. [DOI] [PubMed] [Google Scholar]
- 2.Goodman KJ, Cockburn M. The role of epidemiology in understanding the health effects of Helicobacter pylori. Epidemiology. 2001;12:266–271. doi: 10.1097/00001648-200103000-00023. [DOI] [PubMed] [Google Scholar]
- 3.Setiawan VW, Zhang ZF, Yu GP, Lu QY, Li YL, Lu ML, Wang MR, Guo CH, Yu SZ, Kurtz RC, Hsieh CC. GSTP1 polymorphisms and gastric cancer in a high-risk Chinese population. Cancer Causes Control. 2001;12:673–681. doi: 10.1023/a:1011261602940. [DOI] [PubMed] [Google Scholar]
- 4.Hatakeyama M. Oncogenic mechanisms of the Helicobacter pylori CAGA protein. Nat Rev Cancer. 2004;4:688–694. doi: 10.1038/nrc1433. [DOI] [PubMed] [Google Scholar]
- 5.Rhyu MG, Park WS, Meltzer SJ. Microsatellite instability occurs frequently in human gastric carcinoma. Oncogene. 1994;9:29–32. [PubMed] [Google Scholar]
- 6.Wu MS, Lee CW, Shun CT, Wang HP, Lee WJ, Chang MC, Sheu JC, Lin JT. Distinct clinicopathologic and genetic profiles in sporadic gastric cancer with different mutator phenotypes. Genes Chromosomes Cancer. 2000;27:403–411. [PubMed] [Google Scholar]
- 7.Kim YH, Lee HS, Lee HJ, Hur K, Kim WH, Bang YJ, Kim SJ, Lee KU, Choe KJ, Yang HK. Prognostic significance of the expression of Smad4 and Smad7 in human gastric carcinomas. Ann Oncol. 2004;15:574–580. doi: 10.1093/annonc/mdh131. [DOI] [PubMed] [Google Scholar]
- 8.Attisano L, Wrana JL. Mads and Smads in TGF β signaling. Curr Opin Cell Biol. 1998;10:188–194. doi: 10.1016/s0955-0674(98)80141-5. [DOI] [PubMed] [Google Scholar]
- 9.Heldin C-H, Miyazono K, ten Dijke P. TGF-β signaling from cell membrane to nucleus through Smad proteins. Nature. 1997;390:465–467. doi: 10.1038/37284. [DOI] [PubMed] [Google Scholar]
- 10.Shi Y, Massague J. Mechanisms of TGF-β signaling from cell membrane to nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- 11.Powell SM, Harper JC, Hamilton SR, Robinson CR, Cummings OW. Inactivation of Smad4 in gastric carcinomas. Cancer Res. 1997;57:4221–4224. [PubMed] [Google Scholar]
- 12.Weinstein M, Yang X, Deng C. Functions of mammalian Smad genes as revealed by targeted gene disruption in mice. Cytokine Growth Factor Rev. 2000;11:49–58. doi: 10.1016/s1359-6101(99)00028-3. [DOI] [PubMed] [Google Scholar]
- 13.Xu J, Attisano L. Mutations in the tumor suppressors Smad2 and Smad4 inactivate TGF-β signaling by targeting Smads to the ubiquitin-proteasome pathway. Proc Natl Acad Sci USA. 2000;97:4820–4825. doi: 10.1073/pnas.97.9.4820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miyazono K, Suzuki H, Imamura T. Regulation of TGF-β signaling and its roles in progression of tumors. Cancer Sci. 2003;94:230–234. doi: 10.1111/j.1349-7006.2003.tb01425.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ito K, Liu Q, Salto-Tellez M, Yano T, Tada K, Ida H, Huang C, Shah N, Inoue M, Rajnakova A, et al. Runx3, a novel tumor suppressor, is frequently inactivated in gastric cancer by protein mislocalization. Cancer Res. 2005;65:7743–7750. doi: 10.1158/0008-5472.CAN-05-0743. [DOI] [PubMed] [Google Scholar]
- 16.Tang Y, Katuri V, Dillner A, Mishra B, Deng CX, Mishra L. Disruption of TGF-β signaling in elf beta-spectrin deficient mice. Science. 2003;299:574–577. doi: 10.1126/science.1075994. [DOI] [PubMed] [Google Scholar]
- 17.Mishra L, Cia T, Yu P, Monger P, Mishra B. elf3 encodes a novel 200 KD β-spectrin: role in liver development. Oncogene. 1999;18:353–364. doi: 10.1038/sj.onc.1202313. [DOI] [PubMed] [Google Scholar]
- 18.Katuri V, Tang Y, Cuiling L, Jogunoori W, Deng CX, Rashid A, Sidawy A, Evans S, Mishra B, Mishra L. Critical interactions between TGF signaling/elf and E-Cadherin beta-catenin mediated tumor suppression. Oncogene. 2005 Epub ahead of print. [Google Scholar]
- 19.Redman RS, Katuri V, Tang Y, Dillner A, Mishra B, Mishra L. Orofacial and gastrointestinal hyperplasia and neoplasia in smad4+/− and elf+/−/smad4+/− mutant mice. J Oral Pathol Med. 2005;34:23–29. doi: 10.1111/j.1600-0714.2004.00246.x. [DOI] [PubMed] [Google Scholar]
- 20.Massague J, Blain SW, Lo RS. TGF-β signaling in growth control, cancer and heritable disorders. Cell. 2000;103:295–309. doi: 10.1016/s0092-8674(00)00121-5. [DOI] [PubMed] [Google Scholar]
- 21.Ko TC, Sheng HM, Reisman D, Thompson EA, Beauchamp RD. Transforming growth factor beta 1 inhibits cyclin D1 expression in intestinal epithelial cells. Oncogene. 1995;10:177–184. [PubMed] [Google Scholar]
- 22.Slingerland JM, Hengst L, Pan C, Alexander D, Stampfer MR, Reed SI. A novel inhibitor of cyclin-Cdk activity detected in transforming growth factor beta-arrested epithelial cells. Mol Cell Biol. 1994;14:3683–3694. doi: 10.1128/mcb.14.6.3683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Geng Y, Weinberg RA. TGF-beta effects on expression of G1 cyclins and cyclin-dependent protein kinases. Proc Natl Acad Sci USA. 1993;90:10315–10319. doi: 10.1073/pnas.90.21.10315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ewen ME, Sluss HK, Whitehouse LL, Livingston DM. TGF beta inhibition of Cdk4 synthesis is linked to cell cycle arrest. Cell. 1993;74:1009–1020. doi: 10.1016/0092-8674(93)90723-4. [DOI] [PubMed] [Google Scholar]
- 25.Koff A, Ohtsuki M, Polyak K, Roberts JM, Massague J. Negative regulation of G1 in mammalian cells: inhibition of cyclin E-dependent kinase by TGF-beta. Science. 1993;260:536–539. doi: 10.1126/science.8475385. [DOI] [PubMed] [Google Scholar]
- 26.Datto MB, Li Y, Panus JF, Howe DJ, Xiong Y, Wang XF. Transforming growth factor beta induces the cyclin-dependent kinase inhibitor p21 through a p53-independent mechanism. Proc Natl Acad Sci USA. 1995;92:5545–5549. doi: 10.1073/pnas.92.12.5545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hannon GJ, Beach D. p15INK4B is a potential effector of TGF-β induced cell-cycle arrest. Nature. 1994;371:257–261. doi: 10.1038/371257a0. [DOI] [PubMed] [Google Scholar]
- 28.Polyak K, Kato JY, Solomon MJ, Sherr CJ, Massague J, Roberts JM, Koff A. p27Kip1, a cyclin cdk inhibitor links TGF beta and contact inhibition to cell cycle arrest. Genes Dev. 1994;8:9–22. doi: 10.1101/gad.8.1.9. [DOI] [PubMed] [Google Scholar]
- 29.Gold LI. The role for transforming growth factor beta in human cancer. Crit Rev Oncog. 1999;10:303–360. [PubMed] [Google Scholar]
- 30.Laiho M, Decaprio JA, Ludlow JW, Livingston DM, Massague J. Growth inhibition by TGF-beta linked to suppression of retinoblastoma-protein phosphorylation. Cell. 1990;62:175–185. doi: 10.1016/0092-8674(90)90251-9. [DOI] [PubMed] [Google Scholar]
- 31.Datto MB, Wang X-F. Functional analysis of the TGF-β responsive elements in the WAF1/Cip1/p21 promoter. J Biol Chem. 1995;270:28623–28628. doi: 10.1074/jbc.270.48.28623. [DOI] [PubMed] [Google Scholar]
- 32.Gartenhaus RB, Wang P, Hoffmann P. Induction of the WAF1/CIP1 protein and apoptosis in human T-cell leukemia virus type 1 transformed lymphocytes after treatment with adriamycin by using a p53 independent pathway. Proc Natl Acad Sci USA. 1996;93:265–268. doi: 10.1073/pnas.93.1.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reynisdottir I, Polyak K, Iavarone A, Massague J. Kip/Cip and Ink4 Cdk inhibitors cooperate to induce cell-cycle arrest in response to TGF-β. Genes Dev. 1995;9:1831–1845. doi: 10.1101/gad.9.15.1831. [DOI] [PubMed] [Google Scholar]
- 34.Li JM, Nichols MA, Chandrasekharan S, Xiong Y, Wang XF. TGF-β activates the promoter of cyclin dependent-kinase inhibitor p15INK4B through a Spl consensus site. J Biol Chem. 1995;270:26750–26753. doi: 10.1074/jbc.270.45.26750. [DOI] [PubMed] [Google Scholar]
- 35.Yoo YD, Choi JY, Lee SJ, Kim JS, Min BR, Lee YI, Kang YK. TGF-β induced cell-cycle arrest through the p21 WAF/CIP1-G1 cyclin/CDKS-p130 pathway in gastric carcinoma cells. Int J Cancer. 1999;83:512–517. doi: 10.1002/(sici)1097-0215(19991112)83:4<512::aid-ijc13>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 36.Campisi J. Cancer, aging and cellular senescence. In Vivo. 2000;14:183–188. [PubMed] [Google Scholar]
- 37.Sherwood SW, Rush D, Ellsworth JL, Schimke RT. Defining cellular senescence in IMR-90 cells: a flow cytometric analysis. Proc Natl Acad Sci USA. 1988;85:9086–9090. doi: 10.1073/pnas.85.23.9086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kulju KS, Lehman JM. Increased p53 protein associated with aging in human diploid fibroblasts, Exp. Cell Res. 1995;217:336–345. doi: 10.1006/excr.1995.1095. [DOI] [PubMed] [Google Scholar]
- 39.Atadja P, Wong H, Garkavtsev J, Veillette C, Riabowol K. Increased activity of p53 in senescing fibroblasts. Proc Natl Acad Sci USA. 1995;92:8348–8352. doi: 10.1073/pnas.92.18.8348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Noda A, Ning Y, Venable SF, Pereira-Smith OM, Smith JR. Cloning of senescent cell-derived inhibitors of DNA synthesis using an expression screen. Exp Cell Res. 1994;211:90–98. doi: 10.1006/excr.1994.1063. [DOI] [PubMed] [Google Scholar]
- 41.Sherr CJ, Roberts JM. Inhibitors of mammalian G1 cyclin-dependent kinases. Genes Dev. 1995;9:1149–1163. doi: 10.1101/gad.9.10.1149. [DOI] [PubMed] [Google Scholar]
- 42.Harper JW, Adami GR, Wei N, Keyomarsi K, Elledge SJ. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell. 1993;75:805–816. doi: 10.1016/0092-8674(93)90499-g. [DOI] [PubMed] [Google Scholar]
- 43.Waga S, Hannon GJ, Beach D, Stillman B. The p21 inhibitor of cyclin-dependent kinases controls DNA replication by interaction with PCNA. Nature. 1994;369:574–578. doi: 10.1038/369574a0. [DOI] [PubMed] [Google Scholar]
- 44.Li R, Waga S, Hannon GJ, Beach D, Stillman B. Differential effects by the p21 CDK inhibitor on PCNA-dependent DNA replication and repair. Nature. 1994;371:534–537. doi: 10.1038/371534a0. [DOI] [PubMed] [Google Scholar]
- 45.Merritt AJ, Potten CS, Kemp CJ, Hickman JA, Balmain A, Lane DP, Hall PA. The role of p53 in spontaneous and radiation-induced apoptosis in the gastrointestinal tract of normal and p53 deficient mice. Cancer Res. 1994;54:614–617. [PubMed] [Google Scholar]
- 46.Levine AJ, Momand J, Finlay CA. The p53 tumour suppressor gene. Nature. 1991;351:453–456. doi: 10.1038/351453a0. [DOI] [PubMed] [Google Scholar]
- 47.Brembeck FH, Schreiber FS, Deramaudt TB, Craig L, Rhoades B, Swain G, Grippo P, Stoffers DA, Silberg DG, Rustgi AK. The mutant K-ras oncogene causes pancreatic periductal lymphocytic infiltration and gastric mucous neck cell hyperplasia in transgenic mice. Cancer Res. 2003;63:2005–2009. [PubMed] [Google Scholar]
- 48.Hongyo T, Buzard GS, Palli D, Weghorst CM, Amorosi A, Galli M, Caporaso NE, Jr, Fraumeni JF, Rice JM. Mutations of the K-ras and p53 genes in gastric adenocarcinomas from a high-incidence region around Florence, Italy. Cancer Res. 1995;55:2665–2672. [PubMed] [Google Scholar]
- 49.Yoo J, Park SY, Robinson RA, Kang SJ, Ahn WS, Kang CS. Ras Gene mutations and expression of Ras signal transduction mediators in gastric adenocarcinomas. Arch Pathol Lab Med. 2002;126:1096–1100. doi: 10.5858/2002-126-1096-RGMAEO. [DOI] [PubMed] [Google Scholar]