

Abstract
Drug resistance is a major limitation to the successful treatment of advanced prostate cancer (PCa). Patients who have metastatic, castration-resistant PCa (mCRPC) are treated with chemotherapeutics. However, these standard therapy modalities culminate in the development of resistance. We established paclitaxel resistance in a classic, androgen-insensitive mCRPC cell line (DU145) and, using a suite of molecular and biophysical methods, characterized the structural and functional changes in vitro and in vivo that are associated with the development of drug resistance. After acquiring paclitaxel-resistance, cells exhibited an abnormal nuclear morphology with extensive chromosomal content, an increase in stiffness, and faster cytoskeletal remodeling dynamics. Compared with the parental DU145, paclitaxel-resistant (DU145-TxR) cells became highly invasive and motile in vitro, exercised greater cell traction forces, and formed larger and rapidly growing tumors in mouse xenografts. Furthermore, DU145-TxR cells showed a discrete loss of keratins but a distinct gain of ZEB1, Vimentin and Snail, suggesting an epithelial-to-mesenchymal transition. These findings demonstrate, for the first time, that paclitaxel resistance in PCa is associated with a trans-differentiation of epithelial cell machinery that enables more aggressive and invasive phenotype and portend new strategies for developing novel biomarkers and effective treatment modalities for PCa patients.
Keywords: PROSTATE CANCER, PACLITAXEL, EPITHELIAL MESENCHYMAL TRANSITION, INVASION, CELL TRACTION FORCE, CYTOSKELETAL REMODELING
Prostate cancer (PCa) is the most common cancer in men and is one of the leading causes of cancer-related deaths in U.S. and worldwide [Jemal et al., 2011]. The disease is curable in its early stages; however, it is difficult to eradicate tumors in most men with advanced disease [Howlader et al., 2011]. Treatment for advanced PCa typically involves androgen deprivation to which tumors initially respond but eventually become castrate resistant. Men with metastatic castration-resistant PCa (mCRPC) have a median survival of less than 2 years [Bracarda et al., 2011]. Currently, there is no standard therapeutic modality for this fatal stage of PCa. Broadband chemotherapeutic drugs that target cell division are sometimes used as a last resort.
Taxanes are the only FDA approved chemotherapeutics that have been shown to improve the overall survival of men with PCa [Madan et al., 2011]. Originating from the bark of a yew tree, paclitaxel is an antimitotic agent that promotes apoptosis in cancer cells by stabilizing microtubules [Jordan and Wilson, 2004]. Many derivatives of paclitaxel (i.e., docetaxel and cabazitaxel) have been developed and, in combination with various adjuvants, provided a survival advantage to PCa patients [Petrylak et al., 2004; Tannock et al., 2004; Sartor, 2011]. Unfortunately, tumors often develop resistance to taxanes [Sartor, 2011], however, and the mechanism(s) by which this occurs remain largely unknown.
In this study, we assessed the structural and functional changes in a drug-resistant PCa model (DU145-TxR) that was established by long-term passage of the parental DU145 cell line with increasing concentrations of paclitaxel, the first of the taxanes [Takeda et al., 2007]. We showed that (1) paclitaxel-resistant cells gain mobility and invasiveness through increased epithelial-to-mesenchymal transition (EMT); (2) the increased cell migration and invasion in paclitaxel-resistant cells are facilitated by increased cytoskeleton (CSK) remodeling dynamics, stiffness, and traction forces and by a repression of keratin 8/18/19.
RESULTS
DEVELOPMENT OF A PACLITAXEL RESISTANT PCA CELL LINE
The androgen-insensitive PCa cell line, DU145, was cultured with increasingly higher concentrations of paclitaxel in order to establish the drug resistant cell line (DU145-TxR) [Takeda et al., 2007]. Typically, the parental DU145 cells grew in the form of epithelial cellular aggregates whereas DU145-TxR cells grew in a more dispersed manner with a fibroblast-like phenotype (Fig. 1A). Long term culture in paclitaxel increased the IC50 of DU145-TxR cells to paclitaxel from 7.74 to 3,950 nM (Supplementary Fig. 1). DU145-TxR cells also showed a fourfold increase in IC50 to colchicine. Such characteristic differences suggest that long-term exposure to paclitaxel might confer drug resistance, not only through efflux of paclitaxel, but also by influencing phenotypic changes in DU145-TxR cells.
Fig. 1.

Paclitaxel resistance in PCa is associated with increases in the metastatic potential in vitro and in vivo. A: Representative photos of the cell lines using a light microscope with 10× objective. B: Would healing assays for DU145 and DU145-TxR at a 4× objective view. C: Representative 4× objective images of a soft agar and crystal violet stained Boyden chamber. D: Tumors formed by implanting DU145 or DU145-TxR cells subcutaneously with n = 5 and n = 8 per group, respectively.
DRUG RESISTANCE IS ASSOCIATED WITH INCREASED INVASIVENESS
To contrast the cellular behaviors of DU145 and DU145-TxR, we used various in vitro and in vivo assays. In an in vitro wound healing assay [Tettamanti et al., 2004], DU145-TxR cells exhibited a faster migration and closure of a scratch wound than parental DU145 cells (Fig. 1B). In the Boyden chamber invasion assay, DU145-TxR cells showed an enhanced infiltration into collagen-coated chambers than DU145 cells (Fig. 1C). In addition to cell motility and invasive potential, DU145-TxR cells formed more colonies in soft agar (199 vs. 0) than parental cells (Fig. 1C) [Mani et al., 2008], and produced tumors that were sevenfold larger in a mouse xenograft model (Fig. 1D). In tumors induced by DU145-TxR, there were also more blood vessels near aggregates of cancer cells (Supplementary Fig. 2).
DRUG RESISTANCE CONFERS DISTINCT BIOPHYSICAL FEATURES
At the single cell level, DU145-TxR cells exhibited a larger spreading area and a higher cell traction force than their parental counterparts (Fig. 2). The net contractile moment, which is a scalar measure of the cell’s contractile strength, was threefold higher (P <0.002) in DU145-TxR cells (Fig. 2D). DU145-TxR cells also had significantly higher total strain energy, as well as higher prestress than DU145 cells (Supplementary Table I). These findings demonstrate that paclitaxel resistance in PCa is associated with the ability to exert larger force on the extracellular matrix (ECM), which in turn promotes cell migration through a dense matrix [Koch et al., 2012] and thereby confers a more aggressive and invasive phenotype.
Fig. 2.

Representative traction maps of (A) DU145 parental and (B) paclitaxel-resistant cells. Colors show the magnitude of the tractions in Pascals (Pa). Arrows show the direction and relative magnitude of the tractions. Scale bars: 50 μm. Projected cell area (C) and computed net contractile moment (D) of DU145 (parental vs. TXR) cells. Net contractile moment is a scalar measure of cell’s contractile strength and is expressed in pico-Newton meters (pNm). Data are presented as mean ± SE (n = 23 cells for parental; n = 26 cells for TXR). *, P <0.002; **, P <0.0001.
To probe deeper into the material properties and dynamics of the CSK, forced and spontaneous motions of microbeads anchored to the CSK through integrin cell adhesion receptors were applied [Bursac et al., 2005, 2007; Trepat et al., 2007]. Using magnetic twisting cytometry (MTC), we measured cytoskeletal stiffness (g′) and internal friction (g″) of parental and drug-resistant DU145 cells over a wide frequency range. Over five decades of frequency, stiffness g′ of both DU145 and DU145-TxR cell lines increased with frequency as a weak power law (Fig. 3A). Friction g″ also followed a weak power law at lower frequencies (below ~10 Hz), but showed stronger frequency dependence at higher frequencies (Fig. 3A). The power-law frequency dependence of g′ and g″ differed appreciably between the two cells (DU145, f0.16 and DU145-TxR, f0.19), however. At each probing frequency, g′ and g″ were significantly higher in DU145-TxR cells than in their parental counterparts. Accordingly, although DU145-TxR cells were stiffer than DU145 cells (Fig. 3A), paclitaxel resistance in PCa conferred more fluid-like behavior [Fabry et al., 2001].
Fig. 3.

Rate of cytoskeletal remodeling of DU145 parental and paclitaxel-resistant cells. A: Stiffness g′ and friction g″ of DU145 (parental) and paclitaxel-resistant (TXR) cells were measured over five decades of frequency using MTC. The solid lines are the fit of experimental data to the structural damping equation with addition of a Newtonian viscous term as previously described [Fabry et al., 2001]. Fitting was carried out by nonlinear regression analysis. Closed squares and circles represent g′ of TxR and parental cells, respectively. Open squares and circles represent g″ of TxR and parental cells, respectively. Data are presented as geometric mean ± SE (n = 832 for parental; n = 575 for TXR). B: Unforced, spontaneous bead motions were quantified by their mean square displacements (MSD) as function of time.31,32 The MSDs were computed at intervals that were equally spaced in time (1.3 s).31,32 For both cells, the MSD increases with time according to a power law relationship. The coefficient D* and the exponent a of the bead motion were estimated from a least squares fit of a power law to the MSD data versus time. C: Diffusion coefficient D* of parental versus TXR cells. Data are presented as mean ± SE (n = 1,703 for parental; n = 1,547 for TXR). *, P <0.0001.
Corroborating the increased CSK fluidity in DU145-TxR cells, microbead particle tracking also implied faster remodeling dynamics (Fig. 3B,C). For both cells, mean square displacements (MSD) of unforced particles increased with time as a power law with an exponent greater than unity (Fig. 3B) [Bursac et al., 2007]. Strikingly, these anomalous, super-diffusive bead motions were appreciably greater for DU145-TxR cells than for the parental DU145 cells (Fig. 3B), indicating an increased directional persistence in the remodeling behavior of the CSK[Raupach et al., 2007]. In addition, the apparent diffusion coefficient D* (a measure of the speed of CSK remodeling) was 2.5-fold larger in DU145-TxR cells (Fig. 3C). Together, our findings demonstrate that paclitaxel resistance in PCa cells is associated with increased CSK fluidity and increased persistence and speed of CSK remodeling dynamics—properties that also confer an aggressive and invasive phenotype.
NUCLEAR ABERRATIONS
Nuclear architecture and aneuploidy or aberrations in chromosomes are caused by an elevated rate of chromosomal instability which is associated with poor patient outcome and drug resistance [McGranahan et al., 2012]. Previous studies showed that aneuploid cells mutate to drug resistance at a higher rate than diploid cells in cell line systems [Duesberg et al., 2000]. DNA Ploidy analysis assesses the DNA characteristics of PCa cells and they can be classified as diploid, tetraploid, hypodiploid, or hyperploid according to the amount of DNA in their nuclei. For these reasons, DNA Ploidy analysis might be helpful in predicting patients’ response to specific PCa treatment [Veltri et al., 2012]. As shown in Figure 4, DU145-TxR cells had considerably more DNA than their parental counterparts. In fact, the vast majority of DU145-TxR cells were in the hypertetraploid range indicating an >2 increased DNA content.
Fig. 4.
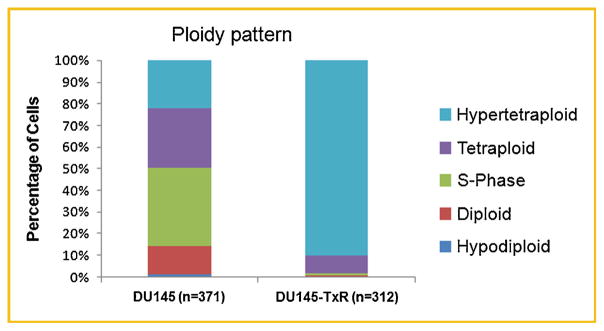
DU145-TxR cells have a higher ploidy than their parental counterparts. Static cytometry was used to categorize (n = 371) DU145 cells into hypoploid (G0), diploid (G1), S-Phase, tetraploid (G2), and hypertetraploid (>G2). The same size parameters were then used to assess (n = 312) DU145-TxR cells. The resulting analysis is displayed in a bar graph.
IDENTIFICATION OF PROTEINS ASSOCIATED WITH RESISTANCE
Due to a dramatically increased DNA ploidy in DU145-TxR, the nuclear proteome matrix was investigated. The nuclear matrix is a well-structured and dynamic network of filaments that performs functions in the nucleus similar to that of microtubules and tubulin in the cytoplasm [Pederson, 2000]. By isolating the nuclear matrix and analyzing the NMPs by 2D-PAGE, keratin 18 (KRT18) was observed to be more highly expressed in the DU145 than in the DU145-TxR cells (Supplementary Fig. 3). This down-regulation in DU145-TxR cells was also seen in mRNA expression and with whole-cell protein lysate, suggesting a global effect rather than a redistribution of KRT18 away from the nucleus due to paclitaxel resistance (Fig. 5). Methylation on the KRT18 promoter was also greater in the case of DU145-TxR (Supplementary Fig. 4).
Fig. 5.

Keratin 8, 18, 19 are repressed in DU145-TxR. A: mRNA expression of keratins 8, 18, and 19, (B) protein expression of keratins 8, 18, and 19, (C) immunofluorescent staining of DAPI (blue) and keratins 8, 18, and 19 (green) in DU145-TxR and its parental DU145.
EMT MARKERS ARE EXPRESSED IN PACLITAXEL RESISTANT CELLS
In the paclitaxel-resistant cell line DU145-TxR, an analysis of the protein expression of several EMT markers revealed a general trend away from epithelium and towards mesenchyme (Fig. 6A). The mRNA level and immunofluorescence intensity of E-cadherin were greater in the parental cell line (DU145), as opposed to the intensity of Vimentin, which was greater in the paclitaxel-resistant cell line (Fig. 6B,C). Interestingly, Vimentin distribution in the resistant cell line was atypical with a small region detected adjacent to the nucleus (Fig. 6C). However, the overall up-regulation of ZEB1/TCF8, Vimentin, and Snail and the down-regulation of E-cadherin and keratins suggest that the paclitaxel resistant cells have undergone an EMT. In contrast, the down-regulation of β-catenin in the DU145-TxR cell line was not consistent with a classically described EMT (Fig. 6A) [Wellner et al., 2009].
Fig. 6.

Paclitaxel resistance induces EMT. A: Classical EMT markers were immunoblotted. B: The mRNA expression of E-Cadherin showing a clear difference between DU145-TxR and DU145. C: immunofluorescence images of E-Cadherin and Vimentin taken at 10× objective magnification. E-Cadherin and Vimentin are marked with red, while DAPI is used to stain the nucleus. D: Luciferase assay shown on four β-catenin responsive promoters.
In general, the activation of Wnt/β-catenin signaling has been shown to trigger an EMT process [Yook et al., 2005; Jiang et al., 2007]. In the paclitaxel-resistant cell line, ZEB1/TCF8, Vimentin, and Snail were up-regulated while E-cadherin was repressed (Fig. 6B,C). In the canonical Wnt signaling pathway, the suppression of E-cadherin expression breaks the complex between β-catenin and E-cadherin at the cell membrane and correlates with a relocalization and high transcriptional activity of β-catenin in the nucleus [Chesire et al., 2002]. However, the expression of β-catenin was reduced in DU145-TxR cells (Fig. 6A). In order to clarify this apparent discrepancy, luciferase reporter assays were performed in both the parental and drug-resistant cell lines to measure the transcriptional activity on various β-catenin responsive promoters. Compared with the parental DU145, transcriptional activity had dramatically increased on four β-catenin responsive promoters (RANKL, Snail, VEGF, and Vimentin) in the DU145-TxR cell line (Fig. 6D). Interestingly when the TCF4/β-catenin binding sites were mutated in the Vimentin promoter, the total transcriptional activity was knocked down by >fivefold in both cell lines, however, the difference between DU145-TxR and its parental cell line was maintained at sixfold (Supplementary Fig. 5). These findings, suggest that chronic treatment of the DU145 cell line with paclitaxel causes resistant cells to emerge which are of the aggressive phenotype and demonstrate the alterations manifested in the EMT pathway.
DISCUSSION
In this study, a paclitaxel resistant cell line, DU145-TxR was created and characterized by molecular and biophysical techniques. Interestingly, DU145-TxR cells also developed resistances to various other antineoplastic agents, such as colchicine, docetaxel, and doxorubicin (Supplementary Fig. 1) [Takeda et al., 2007]. The resistance to both paclitaxel and colchicine initially suggested that DU145-TxR cells had developed resistance by changing the microtubule dynamics or structure. However, such microtubule-related mechanisms were an unlikely occurrence because there was no difference of tubulin in the protein amount and structure of DU145-TxR and those of parental DU145 (Supplementary Fig. 6). This unexpected result suggested that another mechanism was at play such as drug pumping by multidrug resistant protein 1 (MDR1). DU145-TxR had an up-regulation of MDR1 protein (Supplementary Fig. 7). However, MDR1 up-regulation was not the only change the cells underwent. In fact, MDR1 is a β-catenin responsive gene, possibly indicating an EMT in resistant cells mediated by the WNT signaling pathway [Yamada et al., 2000].
The dramatic cell morphology difference between DU145-TxR and its parental cell line under a light microscope indicates that a long-term exposure of paclitaxel transforms the cellular phenotype (Fig. 1A). DU145-TxR displayed increases in cell mobility in wound healing, cell invasion in the Boyden chamber, colony formation in soft agar, and tumor growth in mouse xenografts (Fig. 1). These experiments support the idea that DU145-TxR is a more aggressive phenotype and resistant to anoikis. Together, these results indicate that long term chemotherapy may cause CRPCa to become resistant to chemotherapy and gain invasive biological potential; chemotherapy should instead be intermittent and combined with other forms of therapy such as a mild hyperthermia [Bracarda et al., 2011; Li et al., 2011]. Alternatively, understanding the mechanisms of drug resistance may provide new methods to reverse this effect. Additionally, the increased DNA content is consistent with an advanced PCa phenotype, however, whether changes in the DNA content/ploidy can cause chemotherapeutic resistance and modify EMT marker gene expression or vice versa are not clear [Veltri et al., 2012].
Although these in vitro biological assays represented a general observation of cell movement, more physical quantitative measurements were used to bolster the correlation between drug resistance and aggressiveness. Our biophysical studies with MTC and traction microscopy revealed stunning results that could easily distinguish between DU145-TxR and its parental cell line. In the stress-strain analysis, the storage modulus and loss modulus of DU145-TxR cell line were higher than its parental counterpart (Fig. 3). The storage modulus represents the elastic property of a cell, while the loss modulus indicates the viscous property or the amount of energy dissipated as heat. The analysis of viscoelastic properties indicates a higher CSK stiffness, yet more fluid-like behavior, in DU145-TxR than in parental DU145 (Fig. 3A). These biophysical findings concurred with the results from the biochemical assays. The dramatic decrease in keratin 8, 18, and 19 in DU145-TxR could decrease cell rigidity and help DU145-TxR cells act in a fluid-like manner in order to facilitate cell locomotion (Fig. 5).
Similarly, the measurements of the CSK remodeling rate and cell traction mechanical force matched with the stress-strain analysis. The “fluid-like” cells, DU145-TxR, experienced a faster rate of CSK remodeling than its DU145 counterpart (Fig. 3B,C). One possible explanation for this difference is the loss of keratins 8, 18, and 19 in DU145-TxR cell. Keratins, which start their nucleation at the peripheral zone near the focal adhesions, form networks to maintain epithelial characteristics [Windoffer et al., 2011]. Therefore, the loss of the epithelial phenotype in DU145-TxR correlated well with the increase of EMT markers and contributed to greater wound healing abilities and to the formation of colonies in soft agar (Fig. 1B,C). In addition, the Fourier transform traction microscopy revealed a larger projected cell area as well as higher traction forces for DU145-TxR compared to its parental DU145 (Fig. 2). The ability of DU145-TxR to pull the ECM with a great force could explain how DU145-TxR can invade more effectively than DU145.
Lastly, the invasive potential gained in DU145-TxR was evident by the alterations in the gene expression related with EMT. The up-regulation of ZEB1/TCF8, Vimentin, and Snail by immunoblotting indicated that DU145 transformed into mesenchymal cells due to the long-term exposure of paclitaxel (Fig. 6A). This transition was also supported by the down-regulation of E-Cadherin and Keratin 8, 18, 19 in the immunofluorescence assays (Figs. 5C and 6C). Interestingly, while β-catenin is generally upregulated when a cell becomes more mesenchymal-like, the DU145-TxR showed less expression. This was inconsistent with the expression of E-cadherin and keratins which are thought to be negatively regulated by β-catenin. Furthermore, luciferase assays on four different promoters known to be positively regulated by β-catenin (RANKL, Snail, VEGF, and Vimentin) demonstrate a greater than sixfold activation in the TxR cell line compared to parental (Fig. 6D). Overall, these observations suggest that WNT signaling may be actuated in the DU145-TxR cell line through a non-canonical mechanism.
Therefore, our findings in cellular models demonstrate that paclitaxel resistance in PCa is associated with a gain of tumor aggressiveness, EMT, and resistance to anoikis. The schematic diagram (Fig. 7) illustrates a possible relationship between drug resistance and invasive potential. These observations have profound therapeutic implications for the treatment of PCa and uncover novel opportunities in the search for new biomarkers and potential approaches to regulate drug resistance. In this work, keratin 8, 18, and 19 levels were specifically assayed, since these are the major keratins used in the assessment of circulating tumor cells (CTCs) [Attard et al., 2009]. CTCs are proposed to be the most aggressive cells since they have the ability to invade into the blood vessels and ultimately cause secondary tumors. Therefore, it is somewhat ironic that this work found that the androgen- and chemotherapy-resistant cells, which are the ones that actually kill patients, would be invisible to this method. Perhaps the inclusion of mesenchymal markers and the exclusion of epithelial markers will be helpful in developing better CTC assays. In addition, the measurement of cell traction mechanical force, cytoskeletal remodeling rate, and elastic and viscous properties of cells in a microfluidic chip could be complementary to biomarkers for diagnosing the progress of cancer.
Fig. 7.

Schematic diagram of a proposed chemo-biomechanical pathway related with paclitaxel treatment.
Supplementary Material
Acknowledgments
Grant sponsor: NCI; Grant numbers: U54CA143803, U54CA141868; Grant sponsor: NIH; Grant number: P50CA103175.
This research was funded by NCI U54CA143803 (R.W.V.), U54CA141868 (S.S.A.), and NIH P50CA103175 (S.S.A.). Dr. Donald S. Coffey is thanked for helpful discussions while Yu Zeng is thanked for technical assistance.
Footnotes
Additional supporting information may be found in the online version of this article.
References
- Attard G, Swennenhuis JF, Olmos D, Reid AH, Vickers E, A’Hern R, Levink R, Coumans F, Moreira J, Riisnaes R, Oommen NB, Hawche G, Jameson C, Thompson E, Sipkema R, Carden CP, Parker C, Dearnaley D, Kaye SB, Cooper CS, Molina A, Cox ME, Terstappen LW, de Bono JS. Characterization of ERG, AR and PTEN gene status in circulating tumor cells from patients with castration-resistant prostate cancer. Cancer Res. 2009;69:2912–2918. doi: 10.1158/0008-5472.CAN-08-3667. [DOI] [PubMed] [Google Scholar]
- Bracarda S, Logothetis C, Sternberg CN, Oudard S. Current and emerging treatment modalities for metastatic castration-resistant prostate cancer. BJU Int. 2011;107(Suppl 2):13–20. doi: 10.1111/j.1464-410X.2010.10036.x. [DOI] [PubMed] [Google Scholar]
- Bursac P, Lenormand G, Fabry B, Oliver M, Weitz DA, Viasnoff V, Butler JP, Fredberg JJ. Cytoskeletal remodelling and slow dynamics in the living cell. Nat Mater. 2005;4:557–561. doi: 10.1038/nmat1404. [DOI] [PubMed] [Google Scholar]
- Bursac P, Fabry B, Trepat X, Lenormand G, Butler JP, Wang N, Fredberg JJ, An SS. Cytoskeleton dynamics: Fluctuations within the network. Biochem Biophys Res Commun. 2007;355:324–330. doi: 10.1016/j.bbrc.2007.01.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesire DR, Ewing CM, Gage WR, Isaacs WB. In vitro evidence for complex modes of nuclear beta-catenin signaling during prostate growth and tumorigenesis. Oncogene. 2002;21:2679–2694. doi: 10.1038/sj.onc.1205352. [DOI] [PubMed] [Google Scholar]
- Duesberg P, Stindl R, Hehlmann R. Explaining the high mutation rates of cancer cells to drug and multidrug resistance by chromosome reassortments that are catalyzed by aneuploidy. Proc Natl Acad Sci USA. 2000;97:14295–14300. doi: 10.1073/pnas.97.26.14295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabry B, Maksym GN, Butler JP, Glogauer M, Navajas D, Fredberg JJ. Scaling the microrheology of living cells. Phys Rev Lett. 2001;87:148102. doi: 10.1103/PhysRevLett.87.148102. [DOI] [PubMed] [Google Scholar]
- Howlader N, Krapcho M, Neyman N, Aminou R, Waldron W, Altekruse S, Kosary C, Ruhl J, Tatalovich Z, Cho H, Mariotto A, Eisner M, Lewis D, Chen H, Feuer E, Cronin K, Edwards B. SEER cancer statistics review, 1975–2008 2011 [Google Scholar]
- Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- Jiang YG, Luo Y, He DL, Li X, Zhang LL, Peng T, Li MC, Lin YH. Role of Wnt/beta-catenin signaling pathway in epithelial–mesenchymal transition of human prostate cancer induced by hypoxia-inducible factor-1alpha. Int J Urol. 2007;14:1034–1039. doi: 10.1111/j.1442-2042.2007.01866.x. [DOI] [PubMed] [Google Scholar]
- Jordan MA, Wilson L. Microtubules as a target for anticancer drugs. Nat Rev Cancer. 2004;4:253–265. doi: 10.1038/nrc1317. [DOI] [PubMed] [Google Scholar]
- Koch TM, Munster S, Bonakdar N, Butler JP, Fabry B. 3D Traction forces in cancer cell invasion. PLoS ONE. 2012;7:e33476. doi: 10.1371/journal.pone.0033476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Zeng Y, Mooney SM, Yin B, Mizokami A, Namiki M, Getzenberg RH. Resistance to paclitaxel increases the sensitivity to other microenvironmental stresses in prostate cancer cells. J Cell Biochem. 2011;112:2125–2137. doi: 10.1002/jcb.23134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madan RA, Pal SK, Sartor O, Dahut WL. Overcoming chemotherapy resistance in prostate cancer. Clin Cancer Res. 2011;17:3892–3902. doi: 10.1158/1078-0432.CCR-10-2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, Campbell LL, Polyak K, Brisken C, Yang J, Weinberg RA. The epithelial–mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704–715. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGranahan N, Burrell RA, Endesfelder D, Novelli MR, Swanton C. Cancer chromosomal instability: Therapeutic and diagnostic challenges. EMBO Rep. 2012;13:528–538. doi: 10.1038/embor.2012.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pederson T. Half a century of “the nuclear matrix. Mol Biol Cell. 2000;11:799–805. doi: 10.1091/mbc.11.3.799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrylak DP, Tangen CM, Hussain MH, Lara PN, Jr, Jones JA, Taplin ME, Burch PA, Berry D, Moinpour C, Kohli M, Benson MC, Small EJ, Raghavan D, Crawford ED. Docetaxel and estramustine compared with mitoxantrone and prednisone for advanced refractory prostate cancer. N Engl J Med. 2004;351:1513–1520. doi: 10.1056/NEJMoa041318. [DOI] [PubMed] [Google Scholar]
- Raupach C, Zitterbart DP, Mierke CT, Metzner C, Muller FA, Fabry B. Stress fluctuations and motion of cytoskeletal-bound markers. Phys Rev E Stat Nonlin Soft Matter Phys. 2007;76:011918. doi: 10.1103/PhysRevE.76.011918. [DOI] [PubMed] [Google Scholar]
- Sartor AO. Progression of metastatic castrate-resistant prostate cancer: Impact of therapeutic intervention in the post-docetaxel space. J Hematol Oncol. 2011;4:18. doi: 10.1186/1756-8722-4-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda M, Mizokami A, Mamiya K, Li YQ, Zhang J, Keller ET, Namiki M. The establishment of two paclitaxel-resistant prostate cancer cell lines and the mechanisms of paclitaxel resistance with two cell lines. Prostate. 2007;67:955–967. doi: 10.1002/pros.20581. [DOI] [PubMed] [Google Scholar]
- Tannock IF, de Wit R, Berry WR, Horti J, Pluzanska A, Chi KN, Oudard S, Theodore C, James ND, Turesson I, Rosenthal MA, Eisenberger MA. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N Engl J Med. 2004;351:1502–1512. doi: 10.1056/NEJMoa040720. [DOI] [PubMed] [Google Scholar]
- Tettamanti G, Grimaldi A, Rinaldi L, Arnaboldi F, Congiu T, Valvassori R, de Eguileor M. The multifunctional role of fibroblasts during wound healing in Hirudo medicinalis (Annelida, Hirudinea) Biol Cell. 2004;96:443–455. doi: 10.1016/j.biolcel.2004.04.008. [DOI] [PubMed] [Google Scholar]
- Trepat X, Deng L, An SS, Navajas D, Tschumperlin DJ, Gerthoffer WT, Butler JP, Fredberg JJ. Universal physical responses to stretch in the living cell. Nature. 2007;447:592–595. doi: 10.1038/nature05824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veltri RW, Christudass CS, Isharwal S. Nuclear morphometry, nucleomics and prostate cancer progression. Asian J Androl. 2012;14:375–384. doi: 10.1038/aja.2011.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellner U, Schubert J, Burk UC, Schmalhofer O, Zhu F, Sonntag A, Waldvogel B, Vannier C, Darling D, zur Hausen A, Brunton VG, Morton J, Sansom O, Schuler J, Stemmler MP, Herzberger C, Hopt U, Keck T, Brabletz S, Brabletz T. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat Cell Biol. 2009;11:1487–1495. doi: 10.1038/ncb1998. [DOI] [PubMed] [Google Scholar]
- Windoffer R, Beil M, Magin TM, Leube RE. Cytoskeleton in motion: The dynamics of keratin intermediate filaments in epithelia. J Cell Biol. 2011;194:669–678. doi: 10.1083/jcb.201008095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada T, Takaoka AS, Naishiro Y, Hayashi R, Maruyama K, Maesawa C, Ochiai A, Hirohashi S. Transactivation of the multidrug resistance 1 gene by T-cell factor 4/beta-catenin complex in early colorectal carcinogenesis. Cancer Res. 2000;60:4761–4766. [PubMed] [Google Scholar]
- Yook JI, Li XY, Ota I, Fearon ER, Weiss SJ. Wnt-dependent regulation of the E-cadherin repressor snail. J Biol Chem. 2005;280:11740–11748. doi: 10.1074/jbc.M413878200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.