

Abstract
Correlative fluorescence and electron microscopy (CFEM) is a multimodal technique that combines dynamic and localization information from fluorescence methods with ultrastructural data from electron microscopy, to give new information about how cellular components change relative to the spatiotemporal dynamics within their environment. In this review, we will discuss some of the basic techniques and tools of the trade for utilizing this attractive research method, which is becoming a very powerful tool for biology labs. The information obtained from correlative methods has proven to be invaluable in creating consensus between the two types of microscopy, extending the capability of each, and cutting the time and expense associate with using each method separately for comparative analysis. The realization of the advantages of these methods in cell biology have led to rapid improvement in the protocols and have ushered in a new generation of instruments to reach the next level of correlation – integration.
Keywords: electron microscopy, fluorescence, light microscopy, correlative, cell biology, imaging, cryoEM, immunolabel, tomography, integrated microscope
INTRODUCTION
The goal of correlative techniques is to combine two or more methods to generate and combine data on a single sample to produce a more comprehensive picture of an event or process than could otherwise be obtained by any single method. In the case of correlative fluorescence and electron microscopy (CFEM), spatial and structural data are combined to determine the interactions of cellular components, primarily in living systems (Juette et al., 2008; Zhang, 2013). Fluorescence microscopy (FM) utilizes fluorescent signals or markers to image the interactions and dynamics of chemical species in biological systems. It is commonly used to track the transport and position of chemical species in a cell, tissue sample, or microorganism. In instances involving single proteins, protein complexes, or chemical pathways, methods such as fluorescence resonance energy transfer (FRET) and fluorescence recovery after photobleaching (FRAP) can provide very specific spatial and conformational information about cellular structures (Schultz, 2009). FRET measures the efficiency of energy transfer between donor and acceptor dyes that have been affixed to proteins of interest, and identifies interactions occurring in the 1–10 nm range (Clegg, 2009). FRAP involves bleaching, with high intensity light, a specific area of a fluorescently-labeled sample and tracking the return of fluorescence to the photobleached region, to calculate the diffusion rate of labeled species (Sprague et al., 2004).
During the past several years, the utility of FM has expanded tremendously thanks to the development of complex mathematical transforms that allow high-resolution spatial information to be obtained from diffraction limited samples when visual interpretation using basic light capturing devices is not possible This advance in FM, typically referred to as super-resolution FM, has filled a resolution gap between light microscopy (LM) and electron microscopy (EM) but, more importantly, has provided a precise and direct approach to link LM data with EM data. As reviewed by Leung and Chou, super-resolution techniques in FM arose from mathematical realizations that were achieved long before EM came into existence (Leung and Chou, 2011). During the past two decades, super resolution techniques in FM, such as stimulated emission microscopy (SIM), stimulated emission depletion (STED) microscopy, stochastic optical reconstruction microscopy (STORM,) and photoactivated localization microscopy (PALM), have been refined such that nanoscale spatial resolution can be achieved (Hess et al., 2006; Gustafsson, 2000; Rust et al., 2006; Hell and Wichmann, 1994). SIM and STED are optical techniques that reduce the diffraction barrier through wave geometry and interferometry, respectively, while PALM and STORM overcome the same barrier by reducing or even eliminating the point spread function, via special fluorescent proteins that can be “switched” on or off for sequential activation and time resolved localization (Gustafsson, 2000; Rust et al., 2006; Hess et al., 2006; Hell and Wichmann, 1994).
Despite these advances in FM, electron microscopy still provides the highest resolution, up to sub-nanometer resolution. Scanning (SEM), transmission (TEM), and scanning transmission (STEM) electron microscopy are among the commonly used electron-based imaging devices, employed to obtain detailed information about surfaces, internal structures, and spatial and affinitive properties. SEM is a surface technique in which electrons interact with a sample surface to generate backscattered or secondary electrons, which are collected to create an SEM image (Egerton, 2005). In TEM, electrons penetrate and pass through a thin sample. The transmitted electrons carry information about the sample, and this information is collected on a film or CCD camera to create a projection image of the specimen. Tilts series of thick specimens in TEM allow for internal 3D ultrastructure analysis, termed electron tomography (ET) (Frank, 2006).
RATIONALE
EM provides the most detailed, high resolution images of fixed samples but is limited in terms of the small field that accompanies such high magnifications; FM, on the other hand, is most useful in providing large-field, cellular dynamics information on living systems but with limited resolution. These two imaging modalities are highly complementary. The need to localize targets in EM samples has driven the development of correlative techniques in which a wide-field FM approach is used to localize an area of interest, followed by high-resolution EM. CFEM techniques have been employed since the mid to late 1960s, but, until recently, FM and EM were employed sequentially, on separate instruments (Hayat, 1987). In the past decade, integrated instruments have been custom-built for high throughput analysis (Agronskaia et al., 2008).
The advantage of employing such correlative techniques is found in the precise connections that can be made between distinct aspects of an object or phenomenon such that precise definitions can be asserted. It is not uncommon, in varying fields and among different disciplines within a given field, to find varying definitions for the same term, based on the methodology of research. For example, in cell biology, structures within a cell, such as lipid rafts, have been defined with much different sizes or orientations depending on the imaging technique. By comparing and correlating techniques, these controversies may be resolved (Westphal et al., 2008).
METHODS
CFEM on a single sample can be carried out in several different ways, allowing for correlation of various and distinct data sets. The method chosen will depend on the type of information required (see Figure 1). TEM, SEM, and ET each provide different information and have distinct limitations as to the data provided and how it is obtained.
Figure 1.
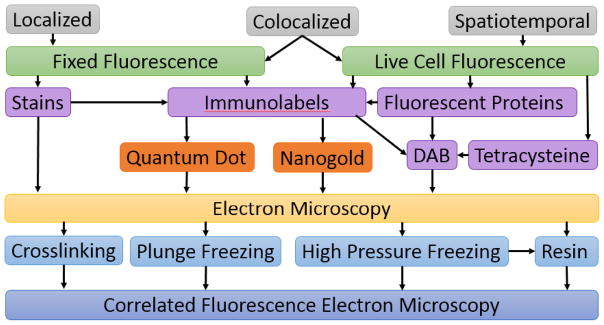
A general overview of methods that are to be considered when approaching a sample for ultrastructural localization and analysis in CFEM. First, the type of information required (localization vs spatiotemporal data) will determine the type of FM that is necessary and will lead to a decision about the best type of label and EM procedure to gain an overall picture of a protein or process using a correlated technique.
The first consideration is the nature of the sample – its size, location (spatial and temporal), and its biochemical makeup. Next, the type of tag(s) needed, and the method required to affix the tag, should be considered. Along with this, the availability of the required instrumentation will dictate the CFEM method chosen and will ultimately limit how and when you fix the sample. Here, we focus our discussion to widely applicable methods.
Although LM can be done with live samples, EM requires the collection of many images to gain enough information to create a single high resolution 3D structure (Zhang, 2013; Volkenandt et al., 2014; Pereira et al., 2014; Giepmans, 2008; Agronskaia et al., 2008). However, biological specimens are fragile, and bombardment with a large dose of high energy electrons causes them to break down before a satisfactory amount of information is obtained to generate a useful image (Zhao et al., 2013; Hoenger, 2014). The necessity of multiple rounds of imaging means the specimen must be fixed, either by rapid freezing or chemical crosslinking, and, also, be generally labelled.
The fixation technique and the labelling technique are cooperative and dependent on the nature of the sample. The labeling technique will depend on several factors. The chemical composition of the structure must be considered; DNA, proteins, and lipids have different affinities, origins, and locales – three properties which play a major role in labeling. The integrity, size, location, and background of the specimen are also important considerations. If very small structures are to be investigated or if the ultrastructure lies directly beneath the surface of a membrane, then large immunolabels might not be ideal. Labels that require genetic intervention are extremely specific but can take time to develop if the loci of the protein has not been defined. A final consideration is whether the structure of interest is visible only under specific cellular conditions (for example, during a specific stage of the cell cycle) or if its configuration is dependent on its associations and the cellular environment – spatiotemporal consideration.
Fixed Sample Fluorescence
Traditionally, CFEM has been carried out on ultrathin sections using affinity probes for detection and localization (Hayat, 1987). Fluorescent dyes can be used but, first, must be modified for EM correlation. Combinatorial affinity labels are most commonly applied since images can be directly correlated by detection of the same label for both FM and EM (Powell et al., 1998). These combinatorial labels have been largely explored and are among the most developed labelling techniques for CFEM (Giepmans, 2008; Sosinsky et al., 2007; Robinson and Takizawa, 2009). Today, the most commonly applied affinity labels are Fluoronano Gold (FNG) and Quantum Dots (QD).
Several fluorescent dyes have been applied to CFEM using photoconversion. These become EM visible either upon illumination, which generates reactive oxygen species for photoconversion, or by coupling them to a peroxidase, such as horseradish peroxidase, which generates the reactive oxygen species (Maranto, 1982; Bushong et al., 2002; Schmued and Snavely, 1993; Von Bartheld et al., 1990). The common problem with such dyes is that they tend to be less specific than affinity labels; however, dyes are robust in that they can be incubated for weeks or even months for diffusion to a target in fixed samples. (Von Bartheld et al., 1990). Fluorescent dyes that have been used in CFEM can be as nonspecific as DiI, which generally targets membrane lipids, or as specific as α-bungarotoxin, a small peptide derived from snake venom that binds exclusively to acetylcholine receptors (Von Bartheld et al., 1990; Modla et al., 2010)
Fluoronano Gold
Fluoronano Gold (FNG) is a combinatorial label that consists of an affinity probe, such as an antibody, bound to a fluorophore, such as FITC, and an electron dense gold nanoparticle, which is a crystalline complex of gold, as opposed to colloidal gold (Robinson et al., 2000b, 2000a). The most obvious advantage of these labels is the fact that they are detected by both FM and EM, so labeling is carried out in a single step and correlation is direct and straightforward even when the instrumentation is separated. This makes it possible to perform both techniques with essentially one sample preparation process, greatly reducing time and effort in the imaging process while also providing a direct correlation (Karreman et al., 2012; Agronskaia et al., 2008). FNG has also been used to restore and enhance fluorescence to GFP-labeled proteins using anti-GFP immunoglobulin-tagged FNG on ultrathin Lowicryl Resin sections (Fabig et al., 2012).
Quantum Dots
The other type of affinity label commonly employed in CFEM is the Quantum dot (QD). QDs are small electron dense crystalline compounds that have been exploited in CFEM, due to their fluorescence properties. The probes are bifunctional and are detectable under both FM and EM. They are of particular interest because the wavelength of fluorescence emitted can be tuned by changing the size of the particles. QDs have been successfully conjugated to biomolecules, such as antibodies, without effecting functional ultrastructure (Alivisatos et al., 2005; Takizawa and Robinson, 2012).
At the core of a QD is an electron dense material, typically made of a Cadmium or Selenium crystal. This core is what makes the dots visible in EM, but it has also raised concerns about their toxicity. Similar to FNG, the major limitation with QD is that they are large labels and the cell must be permeablized to deliver the probe (Takizawa and Robinson, 2012; Sosinsky et al., 2007). For applications where a very specific and highly detailed ultrastructural image is necessary, or when live-cell FM is to be used, genetic labels that do not require cell permeablization or treatment with harsh chemical reagents are always the preferred choice.
Live Cell Fluorescence
Fluorescent proteins
Today, in vivo live-cell fluorescence microscopy commonly utilizes green fluorescent protein (GFP); since its isolation and recombination in the early 1980s, GFP has become the most useful component of several fluorescence techniques, not only microscopy but also spectrometry, genetics, macrobiology and microbiology (Schultz, 2009; Zaidi et al., 2011; Abu-Lail et al., 2006; Sims and Hardin, 2004). More recently, engineered derivatives of GFP have contributed to advancements in FM; these derivatives are characterized by color shifts and lifetime extension (Giepmans, 2006).
The advantage of GFP is its specificity as a genetic tag and its natural ability to sustain its fluorescence in aqueous environments; the fluorophore is protected from bulk water, decreasing the probability of quenching from electrolytes (Sims and Hardin, 2004). Like most proteins, GFP is not readily visible in EM, thus an EM marker must be used in parallel with GFP or its derivatives for CFEM. This is typically done by applying an immunolabel that targets GFP, such as an anti-GFP, after live cell imaging, or by chemically enhancing the protein via photoconversion with diaminobenzidine (DAB) (Grabenbauer et al., 2005). In immunogold labelling after FM, anti-GFP antibodies are used to probe GFP, followed by a secondary antibody carrying a colloidal gold particle (Sims and Hardin, 2004; Giepmans, 2006). GFP photoconversion with DAB is based on the fact that GFP can lose its fluorescence when exposed to various solvents and light frequencies, a phenomenon known as photobleaching. GFP recognition after photobleaching (GRAB) uses oxygen radicals generated by GFP to condense the DAB units into a polymer cluster surrounding the protein, which can be visualized in EM (Grabenbauer et al., 2005).
Tetracysteine Biarsenical
A less invasive system for live cell imaging, in combination with DAB, is the tetracysteine biarsenical system. The term tetracysteine label describes a set of labels that have been developed in the laboratory for live cell CFEM. The name refers to the genetic motif CCPGCC that can be inserted into a protein sequence without disrupting function. Nonfluorescent fluorescein derivatives, FlAsH and ReAsH, bind with high affinity to the CCPGCC motif and become highly green or red fluorescing, respectively (Ellisman et al., 2012). The biarsenical system is used widely in imaging mammalian cell lines, in particular neuronal cells, to track the movement of proteins, such as α-synuclein and connexins (Sosinsky et al., 2003; Gaietta et al., 2011, 2002; Boassa et al., 2013). The advantage of these tags is that they readily produce reactive oxygen for photoconversion of DAB without the need for another dye or enzyme.
Mini-SOG
Flavoprotein miniSOG (Singlet Oxygen Generator) was recently introduced in 2011 by Shu et al. as a genetically encoded tag designed specifically for CFEM. The tag was developed by genetically recombining one domain of phototropin with a flavin mononucleotide and was demonstrated as a CFEM tag in a number of different assays (Shu et al., 2011). The full potential of MiniSOG has been demonstrated in studies of α-actin protein filaments, intranuclear H2B proteins, gap junction channel proteins (Cx43), the neuronal synapse protein SynCAM, mitochondrial Cytochrome-C, as well as a myriad of other proteins. It was even shown to have denser labelling spread than immunogold in Cx43 labelling studies. The major advantage of miniSOG is the SOG component. As a generator of singlet oxygen species, it can readily photoconvert DAB for staining with OsO4 for EM imaging (Shu et al., 2011).
Photoconversion of DAB
DAB is a readily diffusible chemical that does not require membrane permeablization. Photoconversion of DAB occurs in the presence of excited singlet oxygen, and the product can be stained with osmium tetraoxide (OsO4), an electron dense material visible in EM. A number of methods exist to generate singlet oxygen for photoconversion of DAB (Sosinsky et al., 2007). One such method is to use eosin, a singlet oxygen generating fluorescein derivative that can be conjugated to an antibody or various other molecules. Several peroxidases have been used to generate singlet oxygen as well, but these systems are going out of style for live cell imaging due to the invasive constraints of permeablizing the cell and chemically altering the environment. New, genetically encoded tags, such as fluorescent proteins, the tetracysteine biarsenical system, and flavoprotein miniSOG, have become increasingly popular over the last five to ten years, as focus has shifted heavily toward live-cell FM (Giepmans, 2006; Ellisman et al., 2012; Sosinsky et al., 2007; Shu et al., 2011).
Embedding
A number of fixation and embedding techniques have been developed over the years; the most commonly used protocols in CFEM studies involve fixation with crosslinking agents, such as aldehydes in organic solvents, and embedding with resins, adapted from EM. Embedding can be done before or after labelling. Pre-embedding labeling involves adding small amounts of fixative as the cell is permeablized and labelled, to prevent the cell from collapsing during permeation while allowing labels to diffuse into the cell before embedding and sectioning.
Glutaraldehyde, one of the most common fixatives for EM, is a cross-linking agent that forms covalent bonds between cytosolic features (Ellisman et al., 2012; Schnell et al., 2012). Crosslinking is helpful in locking the position of structures but may perturb the structure in the process. It is also lightly fluorescent, antagonizing CFEM by creating substantial amounts of fluorescence background in concentrations as low as 0.4%. Dilution to 0.1% in another aldehyde or organic fixative, such as formaldehyde, acetone, or methanol, has shown great structural preservation while not overpowering the fluorescence signal (Ellisman et al., 2012; Schwarz and Humbel, 2007).
A follow up step that enforces ultrastructural preservation is resin embedding with epoxies or plasticizing agents such as acrylic. Epoxy resins are simple to section, intuitively allowing for EM analysis. However, because epoxy resins can cause a high degree of crosslinking, they do not preserve antigenicity and are not recommended for post-embedding immunolabeling (Stirling, 1990). Lowicryl resins, on the other hand, are noted for preserving antigenicity, have been diversely applied, and come in polar and hydrophobic variations (Carlemalm et al., 1985; Armbruster et al., 1982, 1983); unfortunately, these resins are also highly volatile and toxic (Webster et al., 2008; Morphew, 2007). In CFEM, Lowicryl resins are commonly used in freeze substitution, after cryofixation by high-pressure freezing (Carlemalm et al., 1985; Armbruster et al., 1983; Studer et al., 2008).
Finally, polar and nonpolar types of resins, may be chosen for CFEM, based on their specific properties and the surface affinity or environment of the protein or membrane of interest. LR white and LR Gold, for example, are widely used for their hydrophilic and hydrophobic properties, respectively (Morphew, 2007; Nisman et al., 2004; Watanabe et al., 2011). A more advanced and challenging approach that significantly improves ultrastructure preservation is employing cryo techniques in combination with genetic fluorescent tags.
Cryofixation
Cryofixation, either by plunge-freezing or high pressure freezing (Mcdonald, 2009; Studer et al., 2008), can be performed immediately following FM imaging and is the most reliable technique to preserve ultrastructure for EM. While plunge freezing is applicable for small cells or extracted organelles and proteins, high-pressure freezing (HPF) is suitable for samples larger than several micrometers. The advantage of cryofixation in CFEM is that the cells can be imaged with live-cell FM prior to freezing to obtain spatiotemporal information for EM targeting and analysis. Typically, cells carrying fluorescence labels that are genetically tagged or introduced externally, such as by viral infection, are cultured on EM grids or other substrates and imaged with live cell FM (Figure 2). A standard EM grid can be used, or the sample can be prepared on a finder grid which will aid localizing the area of interest (Valentijn et al., 2008; Rigort, 2010). Immediately after FM imaging, the samples are fixed by plunge freezing, which also embeds the sample in a vitreous ice layer, a two in one process. Because the biological system continues to be dynamic until it is frozen, perfect correlation may not be attained, due to the time lapse between FM and cryofixation. By reimaging the frozen sample using a cryo-FM stage (Robinson et al., 2001; Valentijn et al., 2008, Jun et al 2011, 2013), a tight correlation can be achieved (Figure 2).
Figure 2.

Flow chart for live cell correlation in CFEM. Samples are grown on EM grids (A) and the cultures are imaged in FM (B) prior to being fixed or vitrified and imaged in an EM with correlation (F) or vitrified (C) and reimaged on a cryo-FM (D) for subsequent imaging in EM and correlation (F).
During HPF, samples are pressurized to more than 200 MPa, while being submitted to liquid nitrogen to minimize ice crystal growth (Mcdonald, 2009; Studer et al., 2008). HPF can be carried out immediately after live-cell FM and is typically followed by vitreous sectioning, reimaging in cryo-FM, and correlation with cryoEM, similar to the process with plunge freezing (Figure 2). Freeze substitution followed by resin embedding and sectioning is an alternative to vitreous sectioning (Karreman et al., 2012; McDonald, 2009, Webster et al., 2008). Although sample thickness is a primary limitation for cryofluorescence of vitreous sectioned samples, FM imaging of cryofixed samples has an advantage over standard FM in that photobleaching is decreased, facilitating better correlation with EM (Schwartz et al., 2007).
Correlation with EM
Whether using chemical fixation or cryofixation, the final step in any correlated technique is to take high resolution EM images and correlate the ultrastructural data to the FM images already taken. Correlative methods were developed for both TEM and SEM. Array tomography (AT) is a method developed out of the utility of CFEM by using fluorescent labels to align and correlate TEM images from serial sections for 3D reconstruction (Watanabe et al., 2011). In TEM tomography, instead of stacking the thin serial section images, a tilt series of TEM projection images from a single section is recorded and combined computationally to reconstruct a 3D volume of that section. TEM tomography has become the most commonly used method for acquiring 3D structures with an electron microscope; however, because biological specimens are fragile and tilt series rely on many rounds of imaging of a single sample, sample thickness and size are generally limited (Wacker and Schroeder, 2013; McIntosh et al., 2005).
Serial block SEM (SBF-SEM) is a surface imaging technique. The process is relatively straightforward and is, in some respects, the opposite approach to Array Tomography. Rather than imaging thin sections in series, the surface of the sample is imaged as thin serial sections are removed (Hughes et al., 2014). As discussed, tags for EM can exist exclusively or combinatorial. Localization and enhanced ultrastructure in EM, through high throughput analysis with CFEM, is bringing the dynamic subcellular environment to a new resolution and allows the visualization of rare events and difficult to find structures.
Integrated Microscopy
Because FM and EM are two separate imaging platforms, one of the challenges in CFEM is transferring specimens from one platform to the next with minimum damage and delay; especially for frozen-hydrated samples, the rate of successful transfer is rather low. Automation and integration can greatly enhance this. Currently, a number of teams have developed instruments that integrate the techniques into a single multifunctional machine that is capable of both imaging modalities. With such instruments, the efficiency and accuracy of correlative analysis is greatly enhanced. Such an integrated fluorescence electron microscope (IFEM), which has a laser scanning fluorescence microscope inside a TEM, can conduct experiments at room temperature as well as at cryotemperature. Once an area of interest on a sample grid is localized, the microscope can be quickly switched into the TEM mode for analysis of selected areas (Agronskaia et al., 2008; Faas et al., 2013). Using an IFEM, a correlation scan takes only 20 minutes, compared to several hours using CFEM and separate instruments. The versatility of IFEM for correlation at cryo temperature was demonstrated on bacterial samples, viral proteins, and virus-infected mammalian cells using vitreous sections (Faas et al., 2013).
REMARKS ON THE FUTURE OF CFEM
The utility of CFEM techniques is well-recognized as a means to the reduce time and effort required to find and analyze an area of interest when carrying out EM studies of biological systems. Recently, super resolution techniques have been applied to CFEM to facilitate precise localization and high resolution (on the order of nanometers) depictions of time–lapse processes in several biological specimens (Watanabe et al., 2011; Cotte et al., 2013). Correlation of such fluorescence nanoscopy and electron tomography are at the forefront of 3D imaging, with new instrumentation and developments in application that allow for more precise and even time-resolved CFEM (Lorenz and Zewail 2013, VaLentijn wet al. 2012, Idrissi et al. 2013). Accordingly, a new generation of EM instrument is being ushered in with Direct Electron detectors (DED) and phase plate EM (Mooney et al., 2011; Brnjas-Kraljevi and Pifat-Mrzljak, 2011).
CFEM is bringing dynamic visualization to electron microscopy; 4D cryo-electron microscopy (with time being the fourth dimension) is not only being theorized but put into practice, through correlated approaches and EM technological advances that can increase precision and speed up image processing to make picometer measurements of protein movement within a cell on a nanosecond time scale (Fitzpatrick et al., 2013). The advent of high resolution optimization through new imaging and detection systems, combined with robust image processing carried out by ever increasing computing capabilities, will facilitate precise functional characterization of the subcellular milieu.
Acknowledgments
We thank Dr. Teresa Brosenitsch for critical reading of the manuscript. This work was supported by the National Institutes of Health Grant GM082251 and GM085043 to P.Z.
References
- Agronskaia AV, Valentijn JA, van Driel LF, Schneijdenberg CTWM, Humbel BM, van Ben Henegouwen PMP, Verkleij AJ, Koster AJ, Gerritsen HC. Integrated fluorescence and transmission electron microscopy. Journal of structural biology. 2008;164:183–189. doi: 10.1016/j.jsb.2008.07.003. [DOI] [PubMed] [Google Scholar]
- Alivisatos AP, Gu W, Larabell C. Quantum dots as cellular probes. Annu Rev Biomed Eng. 2005;7:55–76. doi: 10.1146/annurev.bioeng.7.060804.100432. [DOI] [PubMed] [Google Scholar]
- Armbruster BL, Carlemalm E, Chiovetti R, Garavito RM, Hobot JA, Kellenberger E, Villiger W. Specimen preparation for electron microscopy using low temperature embedding resins. Journal of microscopy. 1982;126:77–85. doi: 10.1111/j.1365-2818.1982.tb00358.x. [DOI] [PubMed] [Google Scholar]
- Armbruster BL, Garavito RM, Kellenberger E. Dehydration and embedding temperatures affect the antigenic specificity of tubulin and immunolabeling by the protein A-colloidal gold technique. Journal of Histochemistry & Cytochemistry. 1983;31:1380–1384. doi: 10.1177/31.12.6195215. [DOI] [PubMed] [Google Scholar]
- Von Bartheld CS, Cunningham DE, Rubel EW. Neuronal tracing with DiI: decalcification, cryosectioning, and photoconversion for light and electron microscopic analysis. Journal of Histochemistry & Cytochemistry. 1990;38:725–733. doi: 10.1177/38.5.2185313. [DOI] [PubMed] [Google Scholar]
- Boassa D, Berlanga ML, Yang MA, Terada M, Hu J, Bushong EA, Hwang M, Masliah E, George JM, Ellisman MH. Mapping the Subcellular Distribution of α-Synuclein in Neurons using Genetically Encoded Probes for Correlated Light and Electron Microscopy: Implications for Parkinson’s Disease Pathogenesis. The Journal of Neuroscience. 2013;33:2605–2615. doi: 10.1523/JNEUROSCI.2898-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brnjas-Kraljević J, Pifat-Mrzljak G. Supramolecular Structure and Function. Vol. 10. Springer; 2011. [Google Scholar]
- Bushong EA, Martone ME, Jones YZ, Ellisman MH. Protoplasmic astrocytes in CA1 stratum radiatum occupy separate anatomical domains. The Journal of neuroscience. 2002;22:183–192. doi: 10.1523/JNEUROSCI.22-01-00183.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlemalm E, Villiger W, Hobot JA, Acetarin JD, Kellenberger E. Low temperature embedding with Lowicryl resins: two new formulations and some applications. Journal of microscopy. 1985;140:55–63. doi: 10.1111/j.1365-2818.1985.tb02660.x. [DOI] [PubMed] [Google Scholar]
- Clegg RM. Förster resonance energy transfer—FRET: what is it, why do it, and how it’s done. FRET and FLIM Techniques. 2009;1:1–57. [Google Scholar]
- Cotte Y, Toy F, Jourdain P, Pavillon N, Boss D, Magistretti P, Marquet P, Depeursinge C. Marker-free phase nanoscopy. Nature Photonics. 2013;7:113–117. [Google Scholar]
- Egerton RF. Physical Principles of Electron Microscopy. Springer; US: 2005. [Google Scholar]
- Ellisman MH, Deerinck TJ, Shu X, Sosinsky GE. Picking faces out of a crowd: genetic labels for identification of proteins in correlated light and electron microscopy imaging. Methods in cell biology. 2012;111:139–153. doi: 10.1016/B978-0-12-416026-2.00008-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faas FGA, Bárcena M, Agronskaia AV, Gerritsen HC, Moscicka KB, Diebolder CA, van Driel LF, Limpens RWAL, Bos E, Ravelli RBG. Localization of fluorescently labeled structures in frozen-hydrated samples using integrated light electron microscopy. Journal of structural biology. 2013;181:283–290. doi: 10.1016/j.jsb.2012.12.004. [DOI] [PubMed] [Google Scholar]
- Fabig G, Kretschmar S, Weiche S, Eberle D, Ader M, Kurth T. 5 Labeling of Ultrathin Resin Sections for Correlative Light and Electron Microscopy. Methods in Cell Biology. 2012;111:75–91. doi: 10.1016/B978-0-12-416026-2.00005-4. [DOI] [PubMed] [Google Scholar]
- Frank J. Electron tomography: methods for three-dimensional visualization of structures in the cell. Springer; New York: 2006. [Google Scholar]
- Gaietta G, Deerinck TJ, Adams SR, Bouwer J, Tour O, Laird DW, Sosinsky GE, Tsien RY, Ellisman MH. Multicolor and electron microscopic imaging of connexin trafficking. Science. 2002;296:503–507. doi: 10.1126/science.1068793. [DOI] [PubMed] [Google Scholar]
- Gaietta GM, Deerinck TJ, Ellisman MH. Correlated Live Cell Light and Electron Microscopy Using Tetracysteine Tags and Biarsenicals. Cold Spring Harbor Protocols. 2011;2001 doi: 10.1101/pdb.top94. pdb-prot5548. [DOI] [PubMed] [Google Scholar]
- Giepmans BG. Bridging fluorescence microscopy and electron microscopy. Histochemistry and Cell Biology. 2008;130:211–217. doi: 10.1007/s00418-008-0460-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giepmans BNG. The Fluorescent Toolbox for Assessing Protein Location and Function. Science. 2006;312:217–224. doi: 10.1126/science.1124618. [DOI] [PubMed] [Google Scholar]
- Grabenbauer M, Geerts WJ, Fernadez-Rodriguez J, Hoenger A, Koster AJ, Nilsson T. Correlative microscopy and electron tomography of GFP through photooxidation. Nature methods. 2005;2:857–862. doi: 10.1038/nmeth806. [DOI] [PubMed] [Google Scholar]
- Gustafsson MG. Surpassing the lateral resolution limit by a factor of two using structured illumination microscopy. Journal of microscopy. 2000;198:82–87. doi: 10.1046/j.1365-2818.2000.00710.x. [DOI] [PubMed] [Google Scholar]
- Hayat M. Correlative Microscopy In Biology: Instrumentation and Methods. Elsevier Science 1987 [Google Scholar]
- Hell SW, Wichmann J. Breaking the diffraction resolution limit by stimulated emission: stimulated-emission-depletion fluorescence microscopy. Optics letters. 1994;19:780–782. doi: 10.1364/ol.19.000780. [DOI] [PubMed] [Google Scholar]
- Hess ST, Girirajan TP, Mason MD. Ultra-high resolution imaging by fluorescence photoactivation localization microscopy. Biophysical journal. 2006;91:4258–4272. doi: 10.1529/biophysj.106.091116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoenger A. High-resolution cryo-electron microscopy on macromolecular complexes and cell organelles. Protoplasma. 2014;251:217–227. doi: 10.1007/s00709-013-0600-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes L, Hawes C, Monteith S, Vaughan S. Serial block face scanning electron microscopy—the future of cell ultrastructure imaging. Protoplasma. 2014;251:395–401. doi: 10.1007/s00709-013-0580-1. [DOI] [PubMed] [Google Scholar]
- Idrissi FZ, Geli MI. Zooming in on the molecular mechanisms of endocytic budding by time-resolved electron microscopy. Cellular and Molecular Life Sciences. 2013;1:8211–17. doi: 10.1007/s00018-013-1452-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juette MF, Gould TJ, Lessard MD, Mlodzianoski MJ, Nagpure BS, Bennett BT, Hess ST, Bewersdorf J. Three-dimensional sub–100 nm resolution fluorescence microscopy of thick samples. Nature methods. 2008;5:527–529. doi: 10.1038/nmeth.1211. [DOI] [PubMed] [Google Scholar]
- Jun S, Ke D, Debiec K, Zhao G, Meng X, Ambrose Z, Gibson GA, Watkins SC, Zhang Peijun. Direct Visualization of HIV-1 with Correlative Live-Cell Microscopy and Cryo-Electron Tomography. Structure. 2011;19:1573–1581. doi: 10.1016/j.str.2011.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jun S, Zhao G, Ning J, Gibson GA, Watkins SC, Zhang P. Correlative Microscopy for 3D Structural Analysis of Dynamic Interactions. Journal of Visualized Experiments. 2013 doi: 10.3791/50386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karreman MA, Agronskaia AV, van Donselaar EG, Vocking K, Fereidouni F, Humbel BM, Verrips CT, Verkleij AJ, Gerritsen HC. Optimizing immuno-labeling for correlative fluorescence and electron microscopy on a single specimen. Journal of structural biology. 2012;180:382–386. doi: 10.1016/j.jsb.2012.09.002. [DOI] [PubMed] [Google Scholar]
- Abu-Lail NI, Ohashi T, Clark RL, Erickson HP, Zauscher S. Understanding the elasticity of fibronectin fibrils: unfolding strengths of FN-III and GFP domains measured by single molecule force spectroscopy. Matrix Biology. 2006;25:175–184. doi: 10.1016/j.matbio.2005.10.007. [DOI] [PubMed] [Google Scholar]
- Leung BO, Chou KC. Review of super-resolution fluorescence microscopy for biology. Applied spectroscopy. 2011;65:967–980. doi: 10.1366/11-06398. [DOI] [PubMed] [Google Scholar]
- Lorenz UJ, Zewail AH. Biomechanics of DNA structures visualized by 4D electron microscopy. Proceedings of the National Academy of Sciences. 2013;110:2822–2827. doi: 10.1073/pnas.1300630110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maranto AR. Neuronal mapping: a photooxidation reaction makes Lucifer yellow useful for electron microscopy. Science. 1982;217:953–955. doi: 10.1126/science.7112109. [DOI] [PubMed] [Google Scholar]
- Mcdonald KL. A review of high-pressure freezing preparation techniques for correlative light and electron microscopy of the same cells and tissues. Journal of microscopy-oxford. 2009;235:273–281. doi: 10.1111/j.1365-2818.2009.03218.x. [DOI] [PubMed] [Google Scholar]
- McIntosh R, Nicastro D, Mastronarde D. New views of cells in 3D: an introduction to electron tomography. Trends in cell biology. 2005;15:43–51. doi: 10.1016/j.tcb.2004.11.009. [DOI] [PubMed] [Google Scholar]
- Modla S, Mendonca J, Czymmek KJ, Akins RE. Identification of neuromuscular junctions by correlative confocal and transmission electron microscopy. Journal of neuroscience methods. 2010;191:158–165. doi: 10.1016/j.jneumeth.2010.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mooney P, Contarato D, Denes P, Gubbens A, Lee B, Lent M, Agard D. A high-speed electron-counting direct detection camera for TEM. Microscopy and Microanalysis. 2011;17:1004–1005. [Google Scholar]
- Morphew MK. 3D immunolocalization with plastic sections. Methods in cell biology. 2007;79:493. doi: 10.1016/S0091-679X(06)79019-7. [DOI] [PubMed] [Google Scholar]
- Nisman R, Dellaire G, Ren Y, Li R, Bazett-Jones DP. Application of quantum dots as probes for correlative fluorescence, conventional, and energy-filtered transmission electron microscopy. Journal of Histochemistry & Cytochemistry. 2004;52:13–18. doi: 10.1177/002215540405200102. [DOI] [PubMed] [Google Scholar]
- Pereira CNdB, Daleprane B, Barbosa PF, Moreira AN, de Magalhães CS. Qualitative Evaluation of Scanning Electron Microscopy Methods in a Study of the Resin Cement/Dentine Adhesive Interface. Microscopy and Microanalysis. 2014;20:268–275. doi: 10.1017/S143192761301369X. [DOI] [PubMed] [Google Scholar]
- Powell RD, Halsey CM, Hainfeld JF. Combined fluorescent and gold immunoprobes: reagents and methods for correlative light and electron microscopy. Microscopy research and technique. 1998;42:2–12. doi: 10.1002/(SICI)1097-0029(19980701)42:1<2::AID-JEMT2>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Rigort A, Bäuerlein FJ, Leis A, Gruska M, Hoffmann C, Laugks T, Böhm U, Eibauer M, Gnaegi H, Baumeister W. Micromachining tools and correlative approaches for cellular cryo-electron tomography. Journal of structural biology. 2010;172:169–179. doi: 10.1016/j.jsb.2010.02.011. [DOI] [PubMed] [Google Scholar]
- Robinson JM, Takizawa T. Correlative fluorescence and electron microscopy in tissues: immunocytochemistry. Journal of microscopy. 2009;235:259–272. doi: 10.1111/j.1365-2818.2009.03221.x. [DOI] [PubMed] [Google Scholar]
- Robinson JM, Takizawa T, Pombo A, Cook PR. Correlative fluorescence and electron microscopy on ultrathin cryosections: bridging the resolution gap. Journal of Histochemistry & Cytochemistry. 2001;49:803–808. doi: 10.1177/002215540104900701. [DOI] [PubMed] [Google Scholar]
- Robinson JM, Takizawa T, Vandre DD. Applications of gold cluster compounds in immunocytochemistry and correlative microscopy: comparison with colloidal gold. Journal of microscopy. 2000a;199:163–179. doi: 10.1046/j.1365-2818.2000.00734.x. [DOI] [PubMed] [Google Scholar]
- Robinson JM, Takizawa T, Vandré DD. Enhanced labeling efficiency using ultrasmall immunogold probes: immunocytochemistry. Journal of Histochemistry & Cytochemistry. 2000b;48:487–492. doi: 10.1177/002215540004800406. [DOI] [PubMed] [Google Scholar]
- Rust MJ, Bates M, Zhuang X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM) Nature methods. 2006;3:793–796. doi: 10.1038/nmeth929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmued LARRYC, Snavely LF. Photoconversion and electron microscopic localization of the fluorescent axon tracer fluoro-ruby (rhodamine-dextran-amine) Journal of Histochemistry & Cytochemistry. 1993;41:777–782. doi: 10.1177/41.5.7682231. [DOI] [PubMed] [Google Scholar]
- Schnell U, Dijk F, Sjollema KA, Giepmans BNG. Immunolabeling artifacts and the need for live-cell imaging. Nature methods. 2012;9:152–158. doi: 10.1038/nmeth.1855. [DOI] [PubMed] [Google Scholar]
- Schultz C. Fluorescent Revelations. Chemistry and Biology. 2009;16:107–111. doi: 10.1016/j.chembiol.2009.02.001. [DOI] [PubMed] [Google Scholar]
- Schwartz CL, Sarbash VI, Ataullakhanov FI, Mcintosh JR, Nicastro D. Cryo-fluorescence microscopy facilitates correlations between light and cryo-electron microscopy and reduces the rate of photobleaching. Journal of microscopy. 2007;227:98–109. doi: 10.1111/j.1365-2818.2007.01794.x. [DOI] [PubMed] [Google Scholar]
- Schwarz H, Humbel BM. Correlative light and electron microscopy using immunolabeled resin sections. Electron Microscopy. 2007;369:229–256. doi: 10.1007/978-1-59745-294-6_12. [DOI] [PubMed] [Google Scholar]
- Shu X, Lev-Ram V, Deerinck TJ, Qi Y, Ramko EB, Davidson MW, Jin Y, Ellisman MH, Tsien RY. A genetically encoded tag for correlated light and electron microscopy of intact cells, tissues, and organisms. PLoS biology. 2011;9:e1001041. doi: 10.1371/journal.pbio.1001041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims PA, Hardin J. Visualizing Green Fluorescent Protein and Fluorescence Associated with a Gold Conjugate in Thin Sections with Correlative Confocal and Electron Microscopy. Microscopy and Microanalysis. 2004;10:156–157. [Google Scholar]
- Sosinsky GE, Gaietta GM, Hand G, Deerinck TJ, Han A, Mackey M, Adams SR, Bouwer J, Tsien RY, Ellisman MH. Tetracysteine genetic tags complexed with biarsenical ligands as a tool for investigating gap junction structure and dynamics. Cell communication and adhesion. 2003;10:181–186. doi: 10.1080/cac.10.4-6.181.186. [DOI] [PubMed] [Google Scholar]
- Sosinsky GE, Giepmans BN, Deerinck TJ, Gaietta GM, Ellisman MH. Markers for correlated light and electron microscopy. Methods in cell biology. 2007:79. doi: 10.1016/S0091-679X(06)79023-9. [DOI] [PubMed] [Google Scholar]
- Sprague BL, Pego RL, Stavreva DA, McNally JG. Analysis of binding reactions by fluorescence recovery after photobleaching. Biophysical journal. 2004;86:3473–3495. doi: 10.1529/biophysj.103.026765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stirling JW. Immuno-and affinity probes for electron microscopy: a review of labeling and preparation techniques. Journal of Histochemistry & Cytochemistry. 1990;38:145–157. doi: 10.1177/38.2.2405054. [DOI] [PubMed] [Google Scholar]
- Studer D, Humbel B, Chiquet M. Electron microscopy of high pressure frozen samples: bridging the gap between cellular ultrastructure and atomic resolution. Histochemistry and Cell Biology. 2008;130:877–889. doi: 10.1007/s00418-008-0500-1. [DOI] [PubMed] [Google Scholar]
- Takizawa T, Robinson JM. Correlative fluorescence and transmission electron microscopy in tissues. Methods in cell biology. 2012;111:37–57. doi: 10.1016/B978-0-12-416026-2.00003-0. [DOI] [PubMed] [Google Scholar]
- Valentijn J, van Driel L, Agronskaia A, Knoops K, Koning R, Barcena M, Gerritsen H, Koster A. Novel Methods for Cryo-Fluorescence Microscopy Permitting Correlative Cryo-Electron Microscopy. Microscopy and Microanalysis. 2008;14:1314–1315. [Google Scholar]
- Volkenandt T, Müller E, Gerthsen D. Sample Thickness Determination by Scanning Transmission Electron Microscopy at Low Electron Energies. Microscopy and Microanalysis. 2014;20:111–123. doi: 10.1017/S1431927613013913. [DOI] [PubMed] [Google Scholar]
- Wacker I, Schroeder RR. Array tomography. Journal of microscopy. 2013;252:93–99. doi: 10.1111/jmi.12087. [DOI] [PubMed] [Google Scholar]
- Watanabe S, Punge A, Hollopeter G, Willig KI, Hobson RJ, Davis MW, Hell SW, Jorgensen EM. Protein localization in electron micrographs using fluorescence nanoscopy. Nature methods. 2011;8:80–84. doi: 10.1038/nmeth.1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster P, Schwarz H, Griffiths G. Preparation of cells and tissues for immuno EM. Methods in cell biology. 2008;88:45–58. doi: 10.1016/S0091-679X(08)00403-2. [DOI] [PubMed] [Google Scholar]
- Westphal V, Rizzoli SO, Lauterbach MA, Kamin D, Jahn R, Hell SW. Video-rate far-field optical nanoscopy dissects synaptic vesicle movement. Science. 2008;320:246–249. doi: 10.1126/science.1154228. [DOI] [PubMed] [Google Scholar]
- Yildiz A, Forkey JN, McKinney SA, Ha T, Goldman YE, Selvin PR. Myosin V walks hand-over-hand: single fluorophore imaging with 1.5-nm localization. Science. 2003;300:2061–2065. doi: 10.1126/science.1084398. [DOI] [PubMed] [Google Scholar]
- Zaidi MR, Hornyak TJ, Merlino G. A genetically engineered mouse model with inducible GFP expression in melanocytes. Pigment cell & melanoma research. 2011;24:393–394. doi: 10.1111/j.1755-148X.2011.00832.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P. Correlative cryo-electron tomography and optical microscopy of cells. Current opinion in structural biology. 2013;23:763–770. doi: 10.1016/j.sbi.2013.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao G, Perilla JR, Yufenyuy EL, Meng X, Chen B, Ning J, Ahn J, Gronenborn AM, Schulten K, Aiken C, et al. Mature HIV-1 capsid structure by cryo-electron microscopy and all-atom molecular dynamics. Nature. 2013;497:643–646. doi: 10.1038/nature12162. [DOI] [PMC free article] [PubMed] [Google Scholar]