

Abstract
Bupivacaine is a widely used, local anesthetic agent that blocks voltage-gated Na+ channels when used for neuro-axial blockades. Much lower concentrations of bupivacaine than in normal clinical use, < 10−8 m, evoked Ca2+ transients in astrocytes from rat cerebral cortex, that were inositol trisphosphate receptor-dependent. We investigated whether bupivacaine exerts an influence on the Ca2+ signaling and interleukin-1β (IL-1β) secretion in inflammation-reactive astrocytes when used at ultralow concentrations, < 10−8 m. Furthermore, we wanted to determine if bupivacaine interacts with the opioid-, 5-hydroxytryptamine- (5-HT) and glutamate-receptor systems. With respect to the μ-opioid- and 5-HT-receptor systems, bupivacaine restored the inflammation-reactive astrocytes to their normal non-inflammatory levels. With respect to the glutamate-receptor system, bupivacaine, in combination with an ultralow concentration of the μ-opioid receptor antagonist naloxone and μ-opioid receptor agonists, restored the inflammation-reactive astrocytes to their normal non-inflammatory levels. Ultralow concentrations of bupivacaine attenuated the inflammation-induced upregulation of IL-1β secretion. The results indicate that bupivacaine interacts with the opioid-, 5-HT- and glutamate-receptor systems by affecting Ca2+ signaling and IL-1β release in inflammation-reactive astrocytes. These results suggest that bupivacaine may be used at ultralow concentrations as an anti-inflammatory drug, either alone or in combination with opioid agonists and ultralow concentrations of an opioid antagonist.
Keywords: 5-HT, astrocytes, bupivacaine, glutamate, opioids, rat
Introduction
Local anesthetics may interfere with opioid receptor signaling through their effects on G proteins and Ca2+ channels by decreasing the opioid inhibition of these channels (Komai & McDowell, 2007). Clinically used doses of such agents produce effective analgesia, by blocking voltage-gated Na+ channels. Bupivacaine may have other effects at much lower concentrations where the blocking of Na+ channels does not exist (Toda et al., 2011). Doses far below those that block nerve impulse propagation, at nanomolar concentrations, may have pain-relieving actions via other targets such as neuronal G protein-coupled receptors and binding sites on immune cells (Amir et al., 2006). Bupivacaine has been proposed to attenuate inflammatory responses, although the mechanisms involved and sites of action for such effects of this anesthetic are not known (Bedirli et al., 2011).
Glial cells have been shown to play critical roles in neuropathic, nociceptive and inflammatory pain, which is pain that can be long-lasting and may eventually be considered as chronic (Gao & Ji, 2010; Lundborg et al., 2010; Chiang et al., 2012). The underlying mechanisms of persistent/long-lasting pain involves the presence of neuroinflammation at the damaged or affected nerve(s) (Saadé & Jabbur, 2008; McMahon & Malcangio, 2009; Vallejo et al., 2010), which includes the activation of glial cells.
We have recently shown that Ca2+ signaling in astrocytes is disturbed when the cells are influenced by inflammatory stimuli, and our previous data suggest that some receptor systems are more sensitive to inflammatory stimuli than others, especially opioid-, 5-hydroxytryptamine (5-HT)- and glutamate-receptor systems in the form of increased intracellular Ca2+ transients (Hansson et al., 2008; Lundborg et al., 2011; Block et al., 2012). Astrocyte-propagating Ca2+ waves (Blomstrand et al., 1999; Scemes & Giaume, 2006) can be evoked by transmitters released from neurons and glial cells, for example ATP and glutamate, which can activate G protein-coupled receptors on astrocytes. Inflammatory stimuli disturb the Ca2+ homeostasis of the astrocyte network, which results in Ca2+-evoked oscillations (Hansson & Rönnbäck, 2003; Hansson, 2006, 2010; Forshammar et al., 2011).
The upregulation of interleukin1-β (IL-1β) secretion observed in an inflammatory environment reduces intercellular communication by affecting the passage of inositol 1,4,5-trisphosphate (IP3) through gap junctions in the astrocyte networks (Retamal et al., 2007). This results in inhibition of the intercellular neuroprotective Ca2+ signaling through gap junction channels, and increases another type of Ca2+ signaling providing a pathway for the release and uptake of small molecules, such as ATP, between the extra- and intracellular compartments (Froger et al., 2010).
The first aim of this study was to investigate whether bupivacaine, at ultralow concentrations, < 10−8 m, interacted with the intracellular Ca2+ signaling system in astrocytes. If so, the second aim was to investigate if bupivacaine, at ultralow concentrations, could interact with the opioid-, 5-HT- or glutamate-receptor systems in inflammation-reactive astrocytes through the Ca2+ signaling system. The final aim was to investigate whether bupivacaine could attenuate the IL-1β secretion that is upregulated in reactive astrocytes under inflammatory conditions.
Materials and methods
In this study, an in vitro model involving astrocytes co-cultured with brain endothelial cells was employed (Hansson et al., 2008). The biological rationale for co-culturing astrocytes with endothelial cells is that astrocytes are affected by substances released from the capillary endothelial cells of the blood–brain barrier (BBB) (Huber et al., 2001). These interactions are essential for a functional neurovascular system (Abbott et al., 2006; Willis & Davis, 2008). The endothelial cells are not in direct physical contact with the astrocytes in vitro; rather, the interaction between these two cell types is facilitated by a shared medium. Co-cultured astrocytes are morphologically differentiated from monocultured astrocytes by their long, slender processes; they also exhibit greater Ca2+ responses and cytokine release than monocultured astrocytes. Finally, the μ-opioid receptor is more highly expressed in co-cultured astrocytes (Hansson et al., 2008), as well as the Toll-like receptor 4 (TLR4) (Forshammar et al., 2011).
Chemicals
All chemicals were obtained from Sigma-Aldrich (St Louis, MO, USA) unless otherwise stated.
The experimental protocols were approved by the Ethical Committee in Gothenburg, Sweden, for Laboratory Animals (Nos. 205-2010; 211-2010), and were conducted in accordance with the European Communities Council Directive of 24 November 1986 (86/609/EEC).
Primary astrocytic cultures
Primary astrocytic cultures were prepared from newborn rat cerebral cortices (Charles River, Sulzfeldt, Germany) and cultured on glass coverslips (Nr. 1, 20-mm diameter, BergmanLabora, Stockholm, Sweden) as described by Hansson et al. (2008).
Microvascular endothelial primary cultures
Brain capillary fragments were isolated from male Sprague–Dawley rats; cerebral cortices (Charles River, 225–250 g), and endothelial cells were cultured according to the protocol of Hansson et al. (2008).
Astrocytes co-cultured with adult rat brain microvascular primary cultures
Experimental astrocytes were obtained after the co-cultivation of primary brain microvascular endothelial cultures and primary astrocyte cultures. At 6 days in vitro, astrocyte cultures were co-cultured with newly prepared microvascular cultures. The endothelial cells were grown on inserts above the astrocyte cultures. The cells from the two different cultures were not brought into contact, and they were grown together for 9–11 days. As a result, the experimental astrocytes were cultured for 15–17 days, including 9–11 days of co-cultivation, for the experiments in this study. The endothelial cells were removed before all experimental procedures (Hansson et al., 2008).
Calcium imaging
Astrocytes co-cultured with brain microvascular endothelial cells were incubated at room temperature with the Ca2+-sensitive fluorophore probe Fura-2/AM (Invitrogen Molecular Probes, Eugene, OR, USA) for 20 min [8 μL in 990 μL Hank's HEPES-buffered saline solution (HHBSS), containing 137 mm NaCl, 5.4 mm KCl, 0.4 mm MgSO4, 0.4 mm MgCl2, 1.26 mm CaCl2, 0.64 mm KH2PO4, 3.0 mm NaHCO3, 5.5 mm glucose and 20 mm HEPES dissolved in distilled water, pH 7.4). The fluorophore probe was dissolved in 40 μL dimethyl sulfoxide (DMSO) and 10 μL pluronic acid (Molecular Probes, Leiden, the Netherlands) (Fig.1). After incubation, the cells were rinsed three times with HHBSS before exposure to stimulators for 30 s. To determine the underlying Ca2+ source of the bupivacaine-evoked Ca2+ transients, internal Ca2+ stores were depleted either by pre-incubation with a sarcoendoplasmic reticulum Ca2+ -ATPase inhibitor, thapsigargin (1 μm), and caffeine (20 mm) (Sharma & Vijayaraghavan, 2001) or by incubation in a Ca2+ -free buffer (CaCl2 was replaced by MgCl2 and 1 mm EGTA). The cells were incubated with lipopolysaccharide (LPS, Escherichia coli O111:B4) (10 ng/mL) for 4 or 24 h. Different combinations of bupivacaine (10−12 m), naloxone (10−12 m), and a combination of endomorphin-1 (10−6 m) and β-endorphin (10−6 m) were applied 3.5 min before treatment with endomorphin-1, 5-HT or glutamate (10−4 m). The experiments were performed at room temperature using a Ca2+ imaging system and Simple PCI software (Compix Inc., Imaging Systems, Hamamatsu Photonics Management Corporation, Cranberry Twp., PA, USA) connected to an inverted epifluorescence microscope (Nikon ECLIPSE TE2000-E) with a 20 × (NA 0.45) fluorescence dry lens and a Polychrome V, monochromator-based illumination system (TILL Photonics, CA, USA). Various substances were applied using a peristaltic pump (Instech Laboratories, Plymouth Meeting, PA, USA) at an approximate rate of 600 μL/min. One minute after the start of the experiment, the stimulating substance was pumped into the pump tubes for 30 s. The substance took approximately 60 s to reach the cells through the tubes. HHBSS continued to flow through the pump tubes and onto the cells throughout the experiment. The images were captured with an ORCA-12AG High Res Digital Cooled CCD Camera (C4742-80-12AG, Hamamatsu Photonics).
Figure 1.
Astrocytes incubated with the Ca2+ -sensitive fluorophore probe Fura-2/AM. See Materials and methods for further details.
The total area under the curve (AUC), which reflects the amount of Ca2+ released (Berridge, 2007), was analysed to measure the strength of the Ca2+ responses. The amplitude was expressed as the maximum increase of the 340/380 ratio. The area under the Ca2+ peaks was calculated in Origin (Microcal Software Inc., Northampton, MA, USA). Forty cells were used for each experimental set-up; these cells were taken from four different coverslips and at two different seeding times.
Cytokine release assay
The co-cultured astrocytes were stimulated for 24 h with 10 ng/mL LPS diluted in non-supplemented minimum essential medium (MEM) to measure IL-1β release. The culture inserts containing the endothelial cells were removed immediately before incubation. The cells were incubated with 10−12 or 10−6 m bupivacaine for 30 min prior to LPS treatment, and the bupivacaine remained in the medium throughout the experiment. Control (i.e. untreated) cells were maintained in non-supplemented MEM. Culture supernatants were collected and analysed for protein concentrations. An enzyme-linked immunosorbent assay (ELISA) kit (Nordic Biosite, Täby, Sweden) was used according to the manufacturer's instructions.
Protein determination
The protein content was determined using a detergent-compatible (DC) protein assay (Bio-Rad, Hercules, CA, USA) in accordance with the manufacturer's instructions, based on the method by Lowry et al. (1951) with minor modifications. The standards (0–4 mg/mL BSA) and samples were mixed with the reagents, incubated for 15 min at room temperature, read at 750 nm with a Versa-max microplate reader and analysed using SoftMax Pro 4.8 software (Molecular Devices, Sunnyvale, CA, USA).
Statistics
Differences across different treatments were identified using one-way anova followed by Dunnett's multiple-comparisons test. Error bars represent the standard error of the mean (SEM).
Results
Bupivacaine elicits Ca2+ responses in astrocytes at < 10−8 m
The astrocytes were stimulated with bupivacaine (10−15–10−7 m), and Ca2+ imaging experiments were performed. All samples produced Ca2+ responses when treated with 10−15–10−8 m bupivacaine. No response was observed in cells treated with 10−7 m bupivacaine. The amount of Ca2+ released by each sample was determined by calculating the AUC for each sample (Fig.2).
Figure 2.
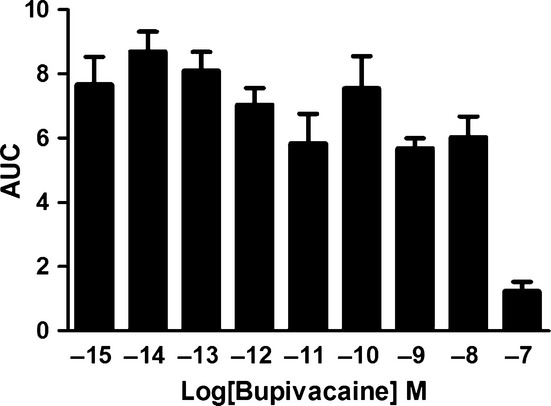
Bupivacaine-induced Ca2+ transients. The astrocytes were stimulated with bupivacaine (10−15–10−7 m). All cells responded to the treatment within the 10−15–10−8 m concentration range. At 10−7 m bupivacaine, only 4/40 cells responded, and the responses were minimal. The areas under the Ca2+ peaks (AUC) were calculated. The data are shown as the mean ± SEM, n = 40.
To investigate whether the increase in intracellular Ca2+ (Fig.3A) was due to Ca2+ crossing the extracellular membrane or Ca2+ originating from intracellular stores, bupivacaine (10−12 m) was first administered in a Ca2+ -free medium, which still evoked Ca2+ transients in most cells (Fig.3B). Cells were also incubated with thapsigargin, a depletor of intracellular Ca2+ stores (Sharma & Vijayaraghavan, 2001), followed by caffeine, which was used to complete the store depletion (Sharma & Vijayaraghavan, 2001); interestingly, this treatment inhibited the Ca2+ response (Fig.3C). Finally, no cells responded when they were treated with thapsigargin and caffeine in a Ca2+ -free buffer (Fig.3D). To determine the involvement of the IP3-sensitive Ca2+ release pathway, astrocytes were incubated with xestospongin C (100 nm), which is a blocker of IP3 receptors (Sharma & Vijayaraghavan, 2001). In these astrocytes, no responsive cells were observed (Fig.3E).
Figure 3.
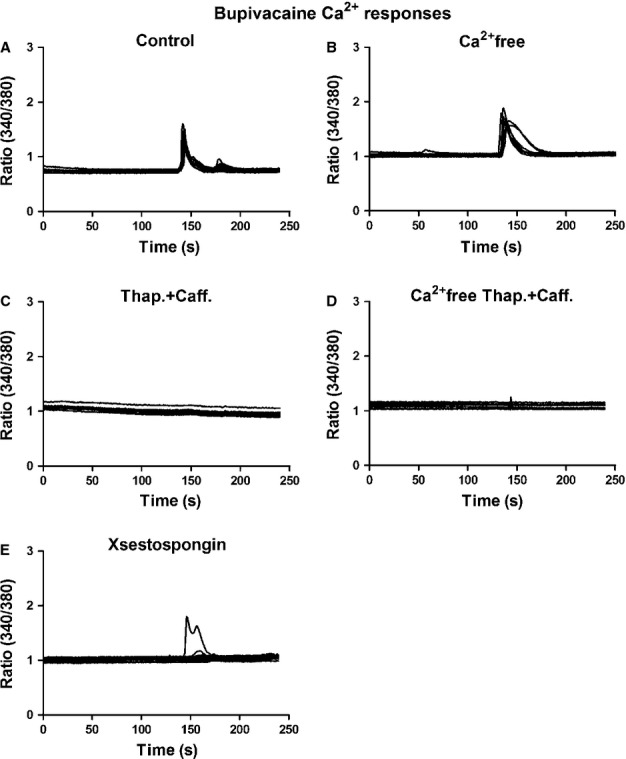
Bupivacaine-induced Ca2+ transients were triggered via intracellular Ca2+ stores. The cells were stimulated and analysed using a Ca2+ imaging system. (A) Bupivacaine (10−12 m) elicited Ca2+ responses in all astrocytes tested. (B) Bupivacaine administered in a Ca2+ -free medium evoked Ca2+ transients in all of the cells tested. (C) To determine the source of the Ca2+ response, internal Ca2+ stores were depleted with thapsigargin followed by caffeine. Interestingly, the Ca2+ responses were inhibited. (D) All cells were inhibited when thapsigargin and caffeine were administered in a Ca2+ -free buffer. (E) To examine the involvement of the IP3-sensitive Ca2+ release system, the astrocytes were incubated with xestospongin C, a blocker of IP3 receptors. Only one of the 40 cells tested responded, n = 40. The results shown are from a typical experiment.
Bupivacaine interacts with endomorphin-1_induced Ca2+ responses in LPS-treated astrocytes
Astrocytes incubated with LPS for 4 h and stimulated with endomorphin-1 did not exhibit significantly different Ca2+ signaling levels compared with the control. Cells incubated with LPS for 24 h showed increased endomorphin-1-induced Ca2+ signaling, as indicated by significantly increased AUC values (P < 0.001). Bupivacaine (10−12 m) attenuated the endomorphin-1-induced Ca2+ transients in all cells, as shown by significantly decreased AUC values for the bupivacaine-treated samples (P < 0.001) (Fig.4).
Figure 4.
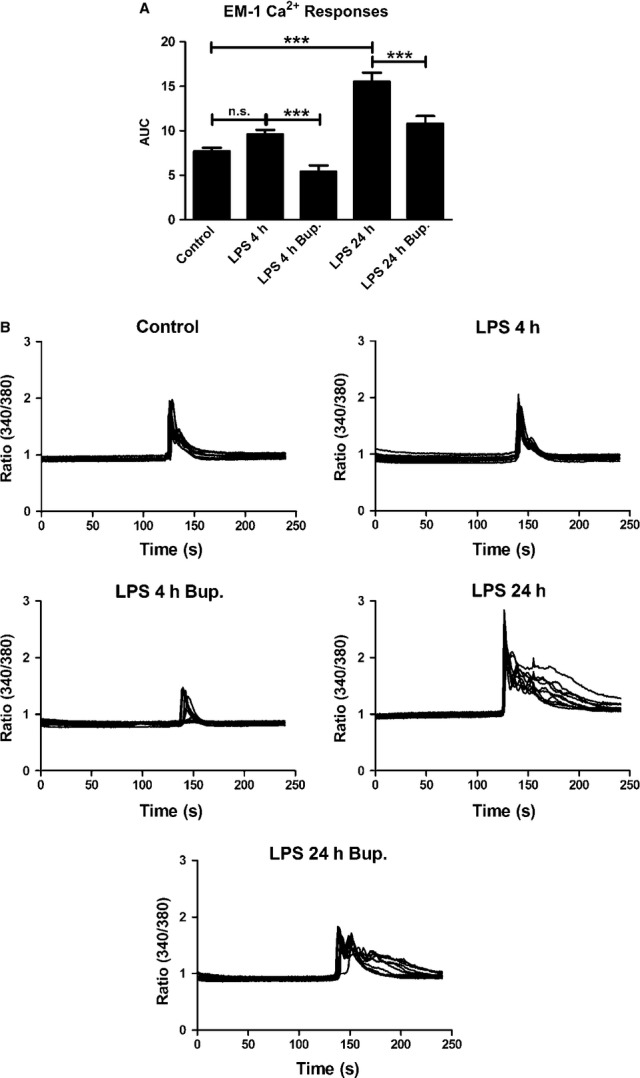
Endomorphin-1 (EM-1) elicited Ca2+ responses in the astrocytes and was used as control. (A) Cells incubated with LPS for 4 h showed no significant changes in Ca2+ level compared with the control. Cells incubated with LPS for 24 h showed increased Ca2+ signaling. Bupivacaine (10−12 m) attenuated the EM-1-induced Ca2+ transients that were upregulated by LPS for 4 and 24 h. The areas under the Ca2+ peaks (AUC) were calculated. (B) The Ca2+ transients were visualized. The results shown are from a typical experiment. Statistical analysis: the level of significance was analysed using one-way anova followed by Dunnett's multiple-comparisons test. ***P < 0.001, n.s. = not significant. n = 40.
Bupivacaine, at ultralow concentration, interacts with 5-HT-induced Ca2+ responses in LPS-treated astrocytes
Cells treated with bupivacaine did not exhibit significantly different 5-HT-induced Ca2+ signaling levels compared with the control. Cells incubated with LPS for 24 h showed increased 5-HT-induced Ca2+ signaling, as indicated by significantly increased AUC values (P < 0.001); all cells exhibited a response. Bupivacaine (10−12 m) attenuated the 5-HT-induced Ca2+ transients that were upregulated by LPS treatment in all cells (P < 0.05) (Fig.5).
Figure 5.
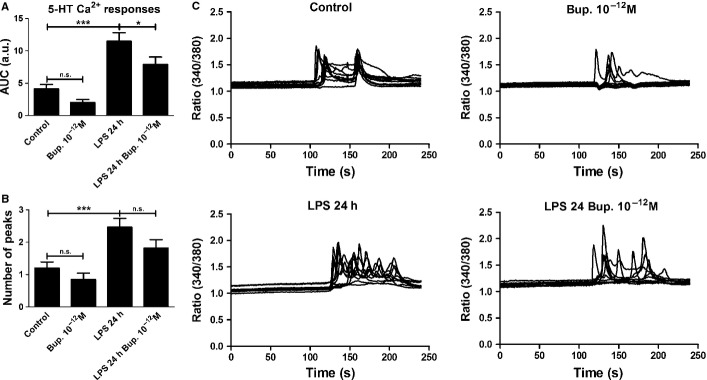
5-HT elicited Ca2+ responses in the astrocytes and was used as control. (A) Bupivacaine showed no significant changes in the level of 5-HT-induced Ca2+ signaling compared with the control. Cells incubated with LPS for 24 h showed an increased level of 5-HT-induced Ca2+ signaling, which were attenuated by bupivacaine (10−12 m). The areas under the Ca2+ peaks (AUC) were calculated. (B) The number of peaks was counted for each cell. (C) The Ca2+ transients were visualized. The results shown are from a typical experiment. Statistical analysis: the level of significance was analysed using one-way anova followed by Dunnett's multiple-comparisons test. ***P < 0.001, *P < 0.05, n.s. = not significant. n = 40.
However, administration of naloxone (10−12 m) together with endomorphin-1 (10−6 m) did not attenuate the 5-HT-induced Ca2+ transients that were upregulated by LPS treatment; all cells responded (Fig.6). No attenuation was observed either for a combination of bupivacaine, naloxone and endomorphin-1 (Fig.6), or a combination of bupivacaine, naloxone and β-endorphin (Fig.7).
Figure 6.
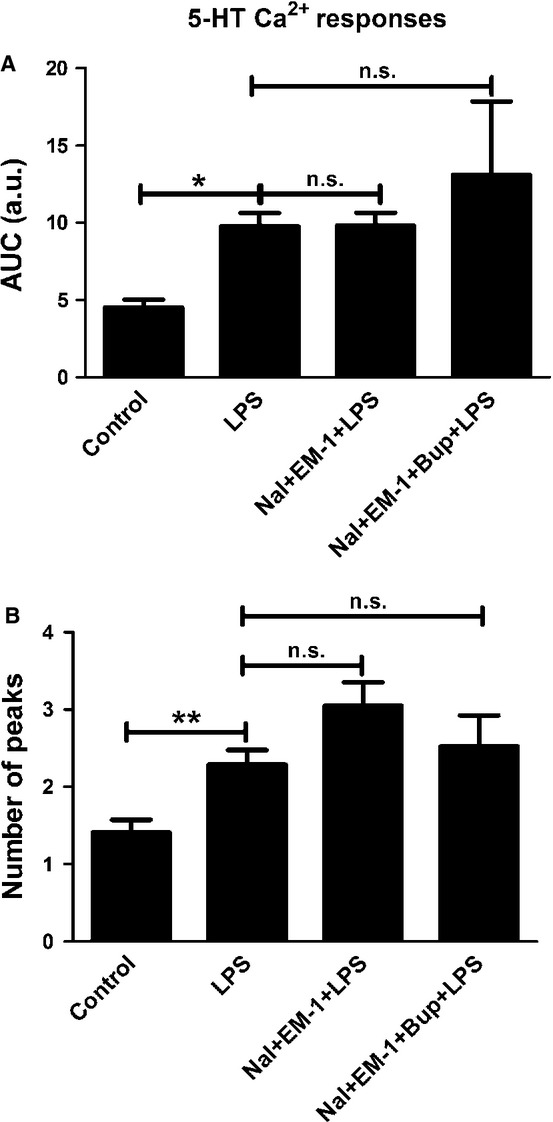
5-HT elicited Ca2+ responses in the astrocytes and was used as control. (A) Cells incubated with LPS for 24 h showed increased 5-HT-induced Ca2+ signaling compared with the control. A combination of naloxone (Nal) (10−12 m) and endomorphin-1 (EM-1) (10−6) did not change the level of 5-HT-induced Ca2+ transients upregulated by LPS, nor did a combination of bupivacaine (Bup), Nal and EM-1. The areas under the Ca2+ peaks (AUC) were calculated. (B) The number of peaks was counted for each cell. Statistical analysis: the level of significance was analysed using one-way anova followed by Dunnett's multiple-comparisons test. **P < 0.01, *P < 0.05, n.s. = not significant. n = 40.
Figure 7.
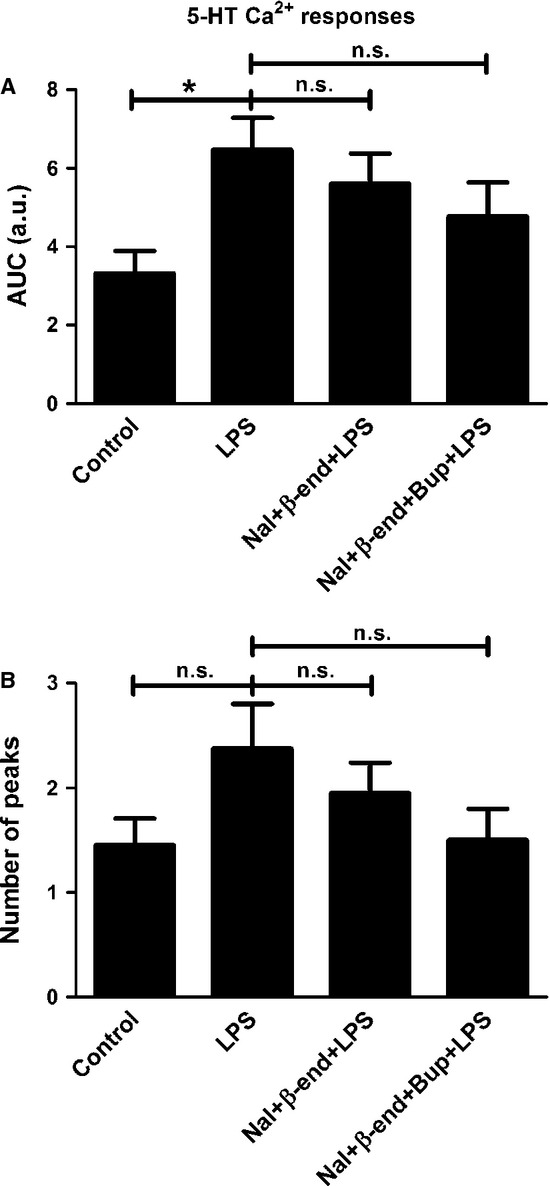
5-HT elicited Ca2+ responses in the astrocytes and was used as control. (A) Cells incubated with LPS for 24 h showed increased 5-HT-induced Ca2+ signaling compared with the control. A combination of naloxone (Nal) (10−12 m) and β-endorphin (β-end) (10−6) did not attenuate the 5-HT-induced Ca2+ transients that were upregulated by LPS, nor did a combination of bupivacaine (Bup), Nal and β-end. The areas under the Ca2+ peaks (AUC) were calculated. (B) The number of peaks was counted for each cell. Statistical analysis: the level of significance was analysed using one-way anova followed by Dunnett's multiple-comparisons test. *P < 0.05, n.s. = not significant. n = 40.
Bupivacaine, at ultralow concentration, interacts with glutamate-induced Ca2+ responses in LPS-treated astrocytes
Bupivacaine (10−12 m) reduced glutamate-induced Ca2+ signaling when compared with the control (P < 0.05), which consisted of cells treated with glutamate alone. All cells exhibited a response. Cells incubated with LPS for 24 h showed increased glutamate-induced Ca2+ signaling, as indicated by a significantly increased AUC value (P < 0.01). Again, all cells responded. Bupivacaine did not restore the glutamate-induced Ca2+ transients that were upregulated by LPS treatment to the control level (Fig.8); all cells responded to these treatments.
Figure 8.
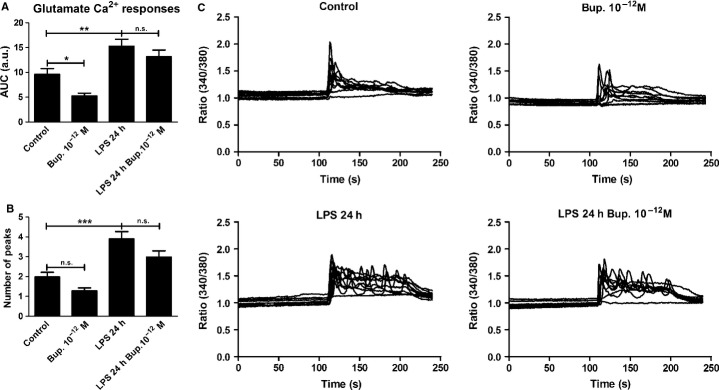
Glutamate elicited Ca2+ responses in the astrocytes and was used as control. (A) Bupivacaine (10−12 m) attenuated the glutamate-induced Ca2+ signaling compared with the control. Cells incubated with LPS for 24 h showed increased glutamate-induced Ca2+ signaling. Bupivacaine did not attenuate the glutamate-induced Ca2+ transients that were upregulated by LPS. The areas under the Ca2+ peaks (AUC) were calculated. (B) The number of peaks was counted for each cell. (C) The Ca2+ transients were visualized. The results shown are from a typical experiment. Statistical analysis: the level of significance was analysed using one-way anova followed by Dunnett's multiple-comparisons test. ***P < 0.001, **P < 0.01, *P < 0.05, n.s. = not significant. n = 40.
Cells treated with naloxone (10−12 m) and endomorphin-1 (10−6) did not restore the glutamate-induced Ca2+ transients that were upregulated by LPS treatment, but a combination of bupivacaine, naloxone and endomorphin-1 did (P < 0.001); all cells responded to these treatments (Fig.9). Furthermore, a combination of bupivacaine, naloxone and β-endorphin attenuated the glutamate-induced signaling that was upregulated by LPS (P < 0.05); again, all cells responded (Fig.10).
Figure 9.
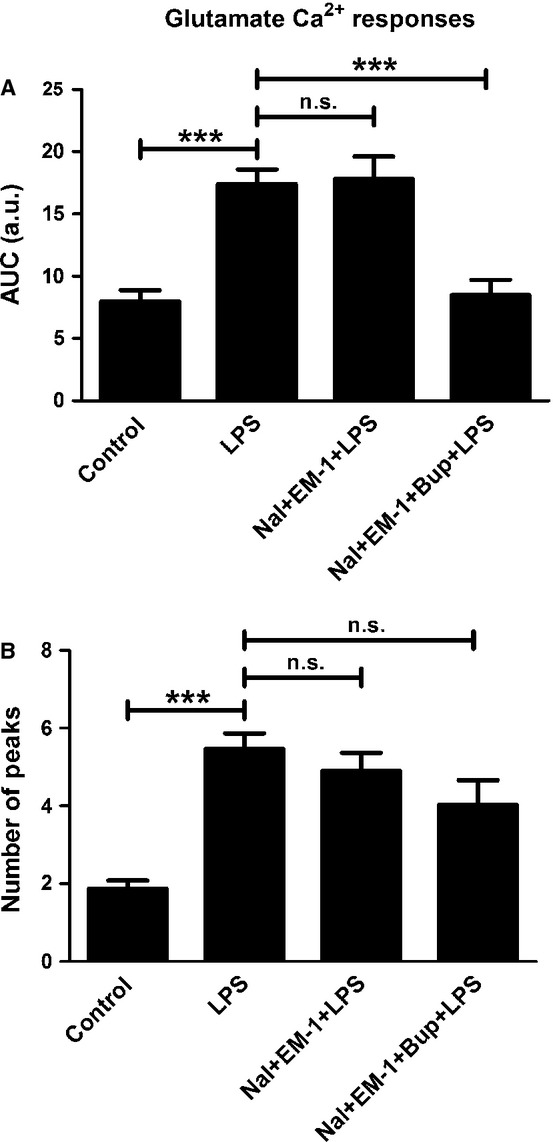
Glutamate elicited Ca2+ responses in the astrocytes and was used as control. (A) Cells incubated with LPS for 24 h showed increased glutamate-induced Ca2+ signaling compared with the control. A combination of naloxone (Nal) (10−12 m) and endomorphin-1 (EM-1) (10−6) did not attenuate the glutamate-induced Ca2+ transients that were upregulated by LPS. However, a combination of bupivacaine (Bup), Nal and EM-1 attenuated the LPS-upregulated glutamate-induced signaling. The areas under the Ca2+ peaks (AUC) were calculated. (B) The number of peaks was counted for each cell. Statistical analysis: the level of significance was analysed using one-way anova followed by Dunnett's multiple-comparisons test. ***P < 0.001, n.s. = not significant. n = 40.
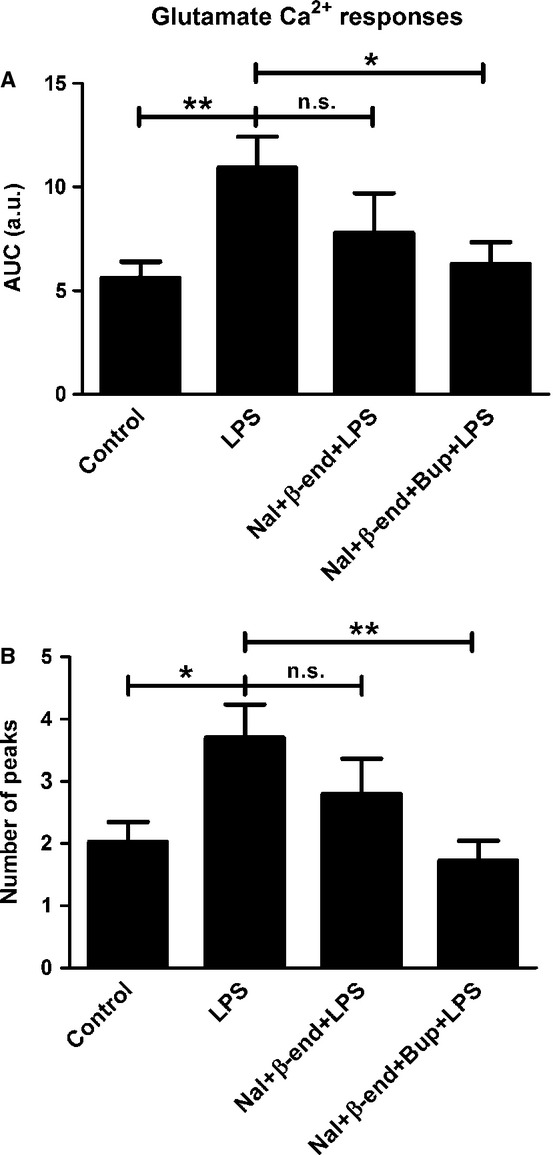
Glutamate elicited Ca2+ responses in the astrocytes and was used as control. (A) Cells incubated with LPS for 24 h showed increased glutamate Ca2+ signaling compared with the control. A combination of naloxone (Nal) (10−12 m) and β-endorphin (β-end) (10−6) did not attenuate the glutamate-induced Ca2+ transients that were upregulated by LPS. However, a combination of bupivacaine (Bup), Nal and β-end attenuated the LPS-upregulated glutamate-induced signaling. The areas under the Ca2+ peaks (AUC) were calculated. (B) The number of peaks was counted for each cell. Statistical analysis: the level of significance was analysed using one-way anova followed by Dunnett's multiple-comparisons test. **P < 0.01, *P < 0.05, n.s. = not significant. n = 40.
Ultralow concentrations of bupivacaine reduce IL-1β release
Astrocytes incubated with LPS for 24 h secreted IL-1β. Other astrocyte samples were treated with bupivacaine (10−12 or 10−6 m) 30 min prior to incubation with LPS and bupivacaine for 24 h. In these cells, the ultralow concentration of bupivacaine (10−12 m) reduced the LPS-induced increase in IL-1β secretion (Fig.11).
Figure 11.
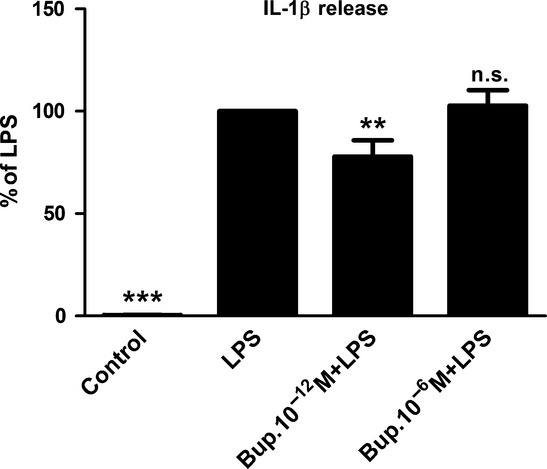
Ultralow concentrations of bupivacaine reduced the amount of IL-1β secreted. The astrocytes were treated with bupivacaine (10−12 or 10−6 m) 30 min prior to incubation with both LPS and bupivacaine for 24 h. The administration of bupivacaine at an ultralow concentration (10−12 m) reduced the LPS-induced IL-1β secretion. Statistical analysis: the level of significance was analysed using one-way anova followed by Dunnett's multiple-comparisons test. ***P < 0.001, **P < 0.01, n.s. = not significant. n = 6.
Discussion
Bupivacaine is clinically used at concentrations that inhibit neuronal voltage-gated Na+ channels, which results in the blockage of both nerve membrane excitation and action potential generation (Hildebrand et al., 2001). We found that bupivacaine at lower concentrations, < 10−8 m, evoked intracellular Ca2+ transients that were IP3 receptor-dependent in astrocytes. The concentration-dependent curve for bupivacaine did not follow a Gaussian curve. For peptides a dose–response curve does not seem to exist in an ordinary way. Endomorpin-1, nicotine and pituitary adenylate cyclase-activating peptide show similar patterns (Delbro et al., 2009; Hansson et al., 2009; Block et al., 2012). For monoamines and amino acids, ordinary dose–response curves are obtained. Therefore, we did not expand this curve to lower concentrations especially as 10−12 m bupivacaine interacted significantly with astroglial endomorphin-, 5-HT- and glutamate-receptor systems. At higher concentrations, > 10−8 m, bupivacaine blocked the Ca2+ release. The absence of a response at higher concentrations of bupivacaine may be due to the inhibition of Na+ channels.
The complexity of Ca2+ signaling has made it difficult to determine the underlying physiology of this phenomenon; however, it probably reflects an important function of astrocytes (Hansson & Rönnbäck, 2003; Hansson, 2010; Forshammar et al., 2011). In addition, we found that ultralow concentrations of bupivacaine decreased the LPS-induced increase in IL-1β release, which is also upregulated under inflammatory conditions. It seems important that bupivacaine has the ability to prevent an increase of IL-1β release under inflammatory conditions.
Given these results, we hypothesized that bupivacaine may have anti-inflammatory properties when used at ultralow concentrations. Under these conditions, bupivacaine could potentially restore the astrocyte network, which is influenced and reorganized by inflammatory stimuli (Hansson, 2006, 2010). The dysregulation of these inflammatory pathways can lead to pathogenic and chronic neuroinflammation and neurodegeneration, which can have deleterious effects on adjacent neurons (Farina et al., 2007).
Previous studies have indicated that opioids, alone or in combination with the μ-opioid antagonist naloxone and either 5-HT or glutamate, affect the astrocytes and the astrocyte network when these cells are inflammation-reactive (Hansson et al., 2008; Forshammar et al., 2011; Block et al., 2012; Gérard & Hansson, 2012).
A neuroinflammatory state is often induced when LPS is used both in vitro and in vivo. LPS is an endotoxin and a potent inflammatory activator (Nakamura, 2002) that stimulates the TLR4 of astrocytes and microglia (Kielian, 2006). The changes associated with neuroinflammation include the downregulation of Na+ transporters, a change in Ca2+ signaling in the astrocyte network and a release of cytokines (Morita et al., 2003; Schmidt et al., 2007; Hansson et al., 2008; Delbro et al., 2009; Vallejo et al., 2010).
In this study, the astrocytes were incubated with LPS, and a resulting increase in intracellular Ca2+ release was observed when the μ-opioid receptor agonist endomorphin-1 was used as a stimulator. Bupivacaine attenuated these Ca2+ oscillations and intracellular Ca2+ release. The mechanisms behind this interaction are currently unknown as it is unclear which cellular systems are affected by bupivacaine. Endomorphin-1 interacts with the Gi/0 protein, which stimulates or activates the sodium transporter Na+/K+-ATPase (Masocha et al., 2002). Bupivacaine may also interact with this system, and it is likely that bupivacaine acts at the receptor and/or ion level in the plasma membrane, given that these reactions occur within seconds. It has been reported that local anesthetics can modulate different subunits of G proteins. G protein-mediated release of IP3-sensitive Ca2+ stores was mediated by the Gαq subunit in Xenopus oocytes (Hollmann et al., 2001). Gαi-mediated inhibition of cAMP induced by activation of adenosine A1 receptors was expressed in CHO cells (Benkwitz et al., 2003). Furthermore, interactions between opioid receptors and Gαo subunits have been reported as well as GTP hydrolysis by Gi and Go proteins (Hageluken et al., 1994), and decreases in opioid inhibition of Ca2+ channels (Komai & McDowell, 2007).
Astrocytes exhibit an increase in intracellular Ca2+ response to a variety of stimuli, including glutamate, γ-amino butyric acid (GABA) and 5-HT (Nilsson et al., 1991; Hansson et al., 1994; Hagberg et al., 1998), which can result in the release of gliotransmitters, such as glutamate, d-serine and ATP (Parpura et al., 1994; Araque et al., 1998, 1999; Fiacco & McCarthy, 2004; Mothet et al., 2005), that bind to pre- and/or postsynaptic neuronal receptors to modulate synaptic transmission and activity (Dani et al., 1992; Nedergaard, 1994; Parri et al., 2001).
In the current study, we investigated whether bupivacaine influenced 5-HT- and/or glutamate-induced intracellular Ca2+ responses. LPS treatment resulted in increased Ca2+ oscillations and intracellular Ca2+ release. This increased Ca2+ signaling activity was attenuated when the astrocytes were also treated with bupivacaine in the 5-HT system. Furthermore, a combination of bupivacaine, ultralow concentrations of naloxone, and either endomorphin-1 or β-endorphin attenuated the LPS-induced upregulation of Ca2+ signaling activity in the glutamate system.
Both 5-HT and glutamate stimulate metabotropic receptors, which stimulate the Gq/11 protein. As bupivacaine interacts with both 5-HT and glutamate, it is possible that bupivacaine interacts with these systems as well. It is also possible that bupivacaine interacts with G proteins that then interfere with the cAMP system and other G proteins that can affect the IP3 system. Currently, there are no blocking agents available that can interact with or block these different systems. It would be very interesting to determine whether different G protein systems can be selectively blocked.
Based on these results, we conclude that low concentrations, < 10−8 m, of bupivacaine evoked Ca2+ transients that are IP3 receptor-dependent. These results suggest that bupivacaine interacts with the astrocyte Ca2+ signaling system. Bupivacaine also interacts with the μ-opioid-, 5-HT- and glutamate-receptor systems in inflammation-reactive astrocytes, and has the ability to restore cellular parameters that are altered by inflammation to their normal, non-inflammatory states. In addition, bupivacaine attenuates the IL-1β secretion that is upregulated in response to LPS. This result indicates that ultralow concentrations of bupivacaine exert anti-inflammatory astrocyte effects. Bupivacaine, when used at ultralow concentrations, might, through such mechanisms, be clinically useful as an anti-inflammatory drug, either alone or in combination with opioid agonists and an opioid antagonist at ultralow concentrations.
Acknowledgments
This work was supported by the Edit Jacobson Foundation, the Lena and Per Sjöberg Foundation, the Göteborg Medical Society, the Sahlgrenska University Hospital (LUA/ALF GBG-11587), Gothenburg, Sweden, and the Göteborg Region Association of Local Authorities (ALFGBG-220311). There are no conflicts of interest to report.
Glossary
Abbreviations
- 5-HT
5-hydroxytryptamine
- AUC
area under the curve
- HHBSS
Hank's HEPES-buffered saline solution
- IL-1
βinterleukin-1β
- IP3
inositol 1,4,5-trisphosphate
- LPS
lipopolysaccharide
- TLR4
Toll-like receptor 4
References
- Abbott NJ, Rönnbäck L. Hansson E. Astrocyte–endothelial interactions at the blood–brain barrier. Nat. Rev. Neurosci. 2006;7:41–53. doi: 10.1038/nrn1824. [DOI] [PubMed] [Google Scholar]
- Amir R, Argoff CE, Bennett GJ, Cummins TR, Durieux ME, Gerner P, Gold MS, Porreca F. Strichartz GR. The role of sodium channels in chronic inflammatory and neuropathic pain. J. Pain. 2006;7(Suppl 3):S1–S29. doi: 10.1016/j.jpain.2006.01.444. [DOI] [PubMed] [Google Scholar]
- Araque A, Parpura V, Sanzgiri RP. Haydon PG. Glutamate-dependent astrocyte modulation of synaptic transmission between cultured hippocampal neurons. Eur. J. Neurosci. 1998;10:2129–2142. doi: 10.1046/j.1460-9568.1998.00221.x. [DOI] [PubMed] [Google Scholar]
- Araque A, Sanzgiri RP, Parpura V. Haydon PG. Astrocyte-induced modulation of synaptic transmission. Can. J. Physiol. Pharm. 1999;77:699–706. [PubMed] [Google Scholar]
- Bedirli N, Akyürek N, Kurtipek O, Kavutcu M, Kartal S. Bayraktar AC. Thoracic epidural bupivacaine attenuates inflammatory response, intestinal lipid peroxidation, oxidative injury, and mucosal apoptosis induced by mesenteric ischemia/reperfusion. Anesth. Analg. 2011;113:1226–1232. doi: 10.1213/ANE.0b013e31822b8984. [DOI] [PubMed] [Google Scholar]
- Benkwitz C, Garrison JC, Linden J, Durieux ME. Hollmann MW. Lidocaine enhances Gαi protein function. Anesthesiology. 2003;99:1093–1101. doi: 10.1097/00000542-200311000-00015. [DOI] [PubMed] [Google Scholar]
- Berridge MJ. Inositol trisphosphate and calcium oscillations. Biochem. Soc. Symp. 2007;74:1–7. doi: 10.1042/BSS0740001. [DOI] [PubMed] [Google Scholar]
- Block L, Forshammar J, Lundborg C, Biber B. Hansson E. Naloxone in ultra-low concentration restores endomorphin-1-evoked Ca2+ signalling in inflammation pre-treated astrocytes. Neuroscience. 2012;205:1–9. doi: 10.1016/j.neuroscience.2011.12.058. [DOI] [PubMed] [Google Scholar]
- Blomstrand F, Khatibi H, Muyderman H, Hansson E, Olsson T. Rönnbäck L. 5-Hydroxytryptamine and glutamate modulate velocity and extent of intercellular calcium signalling in hippocampal astroglial cells in primary cultures. Neuroscience. 1999;88:1241–1253. doi: 10.1016/s0306-4522(98)00351-0. [DOI] [PubMed] [Google Scholar]
- Chiang C-Y, Sessle BJ. Dostrovsky JO. Role of astrocytes in pain. Neurochem. Res. 2012;37:2419–2431. doi: 10.1007/s11064-012-0801-6. [DOI] [PubMed] [Google Scholar]
- Dani JW, Chernjavsky A. Smith SJ. Neuronal activity triggers calcium waves in hippocampal astrocyte networks. Neuron. 1992;8:429–440. doi: 10.1016/0896-6273(92)90271-e. [DOI] [PubMed] [Google Scholar]
- Delbro D, Westerlund A, Björklund U. Hansson E. In inflammatory reactive astrocytes co-cultured with brain endothelial cells nicotine-evoked Ca2+ transients are attenuated due to IL-β release and rearrangement of actin filaments. Neuroscience. 2009;159:770–779. doi: 10.1016/j.neuroscience.2009.01.005. [DOI] [PubMed] [Google Scholar]
- Farina C, Aloisi F. Meinl E. Astrocytes are active players in cerebral innate immunity. Trends Immunol. 2007;28:138–145. doi: 10.1016/j.it.2007.01.005. [DOI] [PubMed] [Google Scholar]
- Fiacco TA. McCarthy KD. Intracellular astrocyte calcium waves in situ increase the frequency of spontaneous AMPA receptor currents in CA1 pyramidal neurons. J. Neurosci. 2004;24:722–732. doi: 10.1523/JNEUROSCI.2859-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forshammar J, Block L, Lundborg C, Biber B. Hansson E. Naloxone and ouabain in ultra-low concentrations restore Na+/K+-ATPase and cytoskeleton in lipopolysaccharide-treated astrocytes. J. Biol. Chem. 2011;286:31586–31597. doi: 10.1074/jbc.M111.247767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Froger N, Orellana JA, Calvo C-F, Amigou E, Kozoriz MG, Naus CC, Sáez J. Giaume C. Inhibition of cytokine-induced connexin43 hemichannel activity in astrocytes is neuroprotective. Mol. Cell. Neurosci. 2010;45:37–46. doi: 10.1016/j.mcn.2010.05.007. [DOI] [PubMed] [Google Scholar]
- Gao Y-J. Ji R-R. Targeting astrocyte signaling for chronic pain. Neurotherapeutics. 2010;7:482–493. doi: 10.1016/j.nurt.2010.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gérard F. Hansson E. Inflammatory activation enhances NMDA-triggered Ca2+ signaling and IL-1β secretion in primary cultures of rat astrocytes. Brain Res. 2012;1473:1–8. doi: 10.1016/j.brainres.2012.07.032. [DOI] [PubMed] [Google Scholar]
- Hagberg GB, Blomstrand F, Nilsson M, Tamir H. Hansson E. Stimulation of 5-HT2A receptors on astrocytes in primary culture opens voltage-independent Ca2+ channels. Neurochem. Int. 1998;32:153–162. doi: 10.1016/s0197-0186(97)00087-9. [DOI] [PubMed] [Google Scholar]
- Hageluken A, Grunbaum L, Nurnberg B, Harhammer R, Schunack W. Seifert R. Lipophilic beta-adrenoceptor antagonists and local anesthetics are effective direct activators of G-proteins. Biochem. Pharmacol. 1994;47:1789–1795. doi: 10.1016/0006-2952(94)90307-7. [DOI] [PubMed] [Google Scholar]
- Hansson E. Could chronic pain and spread of pain sensation be induced and maintained by glial activation? Acta Physiol. 2006;187:321–327. doi: 10.1111/j.1748-1716.2006.01568.x. [DOI] [PubMed] [Google Scholar]
- Hansson E. Long-term pain, neuroinflammation and glial activation. Scand. J. Pain. 2010;1:67–72. doi: 10.1016/j.sjpain.2010.01.002. [DOI] [PubMed] [Google Scholar]
- Hansson E. Rönnbäck L. Glial neuronal signalling in the central nervous system. FASEB J. 2003;17:341–348. doi: 10.1096/fj.02-0429rev. [DOI] [PubMed] [Google Scholar]
- Hansson E, Johansson BB, Westergren I. Rönnbäck L. Glutamate-induced Swelling of single astroglial cells in primary culture. Neuroscience. 1994;63:1057–1066. doi: 10.1016/0306-4522(94)90572-x. [DOI] [PubMed] [Google Scholar]
- Hansson E, Westerlund A, Björklund U. Olsson T. μ-Opioid agonists restore intracellular Ca2+ responses in inflammatory activated astrocytes co-cultured with brain endothelial cells. Neuroscience. 2008;155:1237–1249. doi: 10.1016/j.neuroscience.2008.04.027. [DOI] [PubMed] [Google Scholar]
- Hansson E, Westerlund A, Björklund U. Rönnbäck L. PACAP attenuates 5-HT, histamine, and ATP-evoked Ca2+ transients in astrocytes. NeuroReport. 2009;20:957–962. doi: 10.1097/WNR.0b013e32832ca201. [DOI] [PubMed] [Google Scholar]
- Hildebrand KR, Elsberry DD. Deer TR. Stability, compatibility, and safety of intrathecal bupivacaine administered chronically via an implatable delivery system. Clin. J. Pain. 2001;17:239–244. doi: 10.1097/00002508-200109000-00009. [DOI] [PubMed] [Google Scholar]
- Hollmann MW, Wieczorek KS, Berger A. Durieux ME. Local anesthetic inhibition of G-protein function. Mol. Pharmacol. 2001;59:294–301. doi: 10.1124/mol.59.2.294. [DOI] [PubMed] [Google Scholar]
- Huber JD, Witt KA, Hom S, Egleton RD, Mark S. Davis TP. Inflammatory pain alters blood brain barrier permeability and tight junctional protein expression. Am. J. Physiol. Heart Circ. Physiol. 2001;280:H1241–H1248. doi: 10.1152/ajpheart.2001.280.3.H1241. [DOI] [PubMed] [Google Scholar]
- Kielian T. Toll-like receptors in central nervous system, glial inflammation and homeostasis. J. Neurosci. Res. 2006;83:711–730. doi: 10.1002/jnr.20767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komai H. McDowell TS. Effects of local anesthetics on opioid inhibition of calcium current in rat dorsal root ganglion neurons. Neurosci. Lett. 2007;418:298–303. doi: 10.1016/j.neulet.2007.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowry OH, Rosebrough NJ, Farr AL. Randall RJ. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- Lundborg C, Hahn-Zoric M, Biber B. Hansson E. Glial cell line-derived neurotrophic factor is increased in cerebrospinal fluid but decreased in blood during long-term pain. J. Neuroimmunol. 2010;220:108–113. doi: 10.1016/j.jneuroim.2010.01.007. [DOI] [PubMed] [Google Scholar]
- Lundborg C, Westerlund A, Björklund U, Biber B. Hansson E. Ifenprodil restores GDNF-evoked Ca2+ signalling and Na+/K+-ATPase expression in inflammation-pretreated astrocytes. J. Neurochem. 2011;119:686–696. doi: 10.1111/j.1471-4159.2011.07465.x. [DOI] [PubMed] [Google Scholar]
- Masocha W, Gonzalez LG, Baeyens JM. Agil A. Mechamisms involved in morphine-induced activation of synaptosomal Na+, K+, ATP-ase. Brain Res. 2002;2:311–319. doi: 10.1016/s0006-8993(02)03663-6. [DOI] [PubMed] [Google Scholar]
- McMahon SB. Malcangio M. Current challenges in glia-pain biology. Neuron. 2009;64:46–54. doi: 10.1016/j.neuron.2009.09.033. [DOI] [PubMed] [Google Scholar]
- Morita M, Higuchi C, Moto T, Kozuka N, Susuki J, Itofusa R, Yamashita J. Kudo Y. Dual regulation of calcium oscillation in astrocytes by growth factors and pro inflammatory cytokines via the mitogen-activated protein kinase cascade. J. Neurosci. 2003;23:10944–10952. doi: 10.1523/JNEUROSCI.23-34-10944.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mothet JP, Pollegioni L, Ouanounou G, Martineau M, Fossier P. Baux G. Glutamate receptor activation triggers a calcium-dependent and SNARE protein-dependent release of the gliotransmitter D-serine. Proc. Natl. Acad. Sci. USA. 2005;102:5606–5611. doi: 10.1073/pnas.0408483102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura Y. Regulating factors for microglial activation. Biol. Pharm. Bull. 2002;25:945–953. doi: 10.1248/bpb.25.945. [DOI] [PubMed] [Google Scholar]
- Nedergaard M. Direct signaling from astrocytes to neurons in cultures of mammalian Brain cells. Science. 1994;263:1768–1771. doi: 10.1126/science.8134839. [DOI] [PubMed] [Google Scholar]
- Nilsson M, Hansson E. Rönnbäck L. Adrenergic and 5-HT2 receptors on the same astroglial cell. A microspectrofluorimetric study on cytosolic Ca2+ responses in single cells in primary culture. Brain Res. Dev. Brain Res. 1991;63:33–41. doi: 10.1016/0165-3806(91)90064-p. [DOI] [PubMed] [Google Scholar]
- Parpura V, Basarsky TA, Liu F, Jeftinija K, Jeftinija S. Haydon PG. Glutamate mediated astrocyte-neuron signalling. Nature. 1994;369:744–747. doi: 10.1038/369744a0. [DOI] [PubMed] [Google Scholar]
- Parri HR, Gould TM. Crunelli V. Spontaneous astrocytic Ca2+ oscillations in situ drive NMDAR mediated neuronal excitation. Nat. Neurosci. 2001;4:803–812. doi: 10.1038/90507. [DOI] [PubMed] [Google Scholar]
- Retamal MA, Froger N, Palacios-Prado N, Ezan P, Sáez JC. Giaume C. Cx43 hemichannels and gap junction channels in astrocytes are regulated oppositely by proinflammatory cytokines released from activated microglia. J. Neurosci. 2007;27:13781–13792. doi: 10.1523/JNEUROSCI.2042-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saadé NE. Jabbur SJ. Nociceptive behaviour in animal models for peripheral neuropathy: spinal and supraspinal mechanisms. Prog. Neurobiol. 2008;86:22–47. doi: 10.1016/j.pneurobio.2008.06.002. [DOI] [PubMed] [Google Scholar]
- Scemes E. Giaume C. Astrocyte calcium waves: what they are and what they do. GLIA. 2006;54:716–725. doi: 10.1002/glia.20374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt C, Höcherl K, Schweda F, Kurtz A. Bucher M. Regulation of renal sodium transporters during severe inflammation. J. Am. Soc. Nephrol. 2007;18:1072–1083. doi: 10.1681/ASN.2006050454. [DOI] [PubMed] [Google Scholar]
- Sharma G. Vijayaraghavan S. Nicotinic cholinergic signaling in hippocampal astrocytes involves calcium-induced calcium release from intracellular stores. Proc. Natl. Acad. Sci. USA. 2001;98:4148–4153. doi: 10.1073/pnas.071540198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toda S, Sakaim A, Ikeda Y, Sakamoto A. Suzuki H. A local anesthetic, ropivacaine, suppresses activated microglia via a nerve growth factor-dependent mechanism and astrocytes via a nerve growth factor-independent mechanism in neuropathic pain. Mol. Pain. 2011;7:1–11. doi: 10.1186/1744-8069-7-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallejo R, Tilley DM, Vogel L. Benyamin R. The role of glia and the immune system in the development and maintenance of neuropathic pain. Pain Pract. 2010;10:167–184. doi: 10.1111/j.1533-2500.2010.00367.x. [DOI] [PubMed] [Google Scholar]
- Willis CL. Davis TP. Chronic inflammatory pain and the neurovascular unit: a central role for glia in maintaining BBB integrity? Curr. Pharm. Design. 2008;14:1625–1643. doi: 10.2174/138161208784705414. [DOI] [PubMed] [Google Scholar]