

Abstract
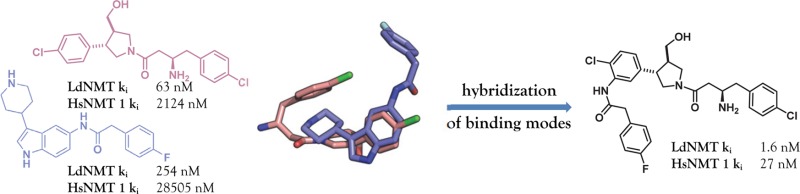
Inhibitors of LeishmaniaN-myristoyltransferase (NMT), a potential target for the treatment of leishmaniasis, obtained from a high-throughput screen, were resynthesized to validate activity. Crystal structures bound to Leishmania major NMT were obtained, and the active diastereoisomer of one of the inhibitors was identified. On the basis of structural insights, enzyme inhibition was increased 40-fold through hybridization of two distinct binding modes, resulting in novel, highly potent Leishmania donovani NMT inhibitors with good selectivity over the human enzyme.
Introduction
The leishmaniases are a spectrum of infectious diseases caused by protozoan parasites of the genus Leishmania. Cutaneous leishmaniasis (CL), caused mainly by Leishmania major (Lm), can lead to permanent scarring and disfiguration, while visceral leishmaniasis (VL), caused mainly by Leishmania donovani (Ld), is often fatal due to failure of the host immune system. The leishmaniases are endemic in 88 countries, 72 of which are low-income,1 and is a major health issue with an estimated 0.2–0.4 million cases of VL, 0.7–1.2 million cases of CL, and a conservative estimate of 20000–40000 deaths per year.2 Treatment of leishmaniasis has previously been dominated by the use of pentavalent antimonials which are toxic, painful to administer, and require long treatment regimens;3 resistance has also developed to these antimonials in India.4 Some progress has been made in the last 10 years in the development of safer, more easily applied therapeutics with the development of lipid formulations of amphotericin B, miltefosine, and paromomycin. However, side effects are common and resistance to these therapies may still be a problem,5 thus the need for new antileishmanials remains high.6,7 Despite these issues, development of new antileishmanial drugs is limited8 and compounded by challenges of cell permeability. The amastigote form of the parasite most relevant to human disease resides within an acidic parasitophorous vacuole inside host cells,9 and the parasite bears a glycoinositolphospholipid coat that could limit uptake of xenoantibiotics.10
N-Myristoyltransferase (NMT), an enzyme ubiquitous in eukaryotes, catalyzes the transfer of myristate (a 14-carbon fatty acid) to the N-terminal glycine of target proteins,11 either cotranslationally12 or post-translationally.13 Between 0.5% and 3% of the cellular proteome is predicted to be N-myristoylated,14 and this modification is vital for multiple regulatory processes, including protein–protein interactions and protein stability.15−17 Inhibition of NMT therefore has pleiotropic effects on cellular function. NMT has been shown to be essential in a range of parasitic organisms including Leishmania,18 and small-molecule cytotoxic inhibitors have been developed for NMTs in parasitic organisms including Trypanosoma brucei(19) and Plasmodium species.20−22 Inhibition of Leishmania NMT therefore represents a rational drug target for development of new therapeutics for this neglected tropical disease.14,23,24
The NMT enzyme operates via a Bi–Bi mechanism, with myristoyl CoA (MyrCoA) binding to the enzyme first and inducing a conformational change before binding of the peptide substrate. The myristate group is then transferred to the N-terminal glycine of the peptide before sequential release of the myristoyl peptide and reduced CoA products.25,26 The structures of several parasitic NMTs have been reported19,27,28 and show a conserved binding site for MyrCoA. The peptide-binding region is less conserved between different species and therefore presents a target for selective inhibition of NMTs from different species.29
A recently published high-throughput screen (HTS) of a diverse subset of the Pfizer corporate collection against LdNMT, Plasmodium falciparum NMT, and the two human isoforms (HsNMT1 and HsNMT2) revealed four novel series of Leishmania-selective NMT inhibitors.30 Here we report the development of highly potent LdNMT inhibitors based on structure-guided fusion of two of these series; piperidinylindoles, exemplified by PF-03393842 1, and aminoacylpyrrolidines, exemplified by PF-03402623 2 and PF-03402619 3 (Chart 1).
Chart 1. Leishmania-Selective Hits from Screening of a Subset of the Pfizer Compound File30.

Results and Discussion
Synthesis and Validation of Hits
To validate the HTS results, synthesis of both piperidinylindole 1 and the most potent aminoacylpyrrolidine 2 was carried out. Synthesis of 1 was achieved in four steps from 5-nitro indole (Scheme 1). Condensation of 5-nitro indole 4 with N-Boc-4-piperidone, followed by concurrent reduction of the resulting double bond and nitro group, yielded amine 6.31 Reaction with para-fluorophenylacetyl chloride followed by Boc deprotection gave piperidinylindole 1.
Scheme 1. Synthesis of 1.

Reagents and conditions: (a) pyrrolidine, N-Boc-4-piperidone, EtOH, rt, 3 days, 80%; (b) NH4HCO2, EtOH, Pd/C, 2 h, 96%; (c) para-fluorophenylacetyl chloride, Et3N, THF, 2 h, 92%; (d) 6 M HCl, IPA, 2 h, 43%.
Compound 2 was synthesized as a mixture of two diastereoisomers, as it was unclear from the original report whether the stereochemistry at the pyrrolidine ring in the HTS hit was relative or absolute. It was envisaged that the preferred stereochemistry could be identified by cocrystallization of the mixture with Leishmania NMT. The pyrrolidine core was accessed as a racemic mixture by cycloaddition of benzyl-(methyoxymethyl)[(trimethylsilyl)methyl]amine 9 and trans-methyl 4-chlorocinnamate 8 in the presence of catalytic TFA to give trans-pyrrolidine 10 (Scheme 2).32,33 The benzyl protecting group was removed using 1-chloroethyl chloroformate34 to avoid the use of reductive methods in the presence of the aromatic chlorine prior to amide coupling with acid 12 to obtain amide 13. Reduction of the ester followed by Boc deprotection proceeded smoothly to afford 2 as a mixture of two diastereoisomers.
Scheme 2. Synthesis of Aminoacylpyrrolidine 2.

Reagents and conditions: (a) 9, TFA, DCM, 0 °C to rt, 24 h, 88%; (b) (i) 1-chloroethyl chloroformate, toluene, 110 °C, 3 h, (ii) MeOH, reflux, 30 min; (c) 12, EDCI, HOBt, DIPEA, DMF, 4 h, 55% over 2 steps; (d) LiBH4, THF, 3 h, 71%; (e) TFA, DCM, 2 h, 31%.
Testing of compounds 1 and 2 against LdNMT and HsNMT1 in our previously reported CPM assay35 confirmed the results of the HTS, with both compounds showing selectivity for LdNMT over HsNMT1 (Table 1). The hits were also tested against related LmNMT, which has been shown to be more amenable to X-ray crystallography19,36 and were shown to have comparable activity to LdNMT.
Table 1. Enzyme Activity Data (Results from HTS in Brackets)30.
| compd | LdNMT IC50 (μM) | LmNMT IC50 (μM) | HsNMT1 IC50 (μM) | EC50a(μM) | LD50b (μM) |
|---|---|---|---|---|---|
| 1 | 0.31 (0.102) | 0.55 | 63 (73) | >30 | >45 |
| 2 | 0.080 (0.093) | 0.031 | 4.7 (5.2) | 10–30 | 8–16 |
EC50 in extracellular Ld amastigotes;
LD50 in bone marrow derived mouse macrophages
Compounds were also tested against extracellular amastigotes of Leishmania donovani and against bone marrow derived macrophages to determine toxicity (Table 1).37 Compound 1 displayed no cell activity up to 30 μM, although no toxicity was observed. Compound 2 showed an EC50 between 10 and 30 μM, however, the compound was also toxic to macrophages at this concentration.
X-ray Crystallography
Our first strategy to optimize these NMT inhibitors was to drive down enzyme potency using structure-guided design. To elucidate the binding mode of the HTS hits and the preferred stereochemistry of 2, crystal structures of ternary complexes of LmNMT (97% sequence homology with LdNMT) and myristoyl-CoA cofactor were obtained for both resynthesized hits, as recently reported.36 Both inhibitors were shown to bind in the peptide binding region. The structure of compound 1 bound to LmNMT revealed a direct interaction between the basic piperidine nitrogen and the C-terminal carboxylate of the enzyme (Leu421) (Figure 1). This type of charge–charge interaction has previously been observed with other NMT inhibitors in Plasmodium NMT20,21 and via a bridging water molecule in LmNMT.19 The indole adopts an equatorial position off the piperidine ring in a hydrophobic pocket, and the amide carbonyl is orthogonal to the indole ring, forming hydrogen bonds to Tyr345 and Asn376.
Figure 1.
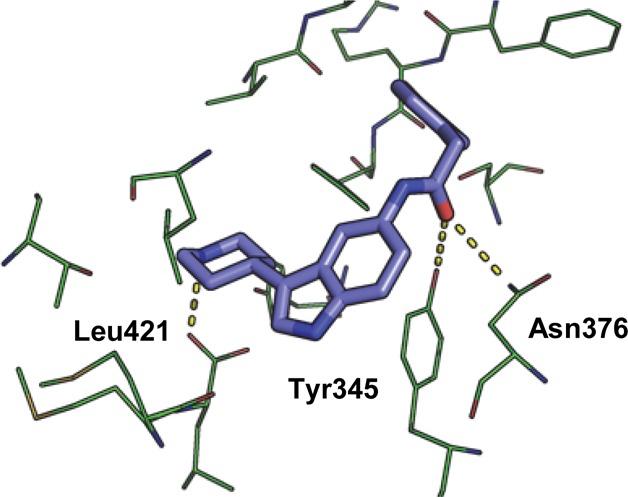
Inhibitor 1 (blue) bound in the peptide binding pocket of LmNMT (green). PDB code: 4cgn.
The cocrystal structure of 2 bound to LmNMT displays a unique binding mode compared to previously reported NMT inhibitors; the conformation of the inhibitor appears to be governed by a hydrophobic collapse38 that folds the aromatic rings into a hairpin conformation about the flexible linker, with the chlorophenyl substituent of the pyrrolidine ring sandwiched between the edge of Tyr345 below and Tyr217 above. The inhibitor takes up a compact conformation in which its surface area is almost completely buried by the protein and MyrCoA. Interestingly, the key charge–charge interaction between the basic amine and Leu421 is not seen (Figure 2). Instead, the primary amine is adjacent to the thioester of MyrCoA and makes bridging contacts with the backbone carbonyl of Thr203 and the side chain of Asn167. The hydroxyl group is actually closest to the C-terminal leucine carboxylate (2.6 Å), and there is a potential hydrogen bond between the amide carbonyl and Thr203 (Figure 2).
Figure 2.
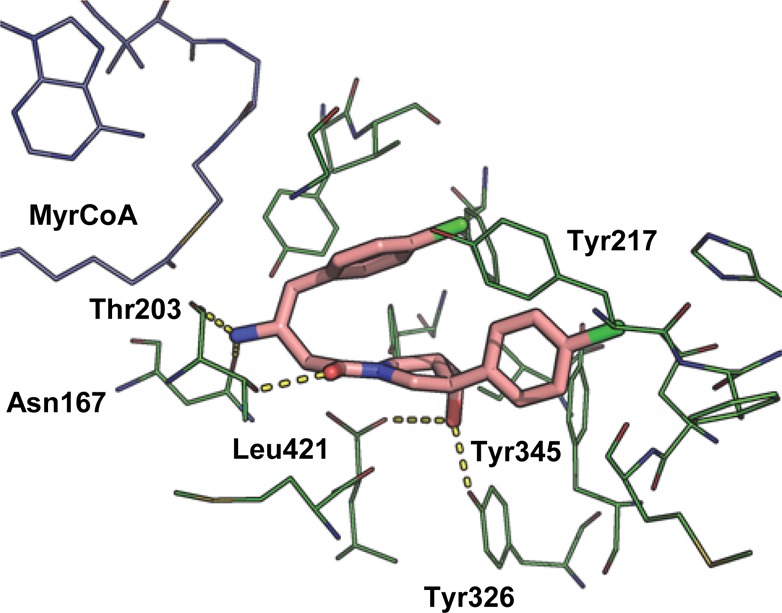
Inhibitor 2a (pink) bound in the peptide binding pocket of LmNMT (green). MyrCoA in blue. PDB code: 4cgl.
As expected, the crystal structure shows a single diastereoisomer (2a, Scheme 3) bound to the enzyme. To confirm that 2a is the most active isomer, both diastereoisomers (2a and 2b) were synthesized separately using enantiopure oxazolidinone 17 for the cycloaddition reaction (Scheme 3). Cycloaddition yielded diasteroisomers 18a and 18b, which could be separated by column chromatography and were assigned by comparison with reported 1H NMR data39 (Scheme 3). Removal of the oxazolidinone gave esters 10a and 10b from which 2a and 2b were synthesized, respectively, using the route detailed in Scheme 2.
Scheme 3. Synthesis of 2a and 2b.

Reagents and conditions: (a) (i) oxalyl chloride, DCM, DMF, 0 °C, 30 min, (ii) 17, Et3N, LiCl, 15 h, 29%; (b) 9, TFA, DCM, 0 °C to rt, 24 h, 18a 36%, 18b 25%; (c) CO(OMe)2, NaOMe, DCM, 15 h, 10a 48%, 10b 40%.
Enzyme inhibition assays confirmed that diastereoisomer 2a was more active with an IC50 of 25 nM against LdNMT, with 2b exhibiting 60-fold lower potency and 4-fold lower selectivity for LdNMT over HsNMT1 (Table 2).
Table 2. Enzyme Activity Data for Diastereoisomers 2a and 2b.
| compd | LdNMT IC50 (μM) | HsNMT1 IC50 (μM) |
|---|---|---|
| 2 | 0.080 | 4.7 |
| 2a | 0.025 | 1.4 |
| 2b | 1.7 | 24 |
A crystal structure was also obtained for 2b bound to LmNMT. The structure shows a similar hydrophobic collapse of the ligand and that the key functional groups (the primary amine and alcohol) of both diastereoisomers are superimposed in the active site (circled, Figure 3). However, as a result of maintaining these interactions, the scaffold is twisted such that the amide carbonyl no longer forms the hydrogen bond with Thr203 seen in the structure of 2a.
Figure 3.
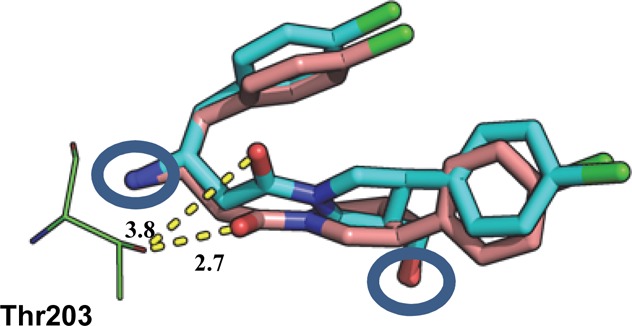
Overlay of I 2b (cyan) and 2a (pink) bound in the peptide binding pocket of LmNMT obtained by alignment of the protein main chain atoms (distances in Å). PDB code for 2b: 4cyn.
Hybridization of Binding Modes
Comparison of the distinct binding modes of hits 1 and 2 showed that the benzo-ring of the indole in 1 and the aromatic substituent of the pyrrolidine in 2 bind in the same region (Figure 4a). For this reason, it was hypothesized that addition of a para-fluorophenyl acetamide ortho- to the chlorine atom in this ring in compound 2 may significantly improve potency (Figure 4b). This could potentially introduce hydrogen bonding between the acetamide carbonyl and Tyr345 and Asn376 and allow 2 to extend into the same hydrophobic pocket as 1. To determine whether an acyl group would be sufficient to increase potency by addition of the hydrogen bond, or whether the hydrophobic bulk of the para-fluorophenyl group would be required, both the acyl and para-fluorophenyl acetamide derivatives of 2 were synthesized (19 and 20, respectively, Figure 4b).
Figure 4.
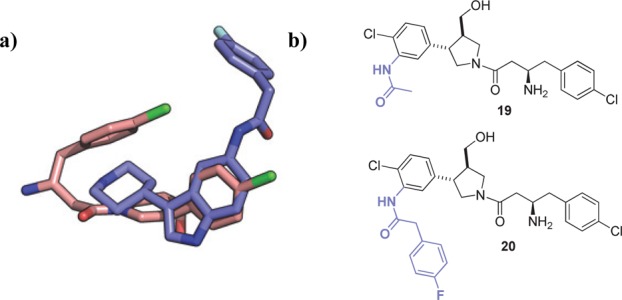
(a) Overlay of binding modes of 1 (blue) and 2a (pink). (b) Proposed hybrid structures 19 and 20.
To allow the addition of an acetamide in the correct position, synthesis of the pyrrolidine core was carried out using 3-nitro, 4-chloro cinnamate 21 (Scheme 4). Selective reduction of the nitro group with tin(II) chloride then provided an aniline moiety for formation of the required amide bond. Both the acyl derivative 19 and para-fluorophenyl derivative 20 were synthesized using the route shown in Scheme 4.
Scheme 4. Synthesis of 19 and 20.

Reagents and conditions: (a) 9, TFA, DCM, 0 °C to rt, 24 h, 79%; (b) SnCl2, EtOH, 2 h, 87% (c) R = H, Ac2O, Et3N, DCM, 2 h, 65%, R = pF-Ph para-fluorophenylacetyl chloride, Et3N, DCM, 2 h, 42%; (d) (i) 1-chloroethyl chloroformate, toluene, 110 °C, 3 h, (ii) MeOH, reflux, 30 min; (e) EDCI, HOBt, DIPEA, 12, DMF, 4 h, R = H 41% over 2 steps, R = cpF-Ph 43% over 2 steps; (f) LiBH4, THF, 3 h; (g) TFA, DCM, 2 h, R = H 61% over 2 steps, R = pF-Ph 34% over two steps.
Testing of the hybrid compound 19 showed that the acyl group was tolerated but did not improve potency. However, addition of the para-fluorophenyl group in compound 20 led to an IC50 at the lower measurement limit of the biochemical assay. In this case, converting to Ki using the Cheng–Prusoff equation for tight binders is more informative40,41 (Table 3). Introduction of the para-fluorophenyl group led to a 40-fold decrease in Ki against LdNMT (Ki = 1.6 nM). Alignment of the X-ray crystal structures of 20, 1, and 2 bound to LmNMT demonstrates that 20 binds as designed and that all interactions with the enzyme are conserved (Figure 5 and Figure S1, Supporting Information).
Table 3. Enzyme and Cell Activity Data.
| compd | pKa | LdNMT Ki (nM) | HsNMT1 Ki (nM) | EC50a (μM) | LD50b (μM) |
|---|---|---|---|---|---|
| 1 | 10.0 | 254 | 28505 | >30 | >45 |
| 2 | 8.9 | 63 | 2124 | 10–30 | 8–16 |
| 2a | 8.9 | 17 | 631 | 10–30 | 12–24 |
| 2b | 8.9 | 1406 | 10857 | 10–30 | 12–24 |
| 19 | 8.9 | 110 | 4910 | >50 | >90 |
| 20 | 8.9 | 1.6 | 27 | 10–30 | 12–24 |
| 43 | 59 | 1710 | 10–30 | >24 |
EC50 in extracellular Ld amastigotes (for comparison, EC50 for the widely used antileishmanial drugs amphotericin B and miltefosine in this assay are 50 and 7850 nM, respectively.)
LD50 in bone marrow-derived mouse macrophages.
Figure 5.
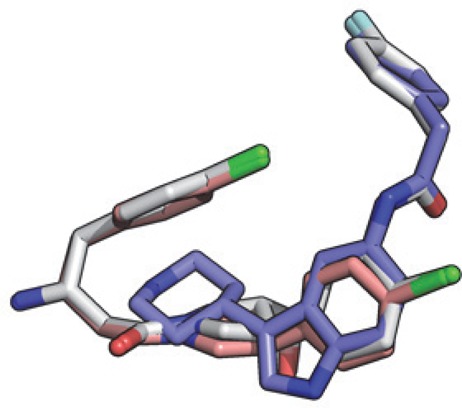
Overlay of hits 1 (blue), 2 (pink), and hybrid 20 (white; PDB code 4cyo) bound to LmNMT obtained by alignment of the protein main chain atoms.
Cell Testing
Despite the greater enzyme inhibition achieved by hybrid compound 20, no improvement in cell-based activity was seen (Table 3). This highlights that simply driving down enzyme potency in this compound series is insufficient to increase cellular activity against this challenging target organism. The diastereoisomers of 2 were also tested separately, and both displayed an EC50 of 10–30 μM and an LD50 of 12–24 μM, demonstrating that both activity and toxicity are unrelated to NMT inhibition for these compounds. We hypothesized that the lack of cell-based activity for this series of compounds is due to lack of cellular uptake and thus, insufficient target engagement. Compounds 1 and 2 and derivatives synthesized here all contain a basic center (pKa 10.0 and 8.9 respectively) which would be charged at physiological pH, with a potentially adverse effect on membrane permeability.
Replacement of the Primary Amine
As the crystal structures of the aminoacylpyrrolidines show that the amine does not make the key charge–charge interaction with the C-terminal carboxylate observed previously in other series, we considered replacing the primary amine with a less basic moiety. It was envisaged that the amine could be replaced with an alcohol without loss of hydrogen bonding, potentially generating a potent, neutral NMT inhibitor. To synthesize this neutral compound, acid 35 was synthesized from 4-chlorophenylacetyl chloride 32. Reaction with Meldrum’s acid followed by hydrolysis gave ketone 33, which was subsequently reduced. Hydrolysis of the resulting ester gave the acid 35 (Scheme 5).
Scheme 5. Synthesis of Acid 35.

Reagents and conditions: (a) (i) Meldrum’s acid, pyridine, DCM, 0 °C, 30 min then rt, 15 h, (ii) EtOH, reflux, 2 h, 70%; (b) NaBH4, MeOH, 0 °C to rt, 1.5 h, 40%; (c) LiOH, MeOH/H2O 99%.
This acid was then used to synthesize the alcohol analogue of hybrid 20 (Scheme 6). As acid 35 was synthesized as a racemic mixture, pyrrolidine 39 was synthesized as a single enantiomer in order to give the hybrid alcohol 43 as a mixture of only two diastereoisomers (Scheme 6). The hybrid alcohol 43 was tested for its enzyme activity and showed reduced activity compared to the corresponding amine (Table 3). However, the activity for this neutral compound is comparable to the original primary amine hit 2, and selectivity over HsNMT1 is maintained.
Scheme 6. Synthesis of Alcohol 43.

Reagents and conditions: (a) (i) oxalyl chloride, DCM, DMF, 0 °C, 30 min, (ii) 16, Et3N, LiCl, 15 h, 45%; (b) 9, TFA, DCM, 0 °C to rt, 24 h, 38a 40%; (c) CO(OMe)2, NaOMe, DCM, 15 h 36%; (d) SnCl2, EtOH, 2 h, quantitative; (e) para-fluorophenylacetyl chloride, Et3N, DCM, 2 h 55%; (f) (i) 1-chloroethyl chloroformate, toluene, 110 °C, 3 h, (ii) MeOH, reflux, 30 min; (g) DMC, 35, Et3N, DCM, 15 h, 27% over 2 steps; (h) LiBH4, THF, 2 h, 25%.
The crystal structure of alcohol 43 bound to LmNMT appeared to overlay well with that of amine 20 (Figure S2, Supporting Information, PDB code 4cyq). However, closer inspection revealed a slight difference in the position of the amine versus the alcohol in these two structures. The primary amine of 20 forms a hydrogen bond to the backbone carbonyl group of Thr203 at a distance of 2.9 Å. When this amine is replaced with an alcohol in compound 43, the corresponding oxygen is 3.5 Å away, reducing its potential to hydrogen bond.
Testing against extracellular amastigotes showed no improvement in activity for compound 43 compared with the original hits or the hybrid amine (Table 3). Metabolic chemical tagging22 in Ld amastigotes (Figure S3, Supporting Information) demonstrated that despite replacement of the primary amine, target engagement consistent with a Ki of 59 nM was not achieved, supporting our hypothesis that the lack of cell-based activity for these compounds is due to poor cellular uptake.
Despite the potential of NMT as a drug target in Leishmania, these organisms are known to be difficult to target, due in part to their cell surface coats, a key component of Leishmania virulence and survival.9,10 Advances in potency and particularly physicochemical properties will be required to progress this series of compounds and to chemically validate Leishmania NMT as a drug target in vivo.
Conclusion
Two Leishmania NMT-selective HTS hits have been resynthesized and their activities validated. Crystal structures of these inhibitors identified their binding modes and, in the case of compound 2, identified the active diastereoisomer. The crystal structures were used to increase enzyme affinity through hybridization of the two independent binding modes, and this led to the discovery of a highly potent inhibitor of LdNMT. The unusual binding mode of the aminoacylpyrrolidines allowed the replacement of the primary amine, leading to compound 43, a potent and neutral NMT inhibitor. Although poor uptake appears to lead to a lack of cell activity for these compounds, elucidation of the binding modes of these inhibitor series along with their hybridization provides a useful starting point for the development of LdNMT inhibitors with improved physicochemical properties.
Experimental Section
The purity of final compounds was determined by reversed-phase LC-MS on a Waters 2767 system and was ≥95% for all tested compounds.
(±)(3R,4S)-Methyl 1-Benzyl-4-(4-chlorophenyl)pyrrolidine-3-carboxylate (10)
TFA (20 μL, 0.20 mmol) was added to a solution of 9 (1.04 mL, 4.06 mmol) and chlorocinnamate 8 (400 mg, 2.03 mmol) in DCM (40 mL) at 0 °C, and the solution was stirred (rt) for 24 h. Saturated aqueous NaHCO3 (40 mL) was added, and the phases were separated. The organic layer was dried over Na2SO4 and the solvent removed under reduced pressure. The crude residue was purified by column chromatography (1:9 EtOAc–hexane, Rf 0.25) to give the product 10 as a colorless oil (590 mg, 88%). 1H NMR (400 MHz,CDCl3) δ 7.41–7.27 (m, 9H), 3.76–3.68 (m, 5H), 3.68–3.63 (m, 1H), 3.16–3.03 (m, 2H), 3.02–2.96 (m, 1H), 2.86 (dd, J = 8.3, 6.4 Hz, 1H), 2.77 (dd, J = 9.4, 5.8 Hz, 1H).
(3R,4S) Methyl 1-((R)-3-((tert-Butoxycarbonyl)amino)-4-(4-chlorophenyl)butanoyl)-4-(4-chlorophenyl)pyrrolidine-3-carboxylate (13)
1-Chloroethyl chloroformate (327 μL, 3.03 mmol) was added to a solution of 10 (500 mg, 1.51 mmol) in toluene (30 mL), and the solution was stirred at 110 °C for 3 h. The reaction was cooled to room temperature, and the solvent was removed under reduced pressure. The residue was dissolved in methanol (30 mL), and the solution was heated at reflux for 30 min before cooling to room temperature. The solvent was removed under reduced pressure to give 513 mg of yellow oil. Then 150 mg of this oil was dissolved in DMF (7.5 mL), and acid 12 (216 mg, 0.69 mmol) was added, followed by EDCI (132 mg, 0.69 mmol), HOBt (93 mg, 0.69 mmol), and DIPEA (120 μL, 0.69 mmol). The solution was stirred at room temperature for 4 h. EtOAc (10 mL) was added, and the mixture was washed with saturated aqueous NaHCO3 (10 mL), water (3 × 10 mL), and brine (10 mL). The solvent was removed under reduced pressure and the residue purified by column chromatography (1:1 EtOAc–hexane, Rf 0.35) to give the product 13 as a colorless oil (186 mg, 55%) as a mixture of diastereoisomers and as a mixture of amide rotamers by 1H NMR at room temperature. 1H NMR (400 MHz, CDCl3) δ 7.38–7.24 (m, 4H), 7.23–7.08 (m, 4H), 5.68 (br s, 1H), 4.18–3.97 (m, 2H), 3.85–3.42 (m, 6H), 3.27–3.10 (m, 1H), 3.10–3.00 (m, 1H), 2.97–2.85 (m, 1H), 2.50–2.40 (m, 2H), 1.45–1.38 (m, 9H).
tert-Butyl ((R)-1-(4-Chlorophenyl)-4-(((3S,4R))-3-(4-chlorophenyl)-4-(hydroxymethyl)pyrrolidin-1-yl)-4-oxobutan-2-yl)carbamate (14)
LiBH4 (3 mg, 0.13 mmol) was added to a solution of 13 (18 mg, 0.04 mmol) in dry THF (1 mL). The solution was stirred for 3 h (rt). Water was added (2 mL), followed by DCM (2 mL), and the phases were separated. The organic layer was dried over MgSO4 and the solvent removed under reduced pressure to give the product 14 as a colorless oil (12 mg, 71%) as a mixture of diastereoisomers and as a mixture of amide rotamers by 1H NMR at room temperature. 1H NMR (400 MHz, CDCl3) δ 7.36–7.24 (m, 4H), 7.17 (m, 4H), 5.89–5.59 (br s, 1H), 4.18–3.86 (m, 2H), 3.77–3.63 (m, 2H), 3.61–3.09 (m, 4H), 3.04 (m, 1H), 2.95–2.84 (m, 1H), 2.54–2.38 (m, 2H), 1.46–1.36 (m, 9H).
(R)-3-Amino-4-(4-chlorophenyl)-1-((3S,4R)-3-(4-chlorophenyl)-4-(hydroxymethyl)pyrrolidin-1-yl)butan-1-one (2)
TFA (11 μL, 0.11 mmol) was added to a solution of 14 (12 mg, 0.02 mmol) in DCM (1 mL), and the reaction was stirred at for 2 h (rt). The solvent was removed under reduced pressure, and the crude residue was purified by preparative LCMS (method B) to give the product 2 as a colorless oil (3 mg, 31%) as a mixture of diastereoisomers. 1H NMR (400 MHz, MeOD) δ 7.43–7.24 (m, 8H), 4.06–3.76 (m, 2H), 3.72 (s, 1H), 3.61–3.55 (m, 1H), 3.52–3.37 (m, 3H), 3.29–3.14 (m, 1H), 2.99–2.85 (m, 2H), 2.72–2.39 (m, 3H). m/z 407 ([M + H]+). HRMS found 407.1313, C21H25N2O2Cl2 requires 407.1293. LCMS Rt = 12.44 min. Complete experimental details including LCMS methods are provided in the Supporting Information.
Acknowledgments
The authors are grateful to Mark Rackham and Zhiyong Yu for valuable discussions, Johan Turkenburg and Sam Hart (York) for help with X-ray data collection, and Diamond Light Source (Harwell, UK) for synchrotron facilities. This work was supported by The Wellcome Trust (grant 087792).
Glossary
Abbreviations Used
- Ld
Leishmania donovani
- Lm
Leishmania major
- NMT
N-myristoyltransferase
- Hs
Homo sapiens
- CPM
7-diethylamino-3-(4′-maleimidylphenyl)-4-methylcoumarin
- MyrCoA
myristoyl-CoA
Supporting Information Available
Experimental procedures and characterization of all compounds, assay procedures, X-ray data collection and statistics, and additional figures. This material is available free of charge via the Internet at http://pubs.acs.org.
Accession Codes
The coordinates and structure factor files have been deposited in the Protein Data Bank with accession codes 4cgn (LmNMT MyrCoA-1), 4cgl (LmNMT-MyrCoA-2a), 4cyn (LmNMT-MyrCoA-2b), 4cyo (LmNMT-MyrCoA-20), and 4cyq (LmNMT-MyrCoA-43).
Author Present Address
∥ For J.A.H.: Wolfson Institute for Biomedical Research, University College London, London WC1E 6BT, UK.
Author Present Address
⊥ For V.G.: Université de Bourgogne, ICMUB UMR 6302 CNRS, 21078 Dijon, France.
Author Present Address
# For D.P.: Wellcome Trust Centre for Molecular Parasitology, University of Glasgow, Glasgow G12 8TA, UK.
Author Present Address
▽ For M.H.W.: TU München, D-85748 Garching, Germany.
Author Present Address
○ For T.M.W.: Department of Chemistry, University College London, London WC1H 0AJ, UK.
Author Present Address
◆ For R.J.L.: Liverpool John Moores University, Liverpool L1 2UA, UK.
Author Contributions
All authors have given approval to the final manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Leishmaniasis: Burden and Distribution; World Health Organization: Geneva, 2013; http://www.who.int/leishmaniasis/burden/en (accessed July 1, 2014).
- Alvar J.; Velez I. D.; Bern C.; Herrero M.; Desjeux P.; Cano J.; Jannin J.; den Boer M. Leishmaniasis worldwide and global estimates of its incidence. PLoS One 2012, 7, e35671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- den Boer M.; Argaw D.; Jannin J.; Alvar J. Leishmaniasis impact and treatment access. Clin. Microbiol. Infect. 2011, 17, 1471–1477. [DOI] [PubMed] [Google Scholar]
- Sundar S. Drug resistance in Indian visceral leishmaniasis. Trop. Med. Int. Health 2001, 6, 849–854. [DOI] [PubMed] [Google Scholar]
- Croft S. L.; Sundar S.; Fairlamb A. H. Drug resistance in leishmaniasis. Clin. Microbiol. Rev. 2006, 19, 111–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- den Boer M. L.; Alvar J.; Davidson R. N.; Ritmeijer K.; Balasegaram M. Developments in the treatment of visceral leishmaniasis. Expert Opin. Emerging Drugs 2009, 14, 395–410. [DOI] [PubMed] [Google Scholar]
- Jain K.; Jain N. K. Novel therapeutic strategies for treatment of visceral leishmaniasis. Drug Discovery Today 2013, 18, 1272–1281. [DOI] [PubMed] [Google Scholar]
- Barrett M. P.; Croft S. L. Management of trypanosomiasis and leishmaniasis. Br. Med. Bull. 2012, 104, 175–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antoine J. C.; Prina E.; Lang T.; Courret N. The biogenesis and properties of the parasitophorous vacuoles that harbour Leishmania in murine macrophages. Trends Microbiol. 1998, 6, 392–401. [DOI] [PubMed] [Google Scholar]
- Novozhilova N. M.; Bovin N. V. Structure, functions, and biosynthesis of glycoconjugates of Leishmania spp. cell surface. Biochemistry 2010, 75, 686–694. [DOI] [PubMed] [Google Scholar]
- Johnson D. R.; Bhatnagar R. S.; Knoll L. J.; Gordon J. I. Genetic and biochemical studies of protein N-myristoylation. Annu. Rev. Biochem. 1994, 63, 869–914. [DOI] [PubMed] [Google Scholar]
- Wilcox C.; Hu J. S.; Olson E. N. Acylation of proteins with myristic acid occurs cotranslationally. Science 1987, 238, 1275–1278. [DOI] [PubMed] [Google Scholar]
- Zha J.; Weiler S.; Oh K. J.; Wei M. C.; Korsmeyer S. J. Posttranslational N-myristoylation of BID as a molecular switch for targeting mitochondria and apoptosis. Science 2000, 290, 1761–1765. [DOI] [PubMed] [Google Scholar]
- Wright M. H.; Heal W. P.; Mann D. J.; Tate E. W. Protein myristoylation in health and disease. J. Chem. Biol. 2010, 3, 19–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resh M. D. Trafficking and signaling by fatty-acylated and prenylated proteins. Nature Chem. Biol. 2006, 2, 584–590. [DOI] [PubMed] [Google Scholar]
- Poulin B.; Patzewitz E. M.; Brady D.; Silvie O.; Wright M. H.; Ferguson D. J.; Wall R. J.; Whipple S.; Guttery D. S.; Tate E. W.; Wickstead B.; Holder A. A.; Tewari R. Unique apicomplexan IMC sub-compartment proteins are early markers for apical polarity in the malaria parasite. Biol. Open 2013, 2, 1160–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price H. P.; Hodgkinson M. R.; Wright M. H.; Tate E. W.; Smith B. A.; Carrington M.; Stark M.; Smith D. F. A role for the vesicle-associated tubulin binding protein ARL6 (BBS3) in flagellum extension in T. brucei. Biochem. Biophys. Acta 2012, 7, 1178–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price H. P.; Menon M. R.; Panethymitaki C.; Goulding D.; McKean P. G.; Smith D. F. Myristoyl-CoA:protein N-myristoyltransferase, an essential enzyme and potential drug target in kinetoplastid parasites. J. Biol. Chem. 2003, 278, 7206–7720. [DOI] [PubMed] [Google Scholar]
- Frearson J. A.; Brand S.; McElroy S. P.; Cleghorn L. A. T.; Smid O.; Stojanovski L.; Price H. P.; Guther M. L. S.; Torrie L. S.; Robinson D. A.; Hallyburton I.; Mpamhanga C. P.; Brannigan J. A.; Wilkinson A. J.; Hodgkinson M.; Hui R.; Qiu W.; Raimi O. G.; van Aalten D. M. F.; Brenk R.; Gilbert I. H.; Read K. D.; Fairlamb A. H.; Ferguson M. A. J.; Smith D. F.; Wyatt P. G. N-Myristoyltransferase inhibitors as new leads to treat sleeping sickness. Nature 2010, 464, 728–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Z.; Brannigan J. A.; Moss D. K.; Brzozowski A. M.; Wilkinson A. J.; Holder A. A.; Tate E. W.; Leatherbarrow R. J. Design and synthesis of inhibitors of Plasmodium falciparumN-myristoyltransferase, a promising target for antimalarial drug discovery. J. Med. Chem. 2012, 55, 8879–8890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rackham M. D.; Brannigan J. A.; Moss D. K.; Yu Z.; Wilkinson A. J.; Holder A. A.; Tate E. W.; Leatherbarrow R. J. Discovery of novel and ligand-efficient inhibitors of Plasmodium falciparum and Plasmodium vivaxN-myristoyltransferase. J. Med. Chem. 2012, 56, 371–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright M. H.; Clough B.; Rackham M. D.; Rangachari K.; Brannigan J. A.; Grainger M.; Moss D. K.; Bottrill A. R.; Heal W. P.; Broncel M.; Serwa R. A.; Brady D.; Mann D. J.; Leatherbarrow R. J.; Tewari R.; Wilkinson A. J.; Holder A. A.; Tate E. W. Validation of N-myristoyltransferase as an antimalarial drug target using an integrated chemical biology approach. Nature Chem. 2014, 6, 112–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowyer P. W.; Tate E. W.; Leatherbarrow R. J.; Holder A. A.; Smith D. F.; Brown K. A. N-Myristoyltransferase: a prospective drug target for protozoan parasites. ChemMedChem 2008, 3, 402–408. [DOI] [PubMed] [Google Scholar]
- Tate E. W.; Bell A. S.; Rackham M. D.; Wright M. H. N-Myristoyltransferase as a potential drug target in malaria and leishmaniasis. Parasitology 2014, 141, 37–49. [DOI] [PubMed] [Google Scholar]
- Towler D. A.; Adams S. P.; Eubanks S. R.; Towery D. S.; Jacksonmachelski E.; Glaser L.; Gordon J. I. Purification and characterization of yeast myristoyl-CoA-protein N-myristoyltransferase. Proc. Natl. Acad. Sci. U. S. A 1987, 84, 2708–2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudnick D. A.; McWherter C. A.; Rocque W. J.; Lennon P. J.; Getman D. P.; Gordon J. I. Kinetic and structural evidence for a sequential ordered bi–bi mechanism of catalysis by Saccharomyces cerevisiae myristoyl-CoA-protein N-myristoyltransferase. J. Biol. Chem. 1991, 266, 9732–9739. [PubMed] [Google Scholar]
- Brannigan J. A.; Smith B. A.; Yu Z.; Brzozowski A. M.; Hodgkinson M. R.; Maroof A.; Price H. P.; Meier F.; Leatherbarrow R. J.; Tate E. W.; Smith D. F.; Wilkinson A. J. N-Myristoyltransferase from Leishmania donovani: structural and functional characterisation of a potential drug target for visceral leishmaniasis. J. Mol. Biol. 2010, 396, 985–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goncalves V.; Brannigan J. A.; Whalley D.; Ansell K. H.; Saxty B.; Holder A. A.; Wilkinson A. J.; Tate E. W.; Leatherbarrow R. J. Discovery of Plasmodium vivaxN-myristoyltransferase inhibitors: screening, synthesis, and structural characterization of their binding mode. J. Med. Chem. 2012, 55, 3578–3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurer-Stroh S.; Eisenhaber B.; Eisenhaber F. N-Terminal N-myristoylation of proteins: refinement of the sequence motif and its taxon-specific differences. J. Mol. Biol. 2002, 317, 523–540. [DOI] [PubMed] [Google Scholar]
- Bell A. S.; Mills J. E.; Williams G. P.; Brannigan J. A.; Wilkinson A. J.; Parkinson T.; Leatherbarrow R. J.; Tate E. W.; Holder A. A.; Smith D. F. Selective inhibitors of protozoan protein N-myristoyltransferases as starting points for tropical disease medicinal chemistry programs. PLoS Negl. Trop. Dis. 2012, 6, e1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macor J. E.Indole derivatives. U.S. Patent US5639752, 1997.
- Padwa A.; Dent W. Use of N-[(trimethylsilyl)methyl]amino ethers as capped azomethine ylide equivalents. J. Org. Chem. 1987, 52, 235–244. [Google Scholar]
- Ujjainawall F.; Warner D.; Snedden C.; Grisson R. D.; Walsh T. F.; Wyvratt M. J.; Kalyani R. N.; MacNeil T.; Tang R.; Weinberg D. H.; Van der Ploeg L.; Goulet M. T. Design and syntheses of melanocortin subtype-4 receptor agonists. Part 2: Discovery of the dihydropyridazinone motif. Bioorg. Med. Chem. Lett. 2005, 15, 4023–4028. [DOI] [PubMed] [Google Scholar]
- Olofson R. A.; Martz J. T.; Senet J. P.; Piteau M.; Malfroot T. A new reagent for the selective, high-yield N-dealkylation of tertiary amines: improved syntheses of naltrexone and nalbuphine. J. Org. Chem. 1984, 49, 2081–2082. [Google Scholar]
- Goncalves V.; Brannigan J. A.; Thinon E.; Olaleye T. O.; Serwa R.; Lanzarone S.; Wilkinson A. J.; Tate E. W.; Leatherbarrow R. J. A fluorescence-based assay for N-myristoyltransferase activity. Anal. Biochem. 2012, 421, 342–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brannigan J. A.; Roberts S. M.; Bell A. S.; Hutton J. A.; Hodgkinson M. R.; Tate E. W.; Leatherbarrow R. J.; Smith D. F.; Wilkinson A. J. Diverse modes of binding in structures of Leishmania majorN-myristoyltransferase with selective inhibitors. IUCrJ 2014, 1, 250–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paape D.; Bell A. S.; Heal W. P.; Hutton J. A.; Leatherbarrow R. J. L.; Tate E. W.; Smith D. F.. Using a non-image-based medium-throughput assay for screening compounds targeting N-myristoylation in intracellular Leishmania amastigotes. Unpublished results. [DOI] [PMC free article] [PubMed]
- Wiley R. A.; Rich D. H. Peptidomimetics derived from natural products. Med. Res. Rev. 1993, 13, 327–384. [DOI] [PubMed] [Google Scholar]
- Calabrese A. A.; Fradet D. S.; Hepworth D.; Lansdell M.. Pharmaceutically active compounds. U.S. Patent US2005/176772, 1995.
- Williams J. W.; Morrison J. F. The kinetics of reversible tight-binding inhibition. Methods Enzymol. 1979, 63, 437–467. [DOI] [PubMed] [Google Scholar]
- Copeland R. A.; Lombardo D.; Giannaras J.; Decicco C. P. Estimating ki values for tight-binding inhibitors from dose–response plots. Bioorg. Med. Chem. Lett. 1995, 5, 1947–1952. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.