

Abstract
Detection of fluorescence provides the foundation for many widely utilized and rapidly advancing microscopy techniques employed in modern biological and medical applications. Strengths of fluorescence include its sensitivity, specificity, and compatibility with live imaging. Unfortunately, conventional forms of fluorescence microscopy suffer from one major weakness, diffraction-limited resolution in the imaging plane, which hampers studies of structures with dimensions smaller than ~250 nm. Recently, this limitation has been overcome with the introduction of super-resolution fluorescence microscopy techniques, such as photoactivated localization microscopy (PALM). Unlike its conventional counterparts, PALM can produce images with a lateral resolution of tens of nanometers. It is thus now possible to use fluorescence, with its myriad strengths, to elucidate a spectrum of previously inaccessible attributes of cellular structure and organization.
Unfortunately, PALM is not trivial to implement, and successful strategies often must be tailored to the type of system under study. In this article, we show how to implement single-color PALM studies of vesicular structures in fixed, cultured neurons. PALM is ideally suited to the study of vesicles, which have dimensions that typically range from ~50-250 nm. Key steps in our approach include labeling neurons with photoconvertible (green to red) chimeras of vesicle cargo, collecting sparsely sampled raw images with a super-resolution microscopy system, and processing the raw images to produce a high-resolution PALM image. We also demonstrate the efficacy of our approach by presenting exceptionally well-resolved images of dense-core vesicles (DCVs) in cultured hippocampal neurons, which refute the hypothesis that extrasynaptic trafficking of DCVs is mediated largely by DCV clusters.
Keywords: Neuroscience, Issue 89, photoactivated localization microscopy, Dendra2, dense-core vesicle, synapse, hippocampal neuron, cluster
Introduction
A number of cellular processes depend on accurate and efficient vesicle-mediated trafficking of biomolecules to specific subcellular destinations. One prominent example is synaptic assembly, which is preceded by long-ranged, vesicle-mediated delivery of synaptic constituents from sites of biogenesis in the neuronal soma to potentially distal pre- and postsynaptic sites1.
Fluorescence microscopy is a powerful and popular method of studying vesicle trafficking. Strengths of the technique include its sensitivity, specificity, and compatibility with live imaging2. Unfortunately, until relatively recently, the technique has suffered from one major weakness, diffraction-limited resolution2, which hampers studies of structures with dimensions smaller than ~250 nm. Recently, lateral resolution in fluorescence microscopy surpassed the diffraction barrier with the introduction of super-resolution fluorescence microscopy techniques, such as PALM3. The lateral resolution of PALM, tens of nanometers, is ideally suited to the study of vesicles, which have dimensions that typically range from ~50-250 nm4. It is thus now possible to use fluorescence, with its myriad strengths, to elucidate a spectrum of previously inaccessible attributes of vesicles, including some aspects of their trafficking to specific subcellular sites.
PALM is not trivial to implement, and successful strategies often must be tailored to the type of system under study. Here we describe how to implement PALM studies of vesicular structures, and we demonstrate the efficacy of our approach for the case of DCVs in hippocampal neurons. In particular, we use PALM to address the hypothesis that trafficking of DCVs to synapses in hippocampal neurons is mediated by DCV clusters5-8.
Cluster-mediated trafficking of vesicles to synapses in developing neurons is an intriguing possibility because it may facilitate rapid synaptic stabilization and assembly9,10. Proponents of clustered trafficking of DCVs cite the large apparent size of extrasynaptic fluorescent puncta harboring exogenous DCV cargo as evidence supporting clustering6. However, these puncta appear in images generated using diffraction-limited fluorescence microscopy techniques, which are not suited to distinguishing size effects arising from diffraction from those arising from clustering.
To resolve this issue, we collected conventional widefield fluorescence and PALM images of hippocampal neurons expressing chimeras targeted to DCVs. Analysis of these images revealed that >92% of putative extrasynaptic DCV clusters in conventional images are resolved as 80 nm (individual DCV-sized)11 puncta in PALM images. Thus, these data largely invalidate the clustering hypothesis as applied to DCVs in developing hippocampal neurons.
Protocol
1. Sample Preparation
Prepare DNA encoding a photoconvertible or photoactivatable chimera targeted to DCVs, using standard molecular biology techniques. Note: One possibility is DNA encoding a tissue plasminogen activator-Dendra2 chimera (tPA-Dendra2). Dendra2 is a photoconvertible protein that switches from green to red emission upon exposure to ultraviolet light12.
Culture hippocampal neurons on high performance #1.5 cover slips for ~5-10 days, following standard protocols13.
- Transfect developing hippocampal neurons using a cationic lipid reagent. Note: Viral-mediated infection is more effective for mature neurons. This alternative approach has been described in detail elsewhere14.
- Prepare a 1.5 ml tube containing ~1 µg of DNA and 250 µl of minimal essential medium (MEM) and a second 1.5 ml tube containing 4 µl of the cationic lipid reagent and 250 µl of MEM.
- Incubate the solutions for 5 min.
- Combine the two solutions and incubate the resulting mixture for an additional 30 min.
- During the incubation, prepare a dish containing several ml of culture medium warmed to 37 °C.
- Gently transfer cover slips containing neurons to the dish containing warm medium and add the transfection mixture.
- Tilt the dish gently and then place it in an incubator at 37 °C for 90 min.
- Return neurons to their "home dish" and wait ~12-18 hr.
Fix cells for 45 min in 4% paraformaldehyde/4% sucrose in phosphate buffered saline (PBS, pH 7.4) warmed to 37 °C. Note: In PALM experiments, it is important to achieve good preservation of ultrastructure, and standard formaldehyde fixation protocols used for immunofluorescence staining typically are not sufficient15. One important reason for poor structural preservation is insufficient fixation time because formaldehyde fixation involves adduct formation and protein crosslinking, and the latter process is quite slow16. A 45 min fixation helps to mitigate this problem with no detectable overfixation-induced loss of fluorescence.
Wash cover slips ~10x with filtered PBS. Note: This reduces fluorescent contaminants. Image samples relatively quickly after fixation. In the interim, store cells at 4 °C in filtered PBS in a light-tight container.
Mount cover slips containing neurons in a chamber in filtered PBS. Note: The sample must be mounted in an aqueous medium, like PBS, with refractive index lower than that of glass, to implement total internal reflection fluorescence (TIRF)-based illumination. TIRF-based illumination is very useful because only cover slip-proximal fluorophores are excited, and there is a large associated drop in background fluorescence17.
2. Image Acquisition
- Turn on the super-resolution imaging system.
- Turn on the arc lamp.
- Flip the "system/pc" and "component" switches (on the power remote switch) to "on."
- Turn on the computer (after the microscope control tablet/docking station comes on). Note: The docking station can be used to control and monitor many attributes of the optics and light path, notably choice of objective and focus.
- Double click the software icon and choose the "start system" option in the login dialog.
- Examine the sample.
- Open the front and top access panels on the laser safety cabinet.
- Tilt the transmitted light arm to access the stage and objectives.
- Put oil on the alpha Plan-Apochromat 100X numerical aperture/NA = 1.46 (TIRF) objective. Note: A 63X high-NA objective can be used in lieu of the 100X.
- Mount the sample on the stage, and raise the objective toward the sample.
- Monitor the lens position using the XYZ function of the docking station. Stop when the lens is in the ballpark of focus (e.g., z ~2.50 mm).
- Fully close the access panels. Note: To engage the laser safety.
- Fine tune the focus using the oculars and buttons in the locate tab. Note: The locate tab provides access to buttons that produce arc lamp-based illumination and facilitates direct observation of the sample with the oculars. Similarly, the acquisition tab provides access to tools that produce laser-based illumination and facilitates image capture by the camera.
- Go to the locate tab, choose "Trans On."
- Click the ocular expansion symbol. Note: This brings up a schematic of the light path. Attributes of the light path can be altered by left clicking appropriate icons in the schematic or by using the touch screen on the docking station. For example, a filter appropriate for viewing emission from Dendra2 can be chosen using the reflector revolver icon.
- Look into the eyepieces, and bring the cells into focus using the docking station as a guide. Note: The sample will be very sparsely populated with fluorescent cells because hippocampal neurons are difficult to transfect, and it thus can be a little tricky to focus using reflected illumination. If this is the case, use transmitted illumination and the z reading of the docking station as a guide.
- Toggle the light "Off" after focusing.
- Identify a cell that is fairly bright, exhibits punctate fluorescence, and has a classic morphology that is indicative of good health.
- Choose reflected light and the filter appropriate for viewing green emission from Dendra2.
- Scan the cover slip for cells expressing Dendra2 chimeras using low intensity excitation light.
- Switch to the acquisition tab.
- Use the "experiment manager" to load a PALM acquisition configuration that is appropriately designed or easily modified. Note: If no such experiment exists, design a starting experimental configuration using the following settings as a guide: Laser power sliders: 405 nm (~0.01% for a 50 mW laser); 488 nm (~3% for a 100 mW laser); 561 nm (~40% for a 100 mW laser); Exposure time: ~50 msec; Camera gain: ~200; Multidimensional acquisition: time series (cycles = 30,000, interval = 0); Tracks: 488 nm with epi-illumination, and 561 nm with TIRF-based illumination (see step 2.5.1); Objective: 100X (NA = 1.46); TIRF angle slider: incidence angle ~67-72°; Pixel size: 100 nm; Field of view: TIRF (the alternative choices yield higher laser intensities and are used for fluorophores that are more difficult to bleach).
- Click "yes" when a pop up menu appears with a query about switching on lasers.
- Bring the image of the cell into focus at the camera plane.
- Choose the 488 nm epi-illumination laser track. Note: Tracks are beam configurations used during imaging. Here, track names are determined by the excitation wavelength that produces the emission that is passed to the camera. For example, the 561 nm track uses both the 405 and 561 nm laser lines, but the filter cube passes only emission from photoconverted Dendra2 excited by 561 nm light to the camera.
- Focus the image of the cell on the camera using the "continuous" acquisition button.
- Collect a conventional widefield image.
- Use the "Snap" button.
- Save the image by going to the "File" menu and choosing "save/save as."
- Set up the TIRF-based illumination.
- Choose the 488 track and switch to "TIRF" using the EPI/TIRF button.
- Choose "continuous" acquisition.
- Adjust the TIRF-based illumination slider. Note: TIRF is achieved when there is a sudden darkening of background AND the specimen can be focused in ONE plane18. If new features become apparent via focusing up into the sample, the incidence angle is too low because in TIRF there is only one plane of focus.
- Double check the TIRF angle using the 561 track with the 405 nm laser "off."
- Turn the 405 nm laser back "on" before initiating a PALM experiment.
- Collect PALM data.
- Hit "start experiment." Note: If desired, open the "online processing options" tab and check "online processing PALM" and "Fit Gauss2D" (before starting the experiment) to monitor the reconstructed PALM image during raw data collection. This will facilitate assessment of the quality of the data and thus the value of continuing the relatively time-consuming data collection process.
- Visually monitor photoconversion.
- Increase the intensity of the 405 nm (photoconverting) laser when Dendra2 photoconversion diminishes (typically after acquisition of ~10,000 raw images).
- Continue the experiment if photoconversion increases. Otherwise, stop the experiment (without loss of data) by hitting the "stop" current step button.
- Save the images when data collection is complete (after ~15 min).
3. Image Processing, Display, and Analysis
- In the absence of online processing, process raw images that were saved to disk.
- Click on the processing tab, open the method tab, and select "PALM."
- Choose "PALM" again from the four suboptions.
- Go to the file menu, open and view the file of interest, and hit "select."
- Hit "apply." Note: This will initiate peak/fluorophore finding and peak localization with default options. If necessary, peaks also can be grouped using the "PAL-Group" tab. Grouping assigns peaks that appear in several consecutive frames to a single molecule if the peak coordinates are sufficiently similar. In addition, the data can be corrected for sample drift using the software’s model-based drift alignment option.
- Filter out poorly localized and poorly sampled components in the peak-localized data.
- Discard fluorophores with localization precisions, σ, >35 nm using the "PAL-Filter" tool.
- Discard fluorophores in areas where the mean distance between peaks, d, is too large to resolve vesicles of diameter, D.
- Extract the mean localization precision, σ, from the localization precision histogram.
- Calculate a maximum acceptable value for d based on σ, and the Shannon-Nyquist criteron19, as embodied in the resolution approximation20 R = D = [(2.35σ)2 + (2d)2]1/2.
- Set the threshold in the "Remove PALM outliers" tool to delete peaks surrounded by fewer than πD2/(4d2) neighbors within a circle of radius D/2.
- Convert the processed PALM data into a PALM image using the "PAL-Rendering" tool; see Figure 1.
- Quantify sizes and separations by choosing the "Profile" function.
- Check the associated line and table options, and draw a line along the desired direction in the image.
- Quantify diameters by determining full widths at half maximal intensity.
- Quantify separations by determining peak-to-peak spacings of intensity profiles.
Representative Results
Figure 1 shows one end product of imaging and processing. In this PALM image, lateral coordinates of localized fluorophores are shown using the centroid display mode, and the super-resolution image of the associated DCV is shown using the Gaussian display mode.
Figure 2A shows analogous widefield and PALM images of the soma and proximal processes of an eight days in vitro hippocampal neuron expressing tPA-Dendra2. Important features of the images include the (1) extensive, one-to-one correspondence, and overlap, between puncta in the conventional and PALM images, (2) significantly smaller size of the PALM puncta, and (3) occasional resolution of a single widefield punctum into multiple PALM puncta.
Figure 2B shows a PALM image of DCVs along part of a process of a second hippocampal neuron expressing tPA-Dendra2. Important features of this image include the (1) small diameter, and homogeneous appearance, of the puncta, and (2) infrequent observation of closely apposed puncta/putative DCV clusters.
Figure 3 outlines a simple quantitative method of determining if two or more DCVs are close enough to comprise a cluster; in systems where clustering is prevalent, more sophisticated methods, based on distribution functions, can be used for quantification21. The simple method is based on generating line profiles of puncta intensities (shown in the graphs), which can be used to quantify DCV separation and DCV width. If separation significantly exceeds width (as discussed in the legend), the DCVs do not "contact," whereas if separation is approximately the same as width, the DCVs might contact. Based on this criterion, the DCVs in Panel A were classified as "not in contact" whereas those in Panel B were classified as in contact. Using this approach, we put an upper bound of ~8% on contact-associated clustering of extrasynaptic DCVs in developing hippocampal neurons. Our DCV size and cluster data are summarized in Table 1.
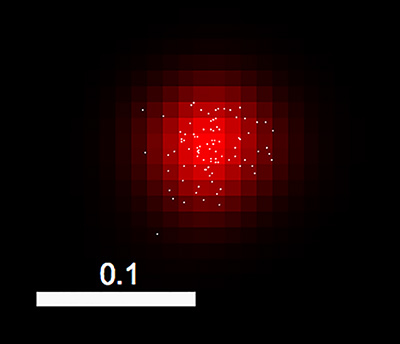 Figure 1. Overlay of a Gaussian-rendered PALM image of a DCV (red) and a centroid-display mode PALM image showing coordinates of individual photoconverted fluorophores (white dots) contained in the DCV. The former mode displays fluorophores rendered to Gaussian functions with widths determined by their localization precisions, and the latter displays fluorophores/peaks as dots localized at their (x,y) coordinates. Bar = 0.1 µm. Click here to view larger image.
Figure 1. Overlay of a Gaussian-rendered PALM image of a DCV (red) and a centroid-display mode PALM image showing coordinates of individual photoconverted fluorophores (white dots) contained in the DCV. The former mode displays fluorophores rendered to Gaussian functions with widths determined by their localization precisions, and the latter displays fluorophores/peaks as dots localized at their (x,y) coordinates. Bar = 0.1 µm. Click here to view larger image.
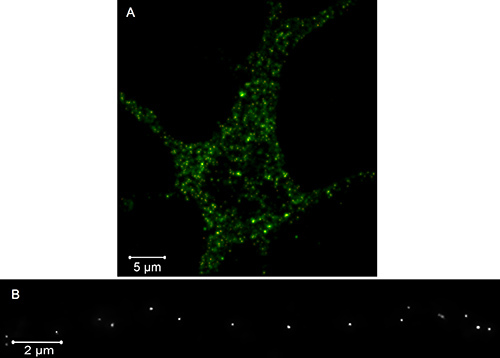 Figure 2. Overlay (A) of summed widefield (green) and PALM (red) images of the soma and proximal processes of a developing hippocampal neuron expressing tPA-Dendra2. Regions of overlap appear yellow/orange. PALM image (B) of DCVs lining a subregion of a process of another cell expressing tPA-Dendra2. Bars = 5 and 2 µm. Click here to view larger image.
Figure 2. Overlay (A) of summed widefield (green) and PALM (red) images of the soma and proximal processes of a developing hippocampal neuron expressing tPA-Dendra2. Regions of overlap appear yellow/orange. PALM image (B) of DCVs lining a subregion of a process of another cell expressing tPA-Dendra2. Bars = 5 and 2 µm. Click here to view larger image.
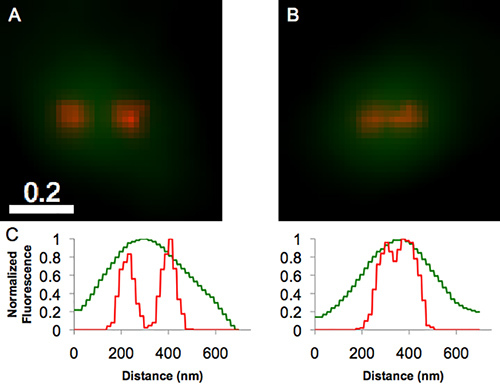 Figure 3. Overlays (A and B) of summed widefield (green) and PALM (red) images of puncta in a developing hippocampal neuron expressing tPA-Dendra2. In both panels, the widefield image shows a single large punctum, whereas the associated PALM image resolves this single punctum into two much narrower puncta. The graphs show analogously color-coded intensity profiles obtained for the puncta in panel A (left graph) and in panel B (right graph). The profiles derived from the PALM image in panel A show that the puncta have a full width at half maximal intensity that is ~40% of their peak-to-peak spacing; thus, these two puncta are not in contact and were not classified as a putative cluster. In contrast, the profiles derived from the PALM image in panel B show that the puncta have a full width at half maximal intensity that is essentially the same as their peak-to-peak spacing; thus, these were classified as a putative cluster. Bar = 0.2 µm. Click here to view larger image.
Figure 3. Overlays (A and B) of summed widefield (green) and PALM (red) images of puncta in a developing hippocampal neuron expressing tPA-Dendra2. In both panels, the widefield image shows a single large punctum, whereas the associated PALM image resolves this single punctum into two much narrower puncta. The graphs show analogously color-coded intensity profiles obtained for the puncta in panel A (left graph) and in panel B (right graph). The profiles derived from the PALM image in panel A show that the puncta have a full width at half maximal intensity that is ~40% of their peak-to-peak spacing; thus, these two puncta are not in contact and were not classified as a putative cluster. In contrast, the profiles derived from the PALM image in panel B show that the puncta have a full width at half maximal intensity that is essentially the same as their peak-to-peak spacing; thus, these were classified as a putative cluster. Bar = 0.2 µm. Click here to view larger image.
| Parameter | Result | Number of DCVs analyzed |
| Upper-bound on clustered DCVs | 7.9 ± 5.9% | 618 |
| Mean DCV diameter | 77 ± 21 nm | 60 |
Table 1. DCV size and cluster data summary.
Discussion
PALM and related super-resolution fluorescence microscopy techniques have recently emerged as a valuable complement to better-established forms of optical microscopy and electron microscopy (EM)22-24. Positive attributes of PALM include relatively simple sample preparation and minimal sample perturbation. In principle, PALM also can be used to study living cells. Probably the main negative attribute of PALM is time-consuming data acquisition, which significantly hampers studies of faster dynamic processes in living cells. In addition, PALM is relatively new, and thus appropriate, robust fluorophores and protocols for sample preparation and for data acquisition and analysis are not fully developed and/or documented3.
Here we have described a protocol that is suited for single-color, PALM-based studies of vesicles in fixed, cultured cells. The protocol also is a good starting point for the development of similarly directed, dual-color PALM studies. The efficacy of our protocol is supported by two key attributes of our PALM data. First, the distribution of puncta in our reconstructed PALM, and complementary widefield, images is very similar. The one significant difference in distribution - occasional resolution of diffraction-limited puncta into multiple puncta by PALM - reflects the markedly enhanced resolution of PALM and further underscores the technique's efficacy and utility.
Second, our PALM results are in good agreement, and/or consistent, with other relevant results. For example, images of hippocampal neurons obtained using EM occasionally reveal slices through DCVs, and the DCV diameter deduced from the slices is ~70-100 nm11. The remarkable agreement between our PALM data and the EM data is reassuring for both approaches. PALM still is being tested, and EM generally is subject to concerns about the requisite harsh sample preparation. Similarly, diffraction-limited movies of living hippocampal neurons coexpressing green and red chimeras targeted to DCVs show that overlapping green and red puncta undergo persistent comovement25. The most straightforward interpretation of this result - the red and green signals arise from the same DCV - is consistent with PALM, which demonstrates that diffraction-limited puncta overwhelmingly represent individual DCVs. A less straightforward interpretation of comovement - the red and green signals arise from clustered DCVs - is largely invalidated by PALM.
The protocol described here has other positive attributes. In particular, sample preparation is relatively straightforward and quite similar to that suited for conventional fluorescence microscopy, with two notable exceptions: (1) vesicles must be labeled with photoconvertible or photoactivatable fluorophores, and (2) fixed cells are best mounted in aqueous media. This relative simplicity reflects the fact that the experiments are short, and thus samples do not need to be labeled with fiducial beads (to facilitate drift correction), which can be tricky26. In addition, vesicles usually are tagged with many fluorophores; thus, achieving adequate fluorophore labeling density is straightforward. In contrast, for other structures, this can be quite difficult3. Image acquisition also is relatively straightforward. However, PALM does require electronic and optical components that are expensive and cutting-edge because PALM signals are intrinsically very weak.
Probably the most sophisticated aspects of PALM center around the processing, analysis, and display of PALM images. As highlighted here, implementing these aspects of PALM requires an understanding of several subtle issues, including the relationship between sampling and resolution, and the distinction between resolution and localization precision. Thus, overall, PALM studies invariably are quite intricate, but this is balanced by resolution enhancement that is likely to be significant.
Disclosures
The authors have nothing to disclose.
Acknowledgments
This work was supported by National Institutes of Health grants 2 R15 GM061539-02 (to B.A.S.), 2 R15 NS40425-03 (to J.E.L.), MH 66179 (to Dr. Gary Banker of Oregon Health & Science University/OHSU), and P30 NS061800 (to Dr. Sue Aicher of OHSU). We thank Barbara Smoody for extensive support with the culture of hippocampal neurons, and Drs. Brian Long and James Abney for a critical reading of this manuscript.
References
- Goldstein AY, Wang X, Schwarz TL. Axonal transport and the delivery of pre-synaptic components. Curr Opin Neurobiol. 2008;18:495–503. doi: 10.1016/j.conb.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Combs CA. Fluorescence microscopy: a concise guide to current imaging methods. Curr Protoc Neurosci. 2010. [DOI] [PMC free article] [PubMed]
- Sengupta P, Van Engelenburg S, Lippincott-Schwartz J. Visualizing cell structure and function with point-localization superresolution imaging. Dev Cell. 2012;23:1092–1102. doi: 10.1016/j.devcel.2012.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alberts B, et al. Molecular Biology of the Cell. Garland Science Publishing; 2007. [Google Scholar]
- Bury LA, Sabo SL. Coordinated trafficking of synaptic vesicle and active zone proteins prior to synapse formation. Neural Dev. 2011;6 doi: 10.1186/1749-8104-6-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda N, et al. Differential activity-dependent secretion of brain-derived neurotrophic factor from axon and dendrite. J Neurosci. 2009;29:14185–14198. doi: 10.1523/JNEUROSCI.1863-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao-Cheng JH. Ultrastructural localization of active zone and synaptic vesicle proteins in a preassembled multi-vesicle transport aggregate. Neuroscience. 2007;150:575–584. doi: 10.1016/j.neuroscience.2007.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai RG, et al. Assembling the presynaptic active zone: a characterization of an active zone precursor vesicle. Neuron. 2001;29:131–143. doi: 10.1016/s0896-6273(01)00185-4. [DOI] [PubMed] [Google Scholar]
- Ahmari SE, Buchanan J, Smith SJ. Assembly of presynaptic active zones from cytoplasmic transport packets. Nat Neurosci. 2000;3:445–451. doi: 10.1038/74814. [DOI] [PubMed] [Google Scholar]
- Garner CC, Zhai RG, Gundelfinger ED, Ziv NE. Molecular mechanisms of CNS synaptogenesis. Trends Neurosci. 2002;25:243–251. doi: 10.1016/s0166-2236(02)02152-5. [DOI] [PubMed] [Google Scholar]
- Chuang JZ, Milner TA, Zhu M, Sung CH. A 29 kDa intracellular chloride channel p64H1 is associated with large dense-core vesicles in rat hippocampal neurons. J Neurosci. 1999;19:2919–2928. doi: 10.1523/JNEUROSCI.19-08-02919.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chudakov DM, Lukyanov S, Lukyanov KA. Using photoactivatable fluorescent protein Dendra2 to track protein movement. Biotechniques. 2007;42 doi: 10.2144/000112470. [DOI] [PubMed] [Google Scholar]
- Kaech S, Banker G. Culturing hippocampal neurons. Nat Protoc. 2006;1:2406–2415. doi: 10.1038/nprot.2006.356. [DOI] [PubMed] [Google Scholar]
- Scalettar BA, et al. Hindered submicron mobility and long-term storage of presynaptic dense-core granules revealed by single-particle tracking. Dev Neurobiol. 2012;72:1181–1195. doi: 10.1002/dneu.20984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippincott-Schwartz J, Manley S. Putting super-resolution fluorescence microscopy to work. Nat Methods. 2009;6:21–23. doi: 10.1038/nmeth.f.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helander KG. Formaldehyde binding in brain and kidney: A kinetic study of fixation. Journal of Histotechnology. 1999;22:317–318. [Google Scholar]
- Axelrod D. Total internal reflection fluorescence microscopy in cell biology. Traffic. 2001;2:764–774. doi: 10.1034/j.1600-0854.2001.21104.x. [DOI] [PubMed] [Google Scholar]
- Toomre D. Alignment and calibration of total internal reflection fluorescence microscopy systems. Cold Spring Harb Protoc. 2012;4:504–509. doi: 10.1101/pdb.prot068668. [DOI] [PubMed] [Google Scholar]
- Shannon C. Communication in the presence of noise. Proc IRE. 1949;37:10–21. [Google Scholar]
- Hess ST, Gould TJ, Gunewardene M, Bewersdorf J, Mason MD. Ultrahigh resolution imaging of biomolecules by fluorescence photoactivation localization microscopy. Methods Mol Biol. 2009;544:483–522. doi: 10.1007/978-1-59745-483-4_32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta P, et al. Probing protein heterogeneity in the plasma membrane using PALM and pair correlation analysis. Nat Methods. 2011;8:969–975. doi: 10.1038/nmeth.1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betzig E, et al. Imaging intracellular fluorescent proteins at nanometer resolution. Science. 2006;313:1642–1645. doi: 10.1126/science.1127344. [DOI] [PubMed] [Google Scholar]
- Schermelleh L, Heintzmann R, Leonhardt H. A guide to super-resolution fluorescence microscopy. J Cell Biol. 2010;190:165–175. doi: 10.1083/jcb.201002018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong H. Photoactivated localization microscopy (PALM): an optical technique for achieving ~10-nm resolution. Cold Spring Harb Protoc. 2010;2010 doi: 10.1101/pdb.top91. [DOI] [PubMed] [Google Scholar]
- Lochner JE, et al. Efficient copackaging and cotransport yields postsynaptic colocalization of neuromodulators associated with synaptic plasticity. Dev Neurobiol. 2008;68:1243–1256. doi: 10.1002/dneu.20650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisler C, et al. Drift estimation for single marker switching based imaging schemes. Optics Express. 2012;20:7274–7289. doi: 10.1364/OE.20.007274. [DOI] [PubMed] [Google Scholar]