

Abstract
Under the influence of a chiral bidentate diphosphine ligand, the Pd-catalyzed asymmetric cross-coupling of allylboron reagents and allylic electrophiles establishes 1,5-dienes with adjacent stereocenters in high regio- and stereoselectivity. . A mechanistic study of the coupling utilizing reaction calorimetry and density functional theory analysis suggests that the reaction operates through an inner-sphere 3,3'-reductive elimination pathway, which is both rate- and stereodefining. Coupled with optimized reaction conditions, this mechanistic detail is used to expand the scope of allyl-allyl couplings to allow the generation of 1,5-dienes with tertiary centers adjacent to quaternary centers as well as a unique set of cyclic structures.
1. INTRODUCTION
The development of highly efficient and selective methods for the construction of new stereocenters is of paramount importance for the continued evolution of organic synthesis.1 Among such developments, palladium-catalyzed allylic substitutions have provided important strategies for selective formation of stereogenic centers by the construction of new C-C or C-X bonds.2 A related process is the coupling of two allylic systems, such as that between an allylmetal reagent (A) and an allyl electrophile (B), to deliver synthetically useful 1,5-diene products (Scheme 1, eq 1).3 In this context, opportunities for stereoselective synthesis emerge when (1) one or both of the allyl fragments are substituted, (2) regioselectivity can be suitably controlled to favor formation of branched 1,5-dienes of type D, and (3) a chiral catalyst is able to enforce stereocontrol during coupling of the prochiral allyl fragments. Under these conditions, allyl-allyl cross-couplings have the power to establish one or two adjacent stereogenic sp3-carbon centers.
Scheme 1. Pd-Catalyzed Allyl-Allyl Couplings.
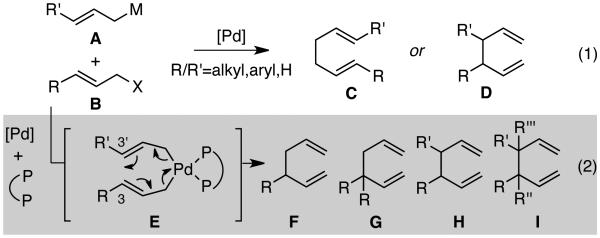
Although efficient methods for the Pd-catalyzed allyl-allyl cross-coupling have been developed, regioselectivity generally favors the thermodynamically more stable achiral linear products of type C.4,5 Recently, however, our group has developed a number of related palladium-catalyzed allyl-allyl cross-couplings that selectively provide the branched isomer of product in high regio- and enantioselectivity.6 A critical design feature is the use of a chiral small bite-angle bidentate phosphine ligand, which appears to promote coupling through an inner-sphere 3,3'-reductive elimination pathway shown in structure E (Scheme 1, eq 2). The coupling applies to substrates bearing substitution on both the allylmetal and the allyl electrophile and thereby allows for the synthesis of 1,5-dienes containing tertiary (F), quaternary (G) and adjacent tertiary (H) stereogenic centers.7
A primary challenge has been the use of more highly substituted coupling partners that allow generation of products containing enhanced steric congestion. We considered that construction of 1,5-dienes bearing adjacent quaternary centers or those with neighboring tertiary and quaternary centers (I) may have significant value in organic synthesis.8 While compounds with such congestion may be accessed by asymmetric intramolecular Heck reactions and cycloadditions, there remain relatively few other strategies for the catalytic asymmetric construction of these compounds.9,10 In this report we describe mechanistic studies that have led to the development of an improved strategy for allyl-allyl cross couplings, particularly those that apply to the selective and efficient construction of more hindered C-C bonds.
2. RESULTS AND DISCUSSION
2.1 Catalyst Optimization
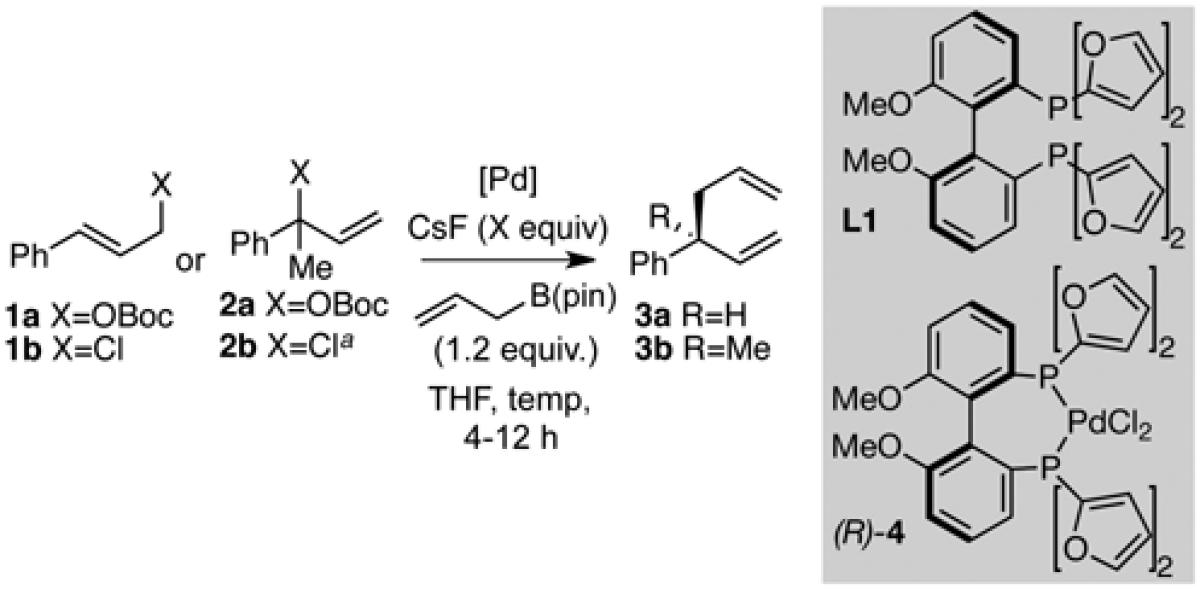
Despite high regio- and stereoselectivity obtained in previous couplings with a catalyst prepared from Pd2dba3 and methoxy(furyl)biphep (L1, Table 1), one drawback was the requirement for relatively high catalyst loadings and long reaction times. For the relatively unhindered coupling of cinnamyl t-butyl carbonate 1a and allylB(pin) to give 3a, 10 % palladium loading and a 12 hour reaction time was necessary for full conversion (Table 1, entry 1). It was suspected that dibenzylideneacetone (dba), a part of the original catalyst formulation, might compete with substrates for binding to palladium and inhibit the reaction.11 To study this feature, the use of a pre-formed PdCl2-ligand complex (R)-4 was evaluated in the reaction. While (R)-4 alone was unable to catalyze the reaction between allylB(pin) and 1a (entry 2), addition of 5% CsF resulted in catalyst activation such that only 0.1 mol % catalyst was required to effect complete conversion of substrates in just 4 hours at 60 °C (entry 3). Remarkably, despite this 100-fold decrease in catalyst loading, the reaction was accompanied by an increase in yield (95% versus 72%) and showed no loss of enantioselectivity. The cesium fluoride is presumed to be necessary for the initial activation of allylB(pin), which is responsible for reduction of (R)-4 to an active Pd(0) catalyst.12
Table 1.
Optimized Catalyst for Allyl-Allyl Couplings
| |||||||
|---|---|---|---|---|---|---|---|
| entry | catalyst | substrate | CsF (equiv) |
t (h) | T (°C) | yield (%)b |
erc |
| 1 | 5% Pd2(dba)3
10% L1 |
1a | 0 | 12 | 60 | 72 | 96:4 |
| 2 | 0.25% (R)−4 | 1a | 0 | 8 | 60 | 0 | - |
| 3 | 0.1% (R)−4 | 1a | 0.05 | 4 | 60 | 95 | 96:4 |
| 4d | 0.1% (R)−4 | 1a | 0.05 | 4 | 60 | 96 | 96:4 |
| 5 | 0.1% (R)−4 | 1b | 3 | 5 | rt | 92 | 97:3 |
| 6e | 2% Pd2(dba)3
4% L1 |
2a | 3 | 12 | 60 | 86 | 96:4 |
| 7e | 0.25% (R)−4 | 2a | 3 | 4 | 60 | 59 | 94:6 |
| 8e | 0.25% (R)−4 | 2b | 5 | 5 | rt | 85 | 98:2 |
a The linear isomer of chloride was used.
Yield of purified material, average of two or more experiments.
Enantiomer ratios were determined by GC analysis on a chiral stationary phase and are an average of two or more experiments.
Reaction set up on benchtop and purged with N2.
Reaction run in 10:1 THF/H2O.
An important added advantage of (R)-4 is that the preformed catalyst is air-stable, allowing the reaction to be set up without the use of a dry-box (entry 4). With the more active catalyst, it was found that reactions of cinnamyl chloride 1b could be conducted at room temperature, leading to a slight increase in enantioselectivity (entry 5). Similar reactivity enhancements were observed when catalyst (R)-4 was employed in the cross-coupling to give products containing all-carbon quaternary centers. For the cross-coupling of allylB(pin) and disubstituted 2a to give 3b, catalyst loadings could be lowered from 4 to 0.25% and the reaction was also complete in 4 hours (entries 6 vs. 7). While an increase in competitive β-hydride elimination was noted at the lower catalyst loadings with carbonate 2a, leading to a decrease in yield (59 versus 86%), both yield and enantioselectivity were improved with use of allylic chloride 2b at room temperature (entry 8).
The differences in catalyst performance between (R)-4 and Pd2(dba)3/(R)-L1 can be seen clearly when the rate of coupling between 1a and allylB(pin) were examined by calorimetry (Figure 1). While the total heat outputs are similar, the reaction with 0.1% of (R)-4 is significantly faster and completes within 2.5 hours (blue line) while the reaction with Pd2(dba)3/(R)-L1 (10 % Pd loading) was less rapid, requiring almost six hours to reach completion despite the 100-fold increase in catalyst loading (red line). Suspecting the probable cause for these differences to be the presence of dba when Pd2(dba)3 is used as a precursor, a reaction of (R)-4 in the presence of added dba was examined. When 15 mol % dba was added to a reaction with 0.1% of (R)-4 (green line), the reaction showed a heat profile that was very similar to the Pd2(dba)3/L1 system. This observation suggests that dba is a non-innocent participant in the reaction, and likely sequesters the catalyst in off-cycle processes.11
Figure 1.

Reaction rates with 0.1% (R)-4 (blue line), 5% Pd2(dba)3 + 10% (R)-L1 (red line), and 0.1% (R)-4 + 15% dba (green line) under standard conditions as described in Table 1.
2.2. Mechanistic Analysis
To better understand the operative mechanism and the origins of regio- and stereocontrol present in the allyl-allyl coupling, mechanistic and computational studies were carried out. Due to its clean and efficient nature, the reaction of cinnamyl t-butyl carbonate (1a) and allylB(pin) to give 3a was chosen as a probe for these studies.
2.2.1. Reaction Progress Kinetic Analysis
Reaction progress kinetic analysis of the model reaction was carried out using calorimetry to follow rates (Figure 2).13 Conditions optimized in Table 1 ([1a] = 0.5 M, [allylB(pin) = 0.6 M], 0.1 mol % (R)-4, 5.0 mol % CsF, 60 °C in THF) were used to provide an accurate picture of the catalytic reaction under conditions employed for typical experiments. Under these conditions, the reaction exhibited the rate versus [1a] profile as shown in A (blue line), which was verified by comparison to NMR analysis of reaction progress.14
Figure 2.

Reaction progress kinetic analysis of the Pd-catalyzed coupling of allylB(pin) and allyl carbonate 1a with (R)-4 as described in Table 1. (A) Reaction rate versus [1a] for 0.1% (R)-4 (blue line) and 0.05% (R)-4 (red line). (B) Heat flow versus time for the standard amount (0.5 M, blue line) versus excess (1.0 M, red line) of 1a. (C) Heat flow versus time for the standard amount (0.6 M, blue line) versus excess (1.2 M, red line) of allylB(pin). (D) Reaction rate versus [1a] for the standard (0.5 M 1a, 0.6 M allylB(pin), ‘excess’ allylB(pin)= 0.1 M, blue line) versus modified initial concentrations (0.3 M 1a, 0.4 M allylB(pin), ‘excess’ allylB(pin)= 0.1 M, red line).
Comparison of reaction rates versus allyl carbonate concentration during the steady-state portion for reactions run with different catalyst loadings (0.1 and 0.05%, A) suggests that the reaction is first-order in catalyst. As depicted in panels B and C, the heat output profile is relatively unchanged when the concentrations of either the carbonate (B), or the allylboron (C) are doubled, suggesting a zero-order dependence on both substrates.15 As demonstrated in D, beginning the reaction with the initial concentrations of allylB(pin) and carbonate at 0.4 and 0.3 M respectively gives a slightly lower rate overall as compared to the standard curve (blue). As both reactions have the same relative proportions of reagents (‘excess’ allylB(pin) = 0.1 M), this study indicated that catalyst deactivation or product inhibition are not complicating factors of the reaction, but could point to a possible acceleration by product or a reaction byproduct.
2.2.2. Proposed Catalytic Cycle
With kinetic studies suggesting a zero-order dependence on both substrates and first order dependence on catalyst, we propose the cycle as pictured in Scheme 2. The cycle is initiated by transmetallation of the fluoride-activated allylB(pin) to (R)-4, which allows reductive elimination to yield the active Pd(0) catalyst K. Oxidative addition of Pd with the allyl carbonate yields the cationic (η3-allyl)Pd(II) complex L, accompanied by loss of the carbonate group which quickly breaks down into CO2 and tert-butoxide.16 The tert-butoxide activates the allylB(pin) towards transmetallation to give the bis(η1-allyl) M. 3,3'-Reductive elimination from M will yield the product-bound Pd complex J, from which the catalyst reenters the cycle upon displacement of the product by starting material.3,5d,e
Scheme 2. Proposed Catalytic Cycle.

The kinetic data suggests that the order of the reaction is zero in both carbonate and allylB(pin), signaling that the rate-determining step is neither oxidative addition nor transmetallation. As the reaction also seems to suffer no product inhibition, it is unlikely that catalyst turnover is rate-determining. These results suggest that the 3,3'-reductive elimination could be the rate-determining step.
2.2.3. Isotope Labeling Studies
The stereochemical features of reductive elimination and transmetallation are readily studied with isotope labeling experiments (Scheme 3). Information about the nature of reductive elimination has been previously gained and is represented here for completeness.6a For this study, substrate (S)-Z-5 was prepared and used in the allyl-allyl cross coupling with allylB(pin) and (R)-L1 to give (S)-E-6 as the sole coupling product (eq 3). This outcome can be explained as follows: oxidative addition via an anti displacement of the carbonate gives intermediate π-allyl complex N.17 Subsequent π-σ isomerization followed by transmetallation gives bis(trans-η1-allyl) complex O. From O, an innersphere reductive elimination leads to the observed product configuration.
Scheme 3. Labeling Studies of Reaction Stereochemistry.

A similar study was also designed to examine the stereochemical aspects of the transmetallation. Allylmetal reagents may undergo SE2' transmetallation through either an anti (open) pathway or a syn (closed) pathway, the latter characterized by coordination between cationic Pd(II) and the allylmetal.18 To distinguish between these two pathways in allyl-allyl couplings, isotopically labeled (S)-E-7 was synthesized and subjected to conditions for diastereoselective allyl-allyl cross-coupling. With (R)-L1 as ligand and 1a as the electrophile, the coupling gave (R,R)-E-8 as the major stereoisomer (eq 4). This outcome suggests that the SE2' anti (open) pathway is favored: (R,R)-E-8 is anticipated after 3,3'-reductive elimination from bis(η1-allyl) complex R. Complex R is accessed via σ-π-σ isomerization of intermediate Q, which is formed after an SE2' anti transmetallation as shown in P.19 Similar product stereochemistry was also observed when the reaction was run with allylic chloride 1b, suggesting that the nature of the counterion has little effect on the preferred mode of transmetallation. Further studies also indicated that the anti pathway was also favored in the absence of CsF as well as when the reaction was run in 10:1 THF/H2O.20 Although this study does not differentiate between SE2' and direct SE2 transmetallation, later experimental results suggest that the SE2' operates (vide infra).
2.2.4 Computational Study of Reaction Mechanism
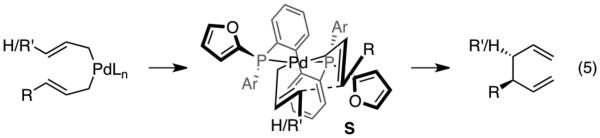
A model that explains the stereochemical outcome of allylallyl coupling is depicted in Scheme 4.6c This is based on an inner-sphere 3,3'-reductive elimination and uses the crystal structure of (R)-421 as a template for the ligand conformation. Central to this model is reaction through bis(η1-allyl)palladium with the allyl groups directed anti to one another in a chair-like array (S, Scheme 4). In this arrangement, the preferred chair-like transition state array is dictated by the pseudoequatorial furyl rings of the ligand scaffold.
Scheme 4. Stereochemical Model for 3,3'-Reductive Elimination.
To probe features of this stereochemical model as well as gain additional understanding of the energetic barriers associated with allyl-allyl coupling, density functional theory (DFT) calculations were carried out.22 Extensive computational studies on the 3,3'-reductive elimination pathway for simple unsubstituted systems with achiral monodentate phosphine ligands were completed by Echavarren and Espinet.23 We focused our analysis on the portion of the reaction coordinate essential to regio- and stereodefinition during formation of the branched (S)-enantiomer of 3a rather than (R)-3a or the linear isomer (the Si-, Re- or linear-pathways, respectively).24,25,26,27,28 Efforts to distinguish between possible reaction pathways began from cationic palladium π-allyl complex GS1 and the activated boronate T (Figure 3). Transmetallation of the allyl unit from boron to the Pd center would initiate the inner-sphere pathway, whereas direct outer-sphere attack at C1 or C3 of the Pd π-allyl would furnish the diene products directly.
Figure 3.

DFT computed potential energy surface to distinguish between the inner- and outer sphere pathways as well as the preferred mode of reductive elimination to give experimentally observed (S)-3a, (Si-pathway, blue), (R)-3a, (Re-pathway, red), or linear product (green). All calculations performed at the B3LYP-PCM(THF)/LANL2DZ-6-31G** level of theory, some hydrogen atoms were removed from the displayed structures for clarity. Energies relative to GS1 + T.
In agreement with the mechanistic experiments, transmetallation through an anti-SE2' (open) pathway (TSTM 1) is calculated to have the lowest free energy of activation, 11.8 kcal/mol with respect to GS1 and T. Reaction through a syn-SE2' (closed) transition state (TSTM 2) in which the methoxide oxygen of boronate T is coordinated to Pd, was calculated to be 5.7 kcal/mol higher in energy.29,30 The three transition states for outer-sphere attack of allylboronate T directly at C1 or C3 of the η3-allyl complex (TSO.S) require activation free energies that are 9.8-14.2 kcal/mol higher than TSTM 1. Additionally, the relative energies between the three outer-sphere pathways predict both regio- and stereochemistry that is contrary to experimental results.
Transmetallation of the allyl unit to the Pd center leads to the bis(η1-allyl)Pd(II) intermediate GS2, from which reductive elimination will furnish branched (3a) and linear products. Previous computational studies by Eschavarren and Espinet on unsubstituted bis(η1-allyl)Pd complexes found that when the complex contains two monodentate phosphine ligands, reductive elimination by coupling the 3 and 3'-carbons was favored significantly compared to direct coupling of the 1 and 1'- carbons.31 As shown in figure 3, the calculated energy difference between the lowest energy conformations for 3,3'- (TS3,3'-Si A) and 1,1'-reductive elimination (TS1,1') of 17.2 kcal/mol is in good agreement with the literature despite the addition of substitution at C3.
In line with our previously proposed stereochemical model, the lowest energy transition state array for 3,3'-reductive elimination (TS3,3'-Si A) is through a chair-like transition state with trans olefin geometry at the substituted allyl fragment (A, Figure 4). Bond formation from this structure leads to the experimentally observed (S)-enantiomer of 3a. Consistent with the crystal structure, the preferred conformation of C2-symmetric (R)-L1 places pseudoequatorial furyl rings in the upper left and lower right quadrants about the bound allyl fragments. We propose the high levels of stereoinduction during reductive elimination occurs due to penalizing interactions between the pseudoequatorial furyl rings with C1 and C2 of each allyl fragment in less favored structures. This interaction favors one chair-like conformer over the other, favoring reaction through TS3,3'-Si A.
Figure 4.

(A) Optimized structure and coordinates for TS3,3’ Si A (B) Activation energies relative to GS2 for other possible allyl conformations during 3,3’-reductive elimination. B3LYP-PCM(THF)/LANL2DZ-6-31G** (333.15 K).
There are seven additional transition states through which the 3,3'-reductive elimination can occur, four of which are relatively close in energy and considered in Panel B of Figure 4.32 Reaction through boat-like TS3,3'-Si B, with the allyl groups syn to each other, could also give (S)-3a, however the activation free energy for this path was calculated to be 4.6 kcal/mol higher in energy. In considering conformers leading to the minor enantiomer (Re pathway), the other possible chair-like arrangement (TS3,3' Re a) is 3.5 kcal/mol higher in energy than TS3,3'-Si A as it places C1 and C2 of both allyl fragments in close proximity to the furyl rings. Also similar in energy are boat-like TS3,3' Re b, and TS3,3' Re c, that bearing Z-alkenes. It is not possible to say which of the Re conformers is preferred because of their similar activation barriers, however the difference in activation barriers between TS3,3'-Si A and any of the TS3,3‘ Re conformers are in line with the high enantioselectivities observed experimentally.
2.3. Formation of Congested C-C Bonds
To begin study of the application of catalyst (R)-4 to more hindered allyl-allyl couplings, allylboron 9 was treated with electrophile 1b (eq 6, Scheme 5). Despite increased catalyst loading, the coupling showed only 17% conversion and delivered none of the desired branched product 10. The product mixture consisted of allylbenzene 11 and undesired γ-regioisomer 12. The undesired products likely stem from the bis(η1-allyl)Pd isomers U or V (Scheme 5). From U, β-hydride elimination followed by reductive elimination will yield 11 (eq 7), whereas 3,3'-reductive elimination from V could yield 12 (eq 8).
Scheme 5. Initial Results for Congested Bond Formations.

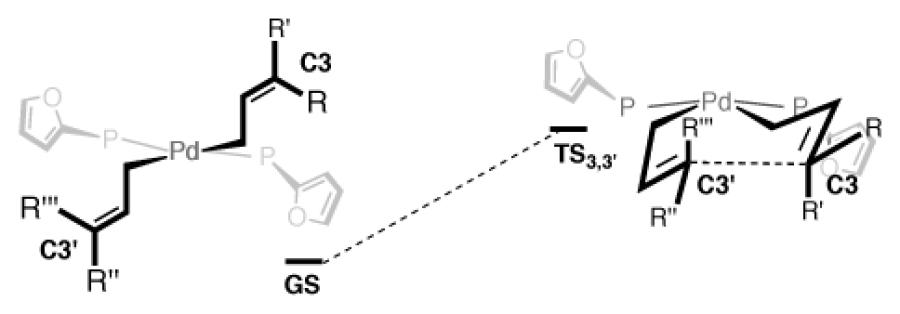
It was postulated that the added substitution at C3' during the 3,3'-reductive elimination might raise the activation barrier for the desired reaction, allowing other pathways to become operative. To probe this possibility, DFT calculations were carried out to determine the effect of added substitution at the 3 and 3' carbons on the calculated energy barrier for reductive elimination (Table 2). To allow comparison, activation barriers for the 3,3'-reductive elimination were calculated for three compounds that have proven accessible through allyl-allyl coupling (13-15), and three classes of more hindered couplings (16-18).
Table 2.
Calculated Activation Free Energies for 3,3’-Reductive Elimination for Hindered Couplings
| |||||
|---|---|---|---|---|---|
| Successful Couplings | Hindered Couplings | ||||
|
|
||||
| 13 | 14 | 15 | 16 | 17 | 18 |
| ΔG* = 16.0 | ΔG‡ = 17.4 | ΔG‡ = 20.0 | ΔG‡ = 20.6 | ΔG‡ = 19.9 | ΔG‡ = 28.0 |
As anticipated, the calculations showed higher activation free energy with increased substitution at the C3 and C3' carbons. Addition of one methyl group at C3 (13) shows a free energy of activation of 16.0 kcal/mol, an increase of 2.4 kcal/mol over the monosubstituted 3a (ΔG‡=13.6). The barrier is raised to 17.4 kcal/mol when the methyl substituent is placed at C3' (14), and increases by an additional 2.6 units to 20.0 kcal/mol with the larger phenyl substituent (15). The calculated activation energies that would be required for 3,3'-reductive elimination to give products 16-18 show a similar trend. Importantly, the barrier to give 16, a compound inaccessible from the reaction in Scheme 5, is only slightly higher (0.6 kcal/mol) than the barrier to give 15, a compound previously prepared in 80% yield with high selectivity.6c
The discrepancy between experimental and computational results suggested that, for the mechanism involving disubstituted allylboron 9 (Scheme 5), 3,3'-reductive elimination may no longer be the rate-determining step. As seen in Figure 5, the addition of two methyl groups at terminus of the activated boronate W has a significant impact on the calculated barrier for SE2' transmetallation. In comparison to the 12.3 kcal/mol barrier for simple allylB(pin), the additional substituents on 9 raise the activation free energy barrier to 30.8 kcal/mol.30 Compared with the barrier for reductive elimination (20.6 kcal/mol, 16, Table 2), transmetallation is now predicted to be the rate-limiting step.
Figure 5.
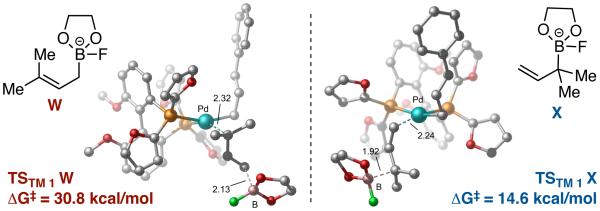
Calculated activation free energy for transmetallation of fluoride-activated 9 (W, left) and 19 (X, right) to GS1. B3LYP-PCM(THF)/LANL2DZ-6-31G** (298.15K).
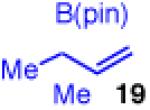
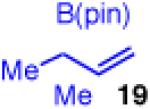
The calculations indicate that modification of the nucleophile could allow for a better reaction. Indeed, if the two methyl groups are moved to the α-position of the allylboron as in X (Figure 5), the calculated energy barrier for transmetallation drops by over 16 kcal/mol, and reductive elimination is again predicted to be the rate determining step. To probe this hypothesis, allyl boronate 19 was prepared and analyed in the cross-coupling with cinnamyl chloride (Scheme 6). This strategy showed a strikingly different outcome.. The coupling now delivers compound 10 in excellent yield, regio- and enantioselectivity.33
Scheme 6. Increased Reactivity with α,α-Disubstituted Allylboron Nucleophiles.
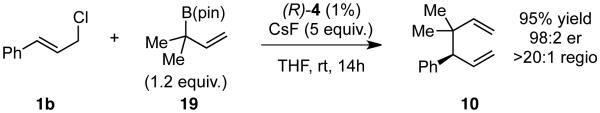
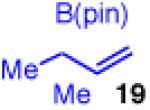
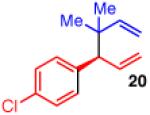
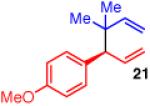
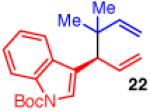
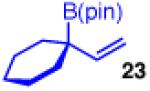
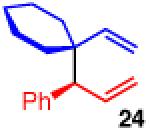
To learn more about the newly modified conditions, a survey of additional coupling partners was undertaken (Table 3). Allyl boronate 19 was effective in couplings with substrates bearing electronically diverse substituents; use of cinnamyl chlorides with para-chloro (entry 1) and para-methoxy (entry 2) groups gave products 20 and 21 in similarly high yields, regio- and enantioselectivities to the parent phenyl compound. Despite lowered regioselectivity, the indole-containing substrate in entry 3 reacts with high enantioselectivity as well. Remarkably, the reaction is also operative when the two sub-stituents are tethered as in 23, giving the 1,1-disubstituted cyclohexane product 24 in high selectivity and yield (entry 4). Although not stereogenic, the reverse prenyl groups in products 10 and 20-22 are a common motif in alkaloid natural products.34
Table 3.
Substrate Scope for Congested Couplingsa
| |||||
|---|---|---|---|---|---|
| entry | allyl boronate |
product | yield (%) |
regio | er |
| 1 |
|
|
94 | >20:1 | 98:2 |
| 2 |
|
|
90 | >20:1 | 98:2 |
| 3 |
|
|
92 | 5:1 | 96:4 |
| 4 |
|
|
85 | >20:1 | 97:3 |
| 5 |
|
|
96 | >20:1 | 98:2 (2:1 dr) |
| 6b |
|
|
92 | >20:1 | 98:2 (3:1 dr) |
| 7c |
|
|
74 | 5:1 | >99:1 (4:1 dr) |
| 8d |
|
|
90 | >20:1 | 99:1 er (3:1 dr) |
| 9 |
|
|
<5 | - | - |
| 10 |
|
|
<5 | - | - |
| 11e |
|
|
<5 | - | - |
Unless otherwise noted reactions were run at 0.5 M concentration of the electrophile with 1.2 equiv of the allylboron reagent. Yield refers to isolated yield of purified material. Enantiomer ratios were determined by GC or SFC analysis on a chiral stationary phase. Diastereomer ratios were determined by 1H NMR analysis. All yield, er and dr values reported are the average of two or more experiments.
Reaction run with 10 equiv CsF for 24 h.
Reaction run in 10:1 THF/H2O.
Reaction run at 60 °C, 0.1% catalyst loading with 0.05 equiv CsF for 4 h.
Reaction run in 10:1 THF/H2O with 1% (S,S)-QuinoxP*PdCl2 in place of (R)−4.
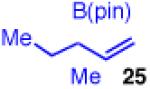
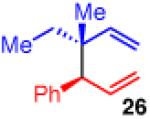
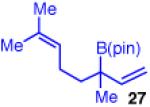
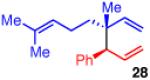
The use of allylboron reagents in which the substitution at the α-carbon is non-symmetric would allow access to adjacent stereocenters. When the allylboron is substituted with one methyl and one ethyl group as in 25 (entry 5, Table 3), the coupling gives product 26 in high yield, regio- and enantioselectivity, but in moderate diastereoselectivity. The selectivity is slightly increased as the size of the substituent is increased, such as with the geraniol-derived allylboron 27; this coupling gives 28 with similarly high enantio- and regioselectivity (entry 6).
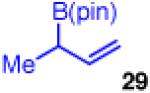
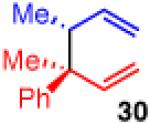
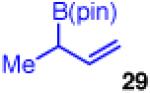
The coupling also furnishes products with adjacent tertiary/quaternary centers when monosubstituted 29 and disubstuted chloride 2b are employed (entry 7). The reaction shows high enantioselectivity and moderate diastereoselectivity, however, the yield suffers slightly due to competitive β-hydride elimination. Use of monosubstituted 29 also allows the previously reported diastereoselective coupling to operate with reduced catalyst and CsF loadings, showing similar yield and enantioselectivity (entry 8).35
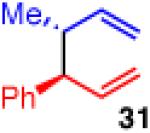
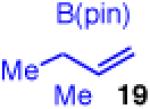
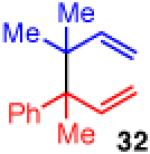
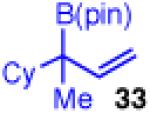
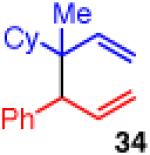
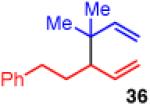
Unfortunately, extension to the construction of products with two adjacent quaternary centers (as in 32) through the coupling of 19 and 2b failed (entry 9). Despite complete conversion, the reaction delivered a mixture of β-hydride elimination product and γ-regioisomer.36 A more encumbered allylboron (33) was prepared, and was also reluctant to react (entry 10). This coupling gave mostly products of β-hydride elimination/reductive elimination pathway similar to that seen in Scheme 5 (eq 7). Aliphatic chlorides (R=CH2CH2Ph, 35), although operative in previous reports, also performed poorly in couplings with 19. The dominant pathway appears to also be the β-hydride elimination/reductive elimination pathway (entry 11).
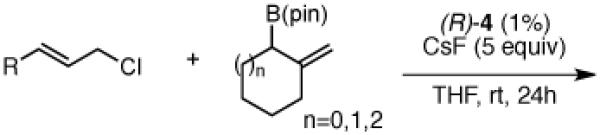
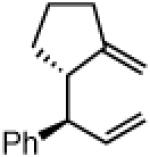
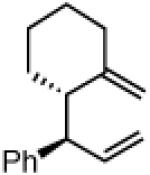
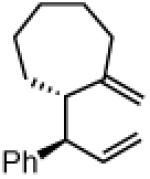
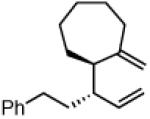
Another class of couplings that can exhibit poor reactivity and selectivity are those in which one of the coupling partners was part of a ring system. Under the optimized conditions, internal α,β-substituted cyclic allylboron reagents were found to be excellent coupling partners (Table 4). Allylboron nucleophiles containing 5-, 6- and 7- membered rings were coupled with cinnamyl chloride to give dienes 37-39 in near-perfect diastereo- and enantioselectivity with high yields and regioselectivities. Additionally, the reaction allowed use of aliphatic chloride 35 to give product 40 with excellent regio- and stereoselectivity.
Table 4.
Scope of Cyclic Substratesa
| |||
|---|---|---|---|
|
|
|
|
|
37
96% yield >99:1 er(>20:1 dr) 15:1 regio |
38b
94% yield >99:1 er(>20:1 dr) >20:1 regio |
39
96% yield >99:1 er(>20:1 dr) >20:1 regio |
40c
82% yield >99:1 er (>20:1 dr) >20:1 regio |
Unless otherwise noted reactions were run at 0.5 M concentration of the electrophile with 1.5 equiv of the allylboron reagent. Yield refers to isolated yield of purified material. Enantiomer ratios were determined by GC analysis on a chiral stationary phase. Diastereomer ratios were determined by 1H NMR analysis. All yield, er and dr values reported are the average of two or more experiments.
Reaction run with 10 equiv CsF.
Reaction run with 10 equiv CsF in 10:1 THF/H2O with 1% (S,S)-QuinoxP*PdCl2 in place of (R)−4.
3. CONCLUSION
In summary, we have described the development of an effective catalyst for the Pd-catalyzed allyl-allyl coupling. Reaction progress kinetic analysis, labeling studies, and DFT analysis of the reaction have provided a deeper understanding of the mechanism and help to explain the origins of high regio- and stereoselectivity. The new reaction conditions and insight have allowed for the extension of the coupling to allow the formation of products containing vicinal quaternary and tertiary centers as well as unique ring systems.
Supplementary Material
ACKNOWLEDGMENT
Frontier Scientific (allylB(pin)) and BASF (pinacolborane) are acknowledged for generous donations of reagents. We thank Dr. Bo Li and Hilan Kaplan for assistance with X-ray analysis, Drs. John Boylan and Thusitha Jayasundera for assistance with NMR, and Dr. Fredrik Haeffner for helpful discussions.
ASSOCIATED CONTENT
Supporting Information. Procedures, characterization, and computational details. This material is available free of charge via the Internet at http://pubs.acs.org.
Funding Sources
The NIH (GM-64451) is acknowledged for financial support. MJA acknowledges support in the form of fellowships from the American Chemical Society (DOC) and AstraZeneca.
Footnotes
Present Addresses
Author Contributions
The manuscript was written through contributions of all authors.
All authors have given approval to the final version of the manuscript.
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).Trost BM. Proc. Natl. Acad. Sci. USA. 2004;101:5348. doi: 10.1073/pnas.0306715101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Kazmaier U, editor. Transition Metal Catalyzed Enantioselective Allylic Substitutions in Organic Synthesis. Vol. 38. Springer: Heidel-berg: 2012. For a general review see. Topics in Organometallic Chemistry. [Google Scholar]; (a) Trost BM, Van Vranken DL. Chem. Rev. 1996;96:395. doi: 10.1021/cr9409804. For reviews regarding prochiral allyl electrophiles, see. [DOI] [PubMed] [Google Scholar]; (b) Helmchen G. J. Organometallic Chem. 1999;576:203. [Google Scholar]
- (3).Negishi E-i., Liao B. In: Handbook of Organopalladium Chemistry for Organic Synthesis. Negishi E-i, de Meijere A., editors. Vol. 1. Wiley-Interscience; West Lafayette: 2002. pp. 591–596. [Google Scholar]
- (4).(a) Trost BM, Keinan E. Tetrahedron Lett. 1980;21:2595. For nonselective or linear selective allyl-allyl couplings, see: [Google Scholar]; (b) Godschalx J, Stille JK. Tetrahedron Lett. 1980;21:2599. [Google Scholar]; (c) van Heerden FR, Huyser JJ, Williams DBG, Holzapfel CW. Tetrahedron Lett. 1998;39:5281. [Google Scholar]; (d) Nakamura H, Bao M, Yamamoto Y. Angew. Chem. Int. Ed. 2001;40:3208. doi: 10.1002/1521-3773(20010903)40:17<3208::AID-ANIE3208>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]; (e) Flegeau EF, Schneider U, Kobayashi S. Chem. –Eur. J. 2009;15:12247. doi: 10.1002/chem.200902221. [DOI] [PubMed] [Google Scholar]; (f) Jiménez-Aquino A, Flegeau EF, Schneider U, Kobayashi S. Chem. Commun. 2011;47:9456. doi: 10.1039/c1cc13348a. [DOI] [PubMed] [Google Scholar]; (g) Trost BM, Pietrusiewicz KM. Tetrahedron Lett. 1985;26:4039. For intramolecular allyl-allyl couplings, see. [Google Scholar]; (h) Cuerva JM, Gómez,-Bengoa E, Méndez M, Echavarren AM. J. Org. Chem. 1997;62:7540. [Google Scholar]; (i) Keinan E, Peretz M. J. Org. Chem. 1983;48:5302. For related couplings, see: [Google Scholar]; (j) Keinan E, Bosch E. J. Org. Chem. 1986;51:4006. [Google Scholar]
- (5).(a) Goliaszewski A, Schwartz J. J. Am. Chem. Soc. 1984;106:5028. For mechanistic studies on allyl-allyl couplings, see: [Google Scholar]; (b) Goliaszewski A, Schwartz J. Tetrahedron. 1985;41:5779. [Google Scholar]; (c) Goliaszewski A, Schwartz J. Organometallics. 1985;4:417. [Google Scholar]; (d) Jolly P. Angew. Chem. Int. Ed. Eng. 1985;24:283. [Google Scholar]; (e) Kraus J, Bonrath W, Pörschke KR. Organometallics. 1992;11:1158. [Google Scholar]
- (6).(a) Zhang P, Brozek LA, Morken JP. J. Am. Chem. Soc. 2010;132:10686. doi: 10.1021/ja105161f. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zhang P, Le H, Kyne RE, Morken JP. J. Am. Chem. Soc. 2011;133:9716. doi: 10.1021/ja2039248. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Brozek LA, Ardolino MJ, Morken JP. J. Am. Chem. Soc. 2011;133:16778. doi: 10.1021/ja2075967. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Le H, Kyne RE, Brozek LA, Morken JP. Org. Lett. 2013;15:1432. doi: 10.1021/ol400088g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Ardolino MJ, Morken JP. J. Am. Chem. Soc. 2012;134:8770. doi: 10.1021/ja302329f. For related allyl-allenyl couplings see: [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Ardolino MJ, Eno MS, Morken JP. Adv. Synth. Catal. 2013;355:3413. doi: 10.1002/adsc.201300720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Hornillos V, Pérez M, Fañanás-Mastral M, Feringa BL. J. Am. Chem. Soc. 2013;135:2140. doi: 10.1021/ja312487r. For Cu-catalyzed asymmetric allyl-allyl coupling, to access monosubstituted dienes of type F, see: [DOI] [PubMed] [Google Scholar]
- (8).(a) Peterson EA, Overman LE. Proc. Natl. Acad. Sci. U.S.A. 2004;101:11943. doi: 10.1073/pnas.0402416101. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Cozzi PG, Hilgraf R, Zimmermann N. Eur. J. Org. Chem. 2007;36:5869. [Google Scholar]; (c) Trost BM, Jiang C. Synthesis. 2006;369 [Google Scholar]; (d) Das JP, Marek I. Chem. Commun. 2011;47:4593. doi: 10.1039/c0cc05222a. [DOI] [PubMed] [Google Scholar]
- (9).(a) Trost BM, Cramer N, Silverman SM. J. Am. Chem. Soc. 2007;129:12396. doi: 10.1021/ja075335w. For transition-metal catalyzed methods to access vicinal allcarbon tertiary and quaternary centers see: [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Trost BM, Zhang Y. J. Am. Chem. Soc. 2007;129:14548. doi: 10.1021/ja0755717. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Streuff J, White DE, Virgil SC, Stoltz BM. Nat. Chem. 2010;2:192. doi: 10.1038/nchem.518. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Trost BM, Miller JR, Hoffman CM., Jr. J. Am. Chem. Soc. 2011;133:8165. doi: 10.1021/ja2029602. [DOI] [PubMed] [Google Scholar]; (e) Trost BM, Silverman SM, Stambuli JP. J. Am. Chem. Soc. 2011;133:19483. doi: 10.1021/ja207550t. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Tan J, Cheon C-H, Yamamoto H. J. Am. Chem. Soc. 2012;51:8264. doi: 10.1002/anie.201203092. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Liu W, Reeves CM, Virgil SC, Stoltz BM. J. Am. Chem. Soc. 2013;135:10626. doi: 10.1021/ja4052075. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Liu W, Reeves CM, Virgil SC, Stoltz BM. J. Am. Chem. Soc. 2013;135:17298. doi: 10.1021/ja4097829. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Krautwald S, Sarlah D, Schafroth MA, Carriera EM. Science. 2013;340:1065. doi: 10.1126/science.1237068. [DOI] [PubMed] [Google Scholar]; (j) Germain N, Guéné L, Maudit M, Alexakis A. Org. Lett. 2014;16:118. doi: 10.1021/ol403104s. [DOI] [PubMed] [Google Scholar]
- (10).(a) Du C, Li L, Li Y, Xie Z. Angew. Chem. Int. Ed. 2009;48:7853. doi: 10.1002/anie.200902908. For transition-metal catalyzed methods to access vicinal quaternary centers see: [DOI] [PubMed] [Google Scholar]; (b) Trost BM, Osipov M. Angew. Chem. Int. Ed. 2013;52:9176. doi: 10.1002/anie.201302805. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Zhang H, Hong L, Kang H, Wang R. J. Am. Chem. Soc. 2013;135:14098. doi: 10.1021/ja408336v. [DOI] [PubMed] [Google Scholar]
- (11).(a) Amatore C, Jutand A. Coord. Chem. Rev. 1998;511:178–180. [Google Scholar]; (b) Fairlamb IJS, Kapdi AR, Lee AF, McGlacken GP, Weissburger F, de Vries AHM, Schmieder-van de Vondervoort LS. Chem. –Eur. J. 2006;12:8750. doi: 10.1002/chem.200600473. [DOI] [PubMed] [Google Scholar]; (c) Fairlamb IJS. Org. Biomol. Chem. 2008;6:3645. doi: 10.1039/b811772a. See also ref 9c. [DOI] [PubMed] [Google Scholar]
- (12).(a) Wright SW, Hageman DL, McClure LD. J. Org. Chem. 1994;59:6095. [Google Scholar]; (b) Littke AF, Fu GC. Angew. Chem. Int. Ed. 1998;37:3387. doi: 10.1002/(SICI)1521-3773(19981231)37:24<3387::AID-ANIE3387>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]; (c) Littke AF, Dai C, Fu GC. J. Am. Chem. Soc. 2000;122:4020. [Google Scholar]
- (13).(a) Blackmond DG. Angew. Chem. Int. Ed. 2005;44:4302. doi: 10.1002/anie.200462544. [DOI] [PubMed] [Google Scholar]; (b) Ferretti AC, Mathew JS, Blackmond DG. Ind. Eng. Chem. Res. 2007;46:8584. [Google Scholar]
- (14). Based on the data shown, it could appear that the reaction operates in two regimes: one initially rapid rate for the first few minutes, followed by transition to a slower reaction rate for the remainder of the reaction. Although this could signal a change the in ratedetermining step, this feature is more likely a result of catalyst activation, an induction period during the onset of the reaction, or imprecise data correction (tau correction) at early stages. Such transient behaviors are sometimes observed during reaction progress kinetic analysis, and, most often, this initial period is ignored and kinetic analysis focused on the steady-state portion of the reaction. See reference 12a for further information and explanation.
- (15). The total heat output for the reaction 1.0 M in carbonate is higher than that of the reaction 0.5 M in carbonate because the allylB(pin) (0.6 M) is now the limiting reagent. Accounting for stoichiometry, the heat outputs are calculated to be similar.
- (16).Tsuji J, Shimizu I, Minami I, Ohashi Y, Sugiura T, Takahashi K. J. Org. Chem. 1985;50:1523. [Google Scholar]
- (17).(a) Hayashi T, Konishi M, Kumada M. J. Am. Chem. Soc. 1983;105:7767. [Google Scholar]; (b) Trost BM, Verhoeven TR. J. Am. Chem. Soc. 1980;102:4730. doi: 10.1021/ja00418a063. [DOI] [PubMed] [Google Scholar]
- (18).(a) Yamamoto Y, Yatagi H, Maruyama K. J. Am. Chem. Soc. 1981;103:1969. [Google Scholar]; (b) Hayashi T, Konishi M, Kumada M. J. Am. Chem. Soc. 1982;104:4963. [Google Scholar]; (c) Hayashi T, Kabeta K, Yamamoto T, Tamao K, Kumada M. Tetrahedron Lett. 1983;24:5661. [Google Scholar]; (d) Wickham G, Kitching W. J. Org. Chem. 1983;48:614. [Google Scholar]; (e) Hayashi T, Konishi M, Kumada M. J. Chem. Soc., Chem. Commun. 1983:736. [Google Scholar]; (f) Naruta Y, Nishigaichi Y, Maruyama K. Tetrahedron Lett. 1989;45:1067. [Google Scholar]; (g) Hiyama T, Matsuhashi H, Fujita A, Tanaka M, Hirabayashi K, Shimizu M, Mori A. Organometallics. 1996;15:5762. [Google Scholar]; (h) Buckle MJC, Flemming I, Gil S, Pang KLC. Org. Biomol. Chem. 2004;2:749. doi: 10.1039/b313446f. [DOI] [PubMed] [Google Scholar]; (i) Yamamoto Y, Takada S, Miyaura N. Organometallics. 2009;28:152. [Google Scholar]; (j) Denmark SE, Werner NS. J. Am. Chem. Soc. 2010;132:3612. doi: 10.1021/ja910804u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19). The π-σ-π isomerization from Q to R would require a 5-coordinate PdL2(η1-allyl)( η3-allyl) intermediate, which has been observed experimentally, see ref. 5d.
- (20). See Supporting Information for experimental details.
- (21). For a .cif file containing the coordinates of this strucutre, see the Supporting Information for reference 6c. This structure may be accessed from the Cambridge Crystallographic Data Centre (CCDC 856101)
- (22).(a) Keith JA, Behenna DC, Mohr JT, Ma S, Marinescu SC, Oxgaard J, Stoltz BM, Goddard WA., III J. Am. Chem. Soc. 2007;129:11876. doi: 10.1021/ja070516j. The use of computational methods has become increasingly useful in predicting reaction mechanism and distinguishing between transition states leading to regio- and stereoisomers in related allylic substitutions, see: [DOI] [PubMed] [Google Scholar]; (b) Keith JA, Behenna DC, Sheridan N, Mohr JT, Ma S, Marinescu SC, Nielsen RJ, Oxgaard J, Stoltz BM, Goddard WA., III J. Am. Chem. Soc. 2012;134:19050. doi: 10.1021/ja306860n. See also reference 17i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).(a) Méndez M, Cuerva JM, GómezBengoa E, Cárdenas DJ, Echavarren AM. Chem.-Eur. J. 2002;8:3620. doi: 10.1002/1521-3765(20020816)8:16<3620::AID-CHEM3620>3.0.CO;2-P. For experimental and computational studies of the 3,3’-reductive elimination, see: [DOI] [PubMed] [Google Scholar]; (b) Cárdenas DJ, Echavarren AM. New J. Chem. 2004;28:338. [Google Scholar]; (c) Perez-Rodriguez M, Braga AAC, de Lera AR, Maseras F, Alvarez R, Espinet P. Organometallics. 2010;29:4983. [Google Scholar]
- (24). All calculations were performed on the full system (with the allyl B(pin) simplified to the glycolato backbone and the tert-butoxide simplified to methoxide) using Gaussian 09 with all geometry optimizations, energies and frequencies calculated at the DFT level utilizing the B3LYP hybrid functional (refs. 25, 26). The 6-31G** basis set was used for the elements C, H, P, B, and O in conjunction with the LANL2DZ relativistic pseudopotential for Pd. All free energies were calculated at 333.15 K. The PCM model was used to estimate the effect of solvation (THF) (ref. 27). The frequency calculations for transition states exhibited one imaginary frequency each. The transition states presented in Figure 3 were connected with the correct ground states through IRC calculations. The three-dimensional structures presented in Figures 3, 4 and 5 were visualized utilizing CYL-view (ref. 28)
- (25).Frisch MJ, et al. Gaussian 09. Gaussian, Inc.; Wallingford, CT: 2009. DFT calculations were performed using Gaussian 09. revision A.02. [Google Scholar]
- (26).(a) Becke AD. Phys. Rev. A. 1988;38:3098. doi: 10.1103/physreva.38.3098. [DOI] [PubMed] [Google Scholar]; (b) Lee C, Yang W, Parr RG. Phys. Rev. B. 1988;37:785. doi: 10.1103/physrevb.37.785. C. [DOI] [PubMed] [Google Scholar]
- (27).(a) Miertus S, Scrocco E, Tomassi J. Chem. Phys. 1981;55:117. [Google Scholar]; (b) Barone V, Cossi M, Tomassi J. Chem. Phys. 1997;107:3210. [Google Scholar]
- (28).CYLview, 1.0b. Legault CY. Université de Sherbrooke. 2009 ( http://www.cylview.org) [Google Scholar]
- (29). These results are in agreement with previous proposed transmetallation pathways of allylboron reagents to Pd by Miyaura in related allyl-aryl couplings. See ref. 18i. Additional conformations of the allyl fragment were considered as well, however showed higher free energies of activation. We were unable to find the direct SE2-transition state.
- (30). The activation barriers for transmetallation by fluoride activated allylB(pin) were also calculated and found to be 12.3 and 14.9 kcal/mol for TSTM 1 and TSTM 2 respectively. See Supporting Information for more detail.
- (31). Formation of the diene through this pathway is favored over coupling between C1 and C1’ by 11.7-12.4 kcal/mol depending on the phosphine ligand used. See refs. 23(a-c)
- (32). The three additional conformers for 3,3’-reductive elimination (the trans olefin isomers of TS3,3’-Re a, TS3,3’-Re b, and TS3,3’-Si B) were calculated to have activation free energies of 21.5, 22.7 and 22.3 kcal/mol respectively and were therefore not included in figure 4.
- (33). In general, use of allyl chlorides in place of the allyl carbonates led to higher yields. In cases where transmetallation is slowed due to the addition of substitution on the allylboron, the tert-butoxide produced with the allyl carbonates can competitively add to the Pd(II)-allyl intermediate and produce ethereal byproducts. See ref. 6c.
- (34).(a) Li S-M. Nat. Prod. Rep. 2010;27:57. doi: 10.1039/b909987p. [DOI] [PubMed] [Google Scholar]; (b) Yang Y, Buchwald SL. J. Am. Chem. Soc. 2013;135:10642. doi: 10.1021/ja405950c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35). Under previous conditions (2.5% Pd2(dba)3/5%L1, 10 equiv CsF, 14h, rt) we observed a 90% yield, >99:1 er and 6:1 dr was observed. See ref. 6c. [Google Scholar]
- (36). Despite modifications to the catalyst as well as the coupling partners, the branched coupling product was never observed when both allyl fragments were disubstituted. Considering the relative increase in calculated barrier heights for these types of couplings in comparison to even the quaternary/tertiary partners (Table 2), it is reasonable that the desired 3,3'-pathway is no longer favored over other decomposition pathways or isomerizations to give regioisomers. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.