

Abstract
Nucleic acid-functionalized gold surfaces have been used extensively for the development of biological sensors. The development of an effective biomarker detection assay requires careful design, synthesis and characterization of probe components. In this feature article, we describe fundamental probe development constraints and provide a critical appraisal of the current methodologies and applications in the field. We discuss critical issues and obstacles that impede the sensitivity and reliability of the sensors to underscore the challenges that must be met to advance the field of biomarker detection.
Keywords: Gold nanoparticles, nucleic acid sensors, hAuNPs, electrochemistry, biobarcode assay, aptamers, DNAzymes
Introduction
The focus of this feature article is the development of robust biological sensors based on nucleic acid-functionalized gold surfaces, with particular emphasis on gold nanoparticles (AuNPs). A brief introduction to the field is provided, including an overview of the properties of gold surfaces, AuNPs and nucleic acids that make them useful components for biosensor development, as well as a review of work that initiated the field. The bulk of the review covers three types of nucleic acid motifs that are commonly conjugated to gold surfaces for biomarker detection: DNA hairpins, DNA biobarcode tags and functional nucleic acids (aptamers and DNAzymes). This feature article is not intended to be an exhaustive review of biological applications for nucleic acid-functionalized gold surfaces, but rather a compilation of useful insights for the development and possible applications of such probes based on experiences in our laboratories and in the recent literature. The discussion focuses primarily on design constraints, synthetic considerations and appropriate methods to characterize the different types of nucleic acid-based probes. Additionally, we offer a critical outlook on the state of the field and highlight important considerations for its advancement.
Because of their unique physical properties, gold surfaces have become a popular platform for the development of biologically relevant detection assays in a variety of settings, including health care,1–4 environmental testing,5, 6 and national security.7, 8 Gold offers many advantages over other available materials; it is biocompatible, essentially nontoxic, readily functionalized with a variety of ligands and relatively stable in many solutions including biological matrices.9 More specifically, AuNPs are frequently used because of their optical properties, ease of functionalization, size and efficient cellular internalization.
The optical properties of AuNPs make them a well-suited platform for biomarker detection applications. When metallic nanoparticles of a size comparable to the electron mean free path in metals (~100 nm) are irradiated with light having specific frequencies, collective oscillations of electrons are observed along the metallic surface. These oscillations are termed surface “plasmons” or localized surface plasmon resonances (SPR).10 For AuNPs (5 – 200 nm), these collective oscillations are in resonance with the incident radiation in the visible region of the electromagnetic spectrum, giving rise to a characteristic plasmon absorption band centered at 520 nm.10 As gold particles increase in size, the increase in diameter is also accompanied by an increase in the extinction coefficient and a red shift in the plasmon band. The net result is that particles with a diameter of around 15 – 100 nm are most visible to the human eye. Particles with an 80 nm diameter have an extinction coefficient of around 9 × 1010 M−1 cm−1. This exceeds the extinction coefficients of organic dyes by four or five orders of magnitude. The aggregation of AuNPs leads to the formation of a new absorption band at longer wavelengths as a result of electric dipole-dipole interaction and coupling between the plasmons of neighboring particles in the formed aggregates. This effect was the basis of some of the earliest types of AuNP-based assays as the coupling between the plasmons gives rise to a noticeable color change of the sample from red to purple. Fluorescence quenching and electron transfer are also a result of SPR, which further increases the diversity of biomarker detection applications using AuNPs. For a more detailed explanation of the optical properties of AuNPs, Myroshnychenko et al. has published an excellent review on the topic.11
Nucleic acids are highly favored as the functional interface between the gold surface and the biological matrix because of their homogenous charge and relatively limited chemical complexity compared to other large biomolecules (e.g. proteins). These characteristics ensure the structural stability and functionality of the nucleic acids on the gold surface. Furthermore, nucleic acids can be designed to be functional sensors because of their high specificity for complementary nucleic acid targets and numerous other biological molecules and because of their diverse repertoire of chemical readout mechanisms.12, 13 Advances in automated solid-phase synthesis using phosphoramidite chemistry have made oligonucleotides of virtually any sequence widely availability. Phosphoramidite building blocks include classic deoxyribo- and ribonucleosides as well as a host of other unnatural or chemically modified nucleosides, providing functional and structural diversity to synthetic oligonucleotides. Modifications incorporating fluorophores, quenchers, locked nucleic acid (LNA) nucleotides, “Click” chemistry reagents, biotin and other molecules have vastly expanded the applications of nucleic acid-based probes. The 5′ hexanethiol nucleoside modification, which allows the oligonucleotide to be easily conjugated to a gold surface via the gold-thiol bond, has facilitated the development of nucleic acid-functionalized gold-based probes.
In 1996 Alivisatos and Mirkin pioneered the use of DNA-AuNP conjugates.14, 15 They independently developed approaches for site-specific attachment of thiolated DNA probes on AuNPs based on gold-thiol chemistry. Alivisatos and coworkers demonstrated the feasibility of DNA-monofunctionalization by using 1.4 nm AuNPs that only allowed for an individual single-stranded DNA (ssDNA) to be attached due to surface area restrictions. These monofunctionalized nanoparticles were used to assemble discrete homodimeric and homotrimeric nanoparticle structures. Mirkin and coworkers used oligonucleotides as synthetically programmable constructs for guiding the assembly of DNA-functionalized AuNPs into macroscopic networks. Two non-complementary, thiol-modified DNA oligonucleotides were each bound to the surface of two batches of 13 nm AuNPs. When an oligonucleotide with ends that are complementary to the two sequences was added to the solution, the AuNPs aggregated, giving rise to a characteristic red-to-blue color change.
Although the primary goal of Alivisato’s and Mirkin’s initial work was to control the assembly of AuNPs for the synthesis of miniaturized electronic components and other nanoscale materials, DNA-gold conjugates have become a popular platform for the development of biological sensors. Along with the characteristic color change upon AuNP aggregation, the conductivity and high quenching efficiency gold have been extensively employed to develop a range of colorimetric, fluorescence and electronic biomolecular detection methods. Table 1 summarizes the results from several formats and applications of nucleic acid-functionalized gold surfaces.
Table 1.
Examples of nucleic acid sensors for the detection of biologically relevant targets
| Design | Biomarker target | Readout | Sample matrix | Limit of detection | Ref |
|---|---|---|---|---|---|
| Hairpin DNA-functionalized AuNPs, chips, wires and electrodes | |||||
| 15 nm hAuNP w/ FAM, Cy5, & Rox | Tumor-suppressor gene cDNA | Fluorescence | Buffer | 500 pM | 22 |
| 15 nm hAuNP w/ Cy5 & CAL Fluor Red 590 | Respiratory syncytial virus & GAPDH mRNA | Fluorescence | Live HEp-2 cells | 0.1 nM | 23 |
| 15 nm hAuNP w/ Quasar 670 | Tyrosinase mRNA | Fluorescence | Live SK-MEL-28 and HEp-2 cells | 0.5 nM | 24 |
| 3 mm × 3 mm Au substrate w/ TAMRA | Synthetic DNA probe target | Fluorescence | Buffer | 10 pM | 25 |
| 100 μm diameter Au-plated filament w/ CAL Fluor Red 590 | Respiratory syncytial virus mRNA | Fluorescence | Buffer | 400 aM | 28 |
| 0.9 mm2 Au electrode w/ methylene blue | Salmonella gyrB gene | Voltammetry | Buffer | 400 nM | 29 |
| 1.6 mm diameter Au disks w/ ferrocene | Synthetic DNA probe target | Voltammetry | Buffer | 10 pM | 30 |
| Biobarcode tag-functionalized AuNPs | |||||
| Antibody-based w/ MMP solid phase | Prostate-specific antigen | Scanometric and PCR | Goat serum | 30 aM with scanometric 3 aM with PCR | 45 |
| DNA-based w/ MMP solid phase | Synthetic anthrax gene | Scanometric | Buffer | 500 zM | 46 |
| Antibody-based w/ well plate solid phase | Hantaan virus nucleocapsid protein | PCR | Human serum | 10 fg/mL (~200 aM) | 47 |
| Antibody-based w/ MMP solid phase | Respiratory syncytial virus fusion protein | PCR | Virus-infected HEp-2 cells | 4.1 pfu/mL | 44 |
| Antibody-based w/ MMP solid phase | Bluetongue virus VP7 protein | PCR | Sheep serum | 0.1 fg/mL (~3 aM) | 48 |
| Antibody-based w/ chip solid phase | Prostate-specific antigen | Scanometric | Goat serum | 500 aM | 49 |
| Antibody-based w/ MMP solid phase | Prostate-specific antigen, human chorionic gonadotropin, & α-fetoprotein | Scanometric | Goat serum and diluted goat serum | 33 pM in serum 170 fM in diluted serum | 50 |
| Aptamer- and DNAzyme-functionalized AuNPs and electrodes | |||||
| DNAzyme AuNP controlled aggregation | Cu(II) | Absorbance | Water | ~5 μM | 60 |
| Aptamer AuNP controlled aggregation | Lymphoblastic leukemia or lymphoma cells | Absorbance | Diluted cell media | 90 cells | 61 |
| DNAzyme AuNP controlled aggregation | Pb(II) | Absorbance | Water and paint | 100 nM in water 0.5% in paint | 5 |
| DNAzyme AuNP controlled aggregation | Adenosine | Absorbance | Buffer | 100 μM | 62 |
| Aptamer AuNP controlled aggregation | Adenosine triphosphate | Absorbance | Buffer | 0.6 μM | 63 |
| DNAzyme AuNP controlled aggregation | Pb(II) | Dynamic light scattering | Water | 35 pM | 64 |
| DNA capture DNAzyme AuNP | Synthetic DNA or telomerase activity | Luminescence | Buffer (DNA) or cell extracts (telomerase) | 100 pM with DNA 20,000 cells with telomerase | 65 |
| DNA capture DNAzyme AuNP | Synthetic Chlamydia gene | Absorbance | Buffer | 50 fM | 66 |
| DNA capture DNAzyme AuNP | Pb(II) | Fluorescence | Buffer | 5 nM | 67 |
| Electrochemical DNAzyme | Pb(II) | Voltammetry | Buffer | 0.3 μM (62 ppb) | 69 |
| Electrochemical Aptamer | Thrombin | Voltammetry | Buffer and human serum | 1 pM in buffer ND in serum | 70 |
| Electrochemical Aptamer w/ biobarcode AuNP | Thrombin | Voltammetry | Buffer and fetal calf serum | 0.1 ng/mL (2.7 pM) in buffer ND in serum | 77 |
| Electrochemical DNAzyme (AuNP enhanced) | Pb(II) | Voltammetry | Buffer | 1 nM | 78 |
DNA Hairpin-Functionalized Gold
There has been ever-increasing demand to develop rapid, sensitive and selective bioassay methods in molecular diagnostics, environmental monitoring and the detection of infectious agents. A novel molecular tool toward this end is the molecular beacon (MB). MBs are end-labeled oligonucleotides that exist in solution as stem-loop structures and utilize the fluorescence of a reporter dye at the 5′ end and a proximal quencher attached to the 3′ end (Scheme 1). The fluorophore/quencher pair produces an “on” or “off” signal dependent on the conformational state of the MB. When they are in close proximity (typically < 5 nm) and the fluorophore is in its excited state, emitted photons are absorbed by the quenching molecule. Conversely, the fluorescence signal is significantly enhanced when the distance between the quencher and the fluorophore exceeds 10 nm. The stem region consists of four to seven base-pairs and functions as a lock to maintain the closed hairpin structure and bring the quencher within a few nanometers of the fluorophore. The loop is the recognition element. Upon hybridization to a complementary nucleic acid strand, the stem region opens, and consequently induces a conformational change of the MB. This increases the physical distance between the fluorophore and the quencher and results in a loss of quenching and an increase in fluorescence. This signal-generation mechanism of MBs enables the analysis of target oligonucleotides without the separation of unbound probes. Because of their signal-on sensing and high sequence specificity resulting from the conformational constraint offered by the stem-loop structure, MBs have found wide application in molecular and cellular biology, pathogen detection and biomedical diagnostics.16
Scheme 1.
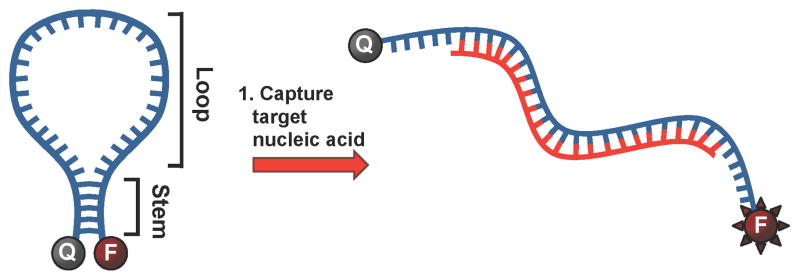
Molecular beacon is held in the closed (“off”) conformation by complementary base-pairing in the stem region. Hybridization of the loop region with the target nucleic acid disrupts the base-pairing of the stem region and the molecular beacon is held in the open (“on”) conformation.
Continued development of MBs has resulted in improvements in various aspects from their original design. The choice of quenchers is of particular interest as it may lead to a decrease in nonspecific background signals and therefore a lower “off” signal. While standard organic quenchers have demonstrated effectiveness in quenching the fluorescence of fluorophores located in their vicinity, their quenching efficiencies vary significantly from one dye to another. For example, the organic quencher 4-((4′-(dimethylamino)-phenyl)azo)benzoic acid (DABCYL) efficiently quenches the fluorescence of fluorescein (FAM), but is much less efficient for dyes emitting at longer wavelengths like Cy5 and Texas Red. Black Hole Quenchers (BHQ) have low background fluorescence and a broad effective range of absorption, but it is still important to match the BHQ with the appropriate dye based on the excitation and emission spectra of the dye.17
Metallic gold, both macroscale gold and AuNPs, is well known for its ultrahigh fluorescence quenching.18 Dubertret et al. formed the first AuNP-based MB by attaching a fluorescently labeled probe in a stem-loop configuration to the surface of 1.4 nm AuNPs, which gave rise to high quenching efficiencies toward four dissimilar organic fluorophores.19 Fluorescein, Rhodamine 6G, Texas Red, or Cy5 was attached on one end and the AuNP was attached to the opposite end. This work detailed the application of AuNPs for organic fluorophore quenching. AuNPs were found to have a quenching efficiency a hundred times higher than DABCYL, giving higher sensitivity for detection of oligonucleotide single mismatches. Maxwell et al. reported a different design that employed a dye-labeled linear oligonucleotide (stemless probe) that curved toward the 2.5 nm AuNP surface because of the strong dye-AuNP adsorption.20 An oligonucleotide molecule was firmly tethered to a particle via a sulfur-metal bond, and the fluorophore at the distal end was able to loop back and adsorb on the same particle. A quantum efficiency of almost 100% was seen when the fluorophore was adsorbed on the particle. They also showed that for larger gold particles (10 – 50 nm diameter), an efficient long-range energy-transfer effect that quenches fluorophores over spatial distances as large as 10 – 20 nm. This long-range feature could allow the development of biosensors and homogeneous bioassays that are not possible using organic dyes. Further optimization of this system has involved the use of 15 nm AuNPs, which have stronger SPR absorption relative to smaller particles. The strong SPR absorption can be used to quench a broad range of fluorophores. Fan et al. reported that AuNPs between 5 – 20 nm diameters absorb strongly between 300 – 500 nm wavelength range, a range in which 2 nm AuNPs absorb hundreds of times less intensely.18 In addition, the greater surface area of the particle is advantageous to functionalize a larger number of oligonucleotides as well as different types of oligonucleotides.
Design & Applications
Mirkin’s group developed a detection method based on oligonucleotide-modified AuNPs, called nanoflares.21 Nanoflares take advantage of the fact that AuNPs functionalized with oligonucleotides efficiently enter live cells without the aid of harsh transfection/permeabilization agents and have limited cytotoxic effects. A nanoflare probe consists of a AuNP conjugated to many sequences of ssDNA, which are complementary to the RNA sequence of interest. Short “reporter” strands of fluorescently-tagged ssDNA are hybridized to the probe DNA. In this state, the organic fluorophore is quenched as a result of its proximity to the AuNP surface. Upon hybridization of the probe strand with the target RNA complement, the reporter strand is displaced, spatially separating it from the AuNP and resulting in a fluorescent signal that can be correlated to the presence of the RNA sequence of interest. While nanoflares overcome many of the challenges faced by previous approaches, their design yields a signal that is separated from the hybridized RNA/nanoflare construct. Consequently, important spatial information is lost in live cell microscopy applications.
A new strategy has been developed, which combines nanotechnology with MB-based nucleic acid detection, using DNA hairpin-gold nanoparticles (hAuNPs).22–24 Because the DNA hairpins are tethered to the gold surface, hAuNPs provide spatiotemporal information about target oligonucleotides when used in live cells. hAuNPs are constructed from 15 nm diameter AuNPs with covalently attached DNA hairpin beacons via a gold-thiol bond. Each DNA hairpin beacon consists of a 5′ hexanethiol linker covalently attached to a 10-thymine extension followed by a 30 base hairpin recognition sequence and a 3′ fluorophore. While in the closed position, this construct utilizes the fluorescence quenching ability of the AuNP resulting in the absence of a fluorescence signal (Scheme 2A). Upon binding of its complementary sequence, the 3′ fluorophore extends past the quenching distance of the AuNP resulting in a positive fluorescence signal; this is referred to as the “on” state.
Scheme 2.

DNA hairpin-functionalized gold surfaces for nucleic acid detection. (A) AuNPs or (B) gold wires quench the fluorophore-modified hairpin DNA. Upon binding target nucleic acid, the hairpin DNA opens and the fluorescence is detected.
Song et al. developed a multiplex nucleic acid detection assay using hAuNPs with multiple DNA hairpin beacons labeled with different fluorophores.22 In this proof-of-principle study, hairpin probes consisting of complementary sequences to three different tumor-suppressor genes were labeled with FAM, Cy5 and Rox, and then they were bound to 15 nm AuNPs. Approximately 40 – 50 probes were conjugated to each AuNP. Using different targets on single particles, these hAuNPs detected each specific mRNA sequence and discriminated against single-base-mismatched DNA.
Due to their efficient cellular internalization and low cytotoxicity, hAuNPs have also been used as in vitro probes for live-cell imaging. Recently, our laboratories reported the first use of hAuNP probes for investigating spatiotemporal RNA trafficking in live cells (Figure 1).22–24 Targets for respiratory syncytial virus (RSV) mRNA and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) mRNA were functionalized onto 15 nm AuNPs and used in live and RSV-infected HEp-2 cells and uninfected HEp-2 cells, respectively, with high target signal to background ratio.23 Due to its stable, constitutive expression in almost all mammalian cell types, GAPDH serves as a useful internal control for normalization in standard quantitative molecular biology assays such as RT-PCR. The detection of RSV mRNA concurrently with GAPDH mRNA as an internal control in our assay enabled real-time analysis of mRNA transport and processing in live cells. Similar hAuNP constructs have been used to positively identify tyrosinase, a common melanoma gene target, in primary melanoma tumor cells.24
Figure 1.
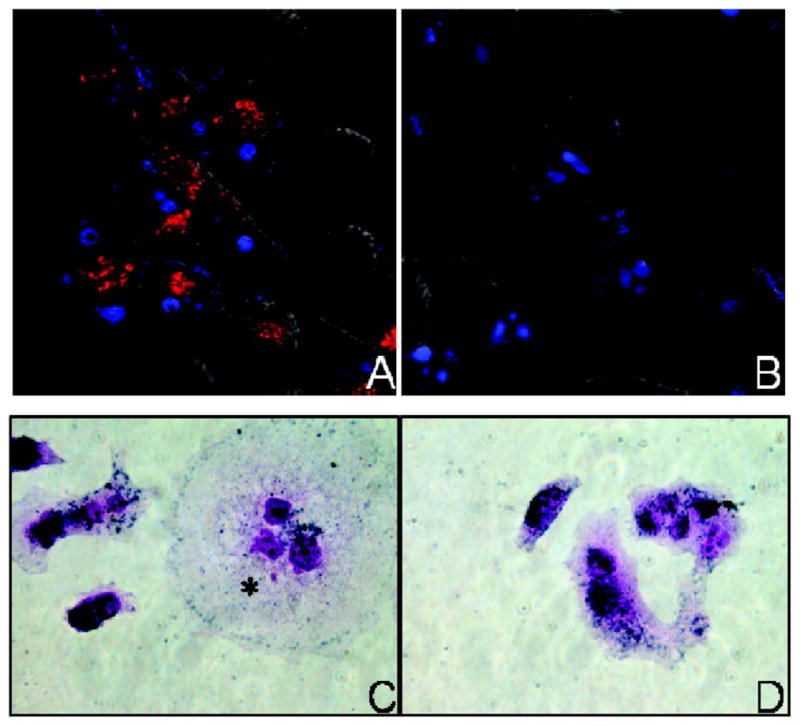
Fluorescence microscopy images of (A) respiratory syncytial virus (RSV)-infected HEp-2 cells and (B) uninfected cells treated with hAuNPs. Red emission from hAuNPs is only observed in RSV-infected cells. Blue emission is from a nuclear counterstain. Intracellular uptake of hAuNPs in both (C) RSV-infected HEp-2 cells and (D) uninfected cells is demonstrated in immunohistochemical stained cells. Reprinted with permission from Ref. 23.
Further studies have utilized DNA in hairpin conformation in coordination with gold surfaces other than AuNPs. Krauss and coworkers reported a surface-confined MB by using macroscopic gold films.25, 26 The fluorescence intensity increased about 100-fold upon hybridization with a complementary DNA, which was detectable at concentrations from 0.2 to 3 μM. An eight-fold lower sensitivity was observed for a singly mismatched target. The sensor was, however, not suitable for multiple measurements in showing an approximately 40% degradation of fluorescence intensity after each regeneration cycle.
The combination of MBs with filament processing retains the simple and rapid processing design of Krauss and improves the lower level of detection significantly. Cederquist et al. used gold nanowires to report the first determination of hybridization efficiency for hairpin DNA immobilized on a surface.27 The hybridization efficiency was highly dependent on probe coverage and dropped to ~20% at both higher and lower coverage, due to steric hindrance and adsorption of the probe to the metal surface, respectively (Figure 2). They also found that the use of thiolated oligoethylene glycol spacers increased the hybridization efficiency at the lowest coverage by displacing nonspecifically bound nucleotides from the surface. Perez et al. designed a viral detection strategy utilizing a MB style hairpin DNA covalently coupled to a moveable gold-clad filament (Scheme 2B).28 This allowed for the viral probes attached to the filament to be easily added to and removed from processing solutions.
Figure 2.

Efficiency of hairpin hybridization to target nucleic acid plotted against surface coverage of hairpins on the gold nanowire. Optimal surface coverage maximizes the hybridization efficiency of the hairpins. Reprinted with permission from Ref. 27.
Recently, Heeger, Plaxco and others developed a series of novel reagentless, sensitive and selective electrochemical DNA sensors that combine the significant advantages of electrochemical detection method with the versatility of surface-attached MBs.29, 30 This electrochemical DNA sensor employs an electrode-attached, stem-loop DNA structure labeled with an electroactive reporter (e.g. ferrocene or methylene blue) as a capture probe. Before hybridization between the probe DNA and its complementary target, the electroactive label is localized to the electrode surface by means of hybridization of the stem region of the probe DNA, allowing facile electron transfer between the electroactive label and the gold electrode. In the presence of a complementary target, the stem-loop is converted into a rigid, linear double helix and the distance between the label and the electrode is significantly expanded, leading to a large, readily measurable signal change.
Methylene blue was also used as a reporter molecule conjugated to the 3′ end of an amino- and thiol-modified stem loop oligonucleotide through succinimide ester coupling for the electrochemical sequence-specific detection of unpurified amplification products of the gyrB gene of Salmonella typhimurium.29 Using asymmetric PCR (aPCR) and alternating current voltammetry as electrochemical transduction techniques, single-stranded amplicons were produced from 90 gene copies and, without subsequent purification, rapidly identified. As illustrated in Scheme 3, the sensor was fabricated by self-assembly of the methylene blue-labeled DNA probes on a gold electrode surface. In the absence of target, the stem-loop structure holds the methylene blue tag close to the electrode surface, thus enabling efficient electron transfer. Upon hybridization with the target PCR amplicon, a large change in the reduction peak of methylene blue was observed. Using a short room temperature deionized water rinse the original sensor signal was almost completely recovered, the sensor being reusable with a synthetic PCR target for more than eight consecutive times without exhibiting unacceptable sensor degradation. Moreover, the sensor did not respond to control oligonucleotides using the gyrB genes of Escherichia coli and various Shigella species. Given the advantages provided by this kind of sensor (i.e. good sensitivity, reproducibility, reusability and no need for expensive optics nor high voltage power suppliers), it is a promising technology for the implementation of portable PCR microdevices for rapid detection of pathogens and other microorganisms.
Scheme 3.
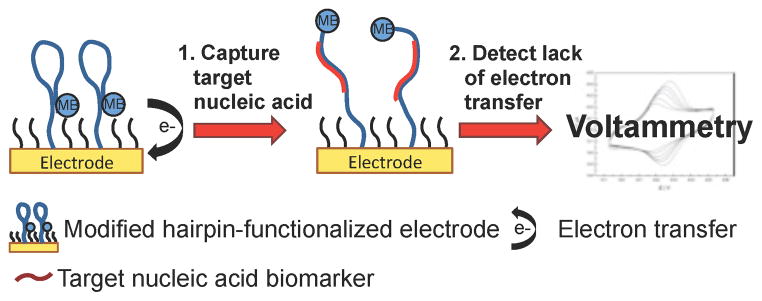
Electrochemistry using methylene blue-functionalized electrodes for nucleic acid detection. The proximity of the methylene blue-modified hairpin DNA induces electron transfer with the electrode surface. Upon binding target nucleic acid, the hairpin DNA opens and the lack of electron transfer is detected by voltammetry.
Detection strategies utilizing DNA in hairpin conformation coordinated to microscale gold surfaces face similar design challenges and require comparable characterization as those based on AuNPs. Surface density of the hairpin DNA strands remains an issue as steric interference between sequences may disrupt hybridization of complementary strands. The oligonucleotide loading may still be determined by subjecting a gold substrate to dithiolthreitol reduction to reduce the gold-thiol bonds; however, small particle analysis techniques such as DLS are not useful for characterization of macroscale surfaces. Instead, cyclic voltammograms for modified gold electrodes may be used to detect surface modifications by detecting peaks in the relevant potential range.
Synthesis
hAuNPs have primarily been constructed using 15 nm AuNPs due to their ease of synthesis and substitution. The method developed by Turkevich et al. in 1951 and refined by Frens in the 1970s is used to produce monodisperse spherical AuNPs between 10 – 20 nm in diameter.31–33 Larger particles can be produced, but this comes at a cost to monodispersity and isotropy. Furthermore, it has been found that thiol-terminated oligonucleotides attached to AuNPs less than 20 nm in diameter have significantly more distance between the neighboring strands moving radially out from the surface than their larger particle counterparts, resulting from the smaller particles’ highly curved surfaces.34 This decreases steric interactions between oligonucleotides attached to small AuNPs, allowing for more oligonucleotides to attach to the highly curved surface than to the larger, flatter surfaces, when equal areas are compared.
The research performed by Song et al. revealed interesting constraints regarding hAuNP synthesis using 15 nm AuNPs.22 Initially, the group prepared the hAuNPs by following the classic self-assembly approaches developed by Mirkin and others.35 A stem-loop oligonucleotide, dually labeled with a 3′ FAM and a 5′ thiol, was incubated with AuNPs, allowed to self-assemble through the gold-thiol bond and then “aged” in salt solution. However, the conjugation reaction turned from red to blue during the aging step, a phenomenon suggesting the formation of large aggregates. This effect is attributed to the low surface density of stem-loop structures achievable on AuNPs modified with hairpin DNA. The low DNA surface density on the AuNPs leads to weak electrostatic/steric repulsion between the DNA strands on the AuNPs that results in the self-aggregation of the DNA-AuNP conjugate at suitable salt concentration due to a screening effect of the salt.
The bulky nature of the self-assembled stem-loop structures significantly lowers the surface density of the probes, leading to unstable DNA-AuNP conjugates. In contrast, AuNPs heavily loaded with linear DNA strands possess strong interparticle electrostatic repulsion, which protects the AuNPs from aggregation. It is also important to consider that there is a maximum number of DNA hairpins that will coat the surface of a gold sphere before steric interference of hairpins with one another becomes a hindrance to the hairpin DNA recognition and opening mechanism.
To address the issues with low or high DNA surface density, Song et al. modified the original assembly protocol with the introduction of short “helper” oligonucleotides, that is, 3′-thiolated 10 base oligothymines (10-T).22 Also of note, since the stem-loop oligonucleotide (35 base) employed is appended with a 10-T spacer at the 3′ end, the presence of helper strands of equivalent length does not sterically prevent the formation of the stem-loop structure. The 10-Ts were co-assembled with the stem-loop oligonucleotide to form a mixed monolayer at the surface of 15 nm AuNPs. These short oligonucleotides form a dense layer at the nanoparticle surface and prevent the steric interference of DNA hairpins with one another while also protecting the AuNPs from aggregating during the aging step. The helper oligonucleotides significantly increased the stability of the hAuNPs, which remained dispersed even at high ionic strength (e.g., 1 M NaCl).22
The overall performance of hAuNPs is critically dependent on the ratio between the DNA hairpins and the helper oligonucleotides in the assembly solution. hAuNPs were not stable at low ratio of helper to hairpin oligonucleotides (e.g., 1:10), which resulted in the aggregation of the nanoparticles. On the other hand, although hAuNPs were successfully prepared at high ratio of helper to hairpin oligonucleotides (e.g., 10:1), the signal of the hAuNPs at the “on” state was low because of the low surface density of the stem-loop probe. A ratio of 2:1 was experimentally chosen for the preparation of hAuNPs, which provides a balance between the stabilization and the signal intensity. By following a well-established displacement-based fluorescence assay protocol,36 it was found that each hAuNP carried approximately 40 – 50 strands (44 ± 5) of probe DNA.
Characterization
Careful characterization of hAuNPs is necessary for proper analysis of successful design and synthesis of the probes. UV-visible absorbance spectra of hAuNPs exhibit absorbance peaks for nucleic acids and AuNPs at 260 and 520 nm, respectively. The oligonucleotide loading for each particle can be determined by subjecting a solution of hAuNPs to dithiolthreitol for reduction of the gold-thiol bonds and measuring the number of released oligonucleotides. Dynamic light scattering (DLS) analysis of the hAuNPs compared to unconjugated AuNPs is a powerful, qualitative characterization technique. An increase in hydrodynamic diameter when AuNP are coupled to hairpin DNA is indicative of successful conjugation.
Fluorescence spectrophotometry studies are used to confirm that hAuNPs are specific for synthetic target sequences, with emission of fluorescence consistent with the fluorophore coupled to the 3′ end of the hairpin DNA. Jayagopal et al. demonstrated that hAuNPs targeted against RSV or GAPDH both exhibited dose-specific increases in fluorescence intensity in response to complement concentration. RSV- or GAPDH-hAuNPs, in contrast, did not react to an appreciable level when incubated with a concentration of mismatched cDNA featuring similar C-G content to the natural target.
For some applications (i.e. live cell imaging), it is important to demonstrate stability against nuclease-mediated degradation as degradation gives rise to false-positive signals. While Song et al.’s hAuNPs were not tested for stability, Jayagopal et al. determined hAuNP stability against degradation via a fluorimetric assay using DNAse I.22, 23 The degradation rate of hAuNPs targeted toward RSV was 0.072 nmol min−1, and the degradation rate for GAPDH-specific hAuNPs was 0.094 nmol min−1 compared to fluorophore-quencher paired MBs with exhibited degradation rates of up to 1.25 nmol min−1. While this is an improved method for determining stability against nuclease degradation compared to previous approaches, an assay determining the stability in cell lysates might be more representative of degradation in a cellular environment.
Outlook
The combination of hairpin DNA and gold surfaces is rapidly gaining popularity in oligonucleotide sensing-based technologies. The increase in distance of an electroactive reporter and the gold surface upon hybridization of a complementary sequence has been shown to lead to a large, readily measurable signal change which could potentially pave the way for the development of multiplexed point-of-care diagnostic device and defense-related applications. However, there is significant room for improvement, as these designs still operate under many of the constraints faced when using MB probes. From the synthesis perspective, each particular hAuNP application requires that the loading of the probe ligands and their ratio to potential backfill ligands be optimized. This remains, for now, an empirical problem. But with proper characterization and reporting, the appropriate ligand ratios for a AuNP of a given size may emerge. In the context of assay development, the temperature and salt conditions must be designed to match with what is required for efficient hybridization. The melting temperature of the stem-loop structure should be higher than the detection temperature, although stem-loop dynamics lead to fluctuations with temperature. In practice, stems of 5 – 6 bases and probe-loop sequences of 16 – 22 bases are most commonly used on AuNPs, although to our knowledge a detailed study of the effect of varying the number of bases in either region has on specificity has not been done. Finally, the signal-to-noise ratio for MB systems that use a solid or immobilized phase often falls in a range of 2 – 5,37, 38 which is lower than ratios typically seen in solution.39, 40 The optimization of these design parameters will be important for the future of molecular diagnostics and facilitate advances in real-time monitoring of gene expression.
DNA Biobarcode Tag-Functionalized Gold
Analytical methods for protein and nucleic acid detection have advanced tremendously in recent years. Antibody-based protein detection strategies, or immunoassays, are an attractive option for detection because of their specificity and versatility to detect a variety of pathogens and toxins. Enzyme-linked immunosorbent assays (ELISAs) use antibodies conjugated to fluorophores or enzymes to detect target proteins fixed to a solid surface and has been the gold standard of protein biomarker detection because of its relatively high specificity and sensitivity.41 Various ELISA formats have been developed that increase the flexibility of the assay. Sandwich ELISA, which employs capture antibodies to attach the antigen to the solid phase, increases specificity and reproducibility over standard ELISA and has provided a way to perform ELISA on the surface of antibody-functionalized magnetic beads. Immuno-PCR (IPCR) has taken advantage of the robustness and versatility of ELISA coupled with the high specificity and sensitivity of PCR to improve the limit of detection of immunoassays by several orders of magnitude.42, 43 Instead of using fluorescence or enzymatic turnover as in ELISA, IPCR employs DNA tags, which can be detected by PCR or other DNA recognition assays, as the readout mechanism. The DNA tags take advantage of the inherent function of nucleic acids to store information, thereby functioning as “biobarcodes.” The sensitivity of IPCR has been further enhanced with the use of AuNPs to mediate the association of many DNA biobarcode tags with a single target recognition element.44
The biobarcode format employs AuNPs functionalized with a capture moiety, such as an antibody or DNA recognition sequence, along with multiple DNA biobarcode tags to provide intrinsic signal amplification (Scheme 4 and Scheme 5).44–46 Anchored to a solid phase, such as a magnetic particle or the bottom of a 96-well plate, a second capture moiety is used to isolate the probe from the analyte solution. The capture moiety on the AuNP binds a different region of the target biomarker, forming a sandwich structure. The biobarcode DNA tags are then removed from the AuNP and detected using quantitative PCR (qPCR), a microarray assay, or fluorescence.
Scheme 4.

Antibody-based biobarcode assay for antigen detection. (A) Capture antibody-functionalized magnetic microparticles (MMPs) or (B) well plates bind target antigen biomarker to separate it from the analyte solution. DNA tag and antibody-functionalized AuNPs bind and form a sandwich structure around the target antigen. The DNA tags are removed and detected by quantitative polymerase chain reaction (qPCR), a microarray assay, or fluorescence.
Scheme 5.

DNA-based biobarcode assay for nucleic acid detection. (A) Capture DNA-functionalized magnetic microparticles (MMPs) or (B) well plates hybridize to the target nucleic acid biomarker to separate it from the analyte solution. DNA tag-functionalized AuNPs hybridize to the target nucleic acid. The DNA tags are removed and detected by quantitative polymerase chain reaction (qPCR), a microarray assay, or fluorescence.
Design & Applications
The design constraints for the components of the biobarcode assay are dependent on the application, the type of target biomarker, the equipment available for detection and the environment in which the assay will be performed. Consequently, to develop an effective biobarcode assay the capture moieties, DNA tags and detection format must be carefully considered.
The type of capture moiety used in the assay depends on whether the target biomarker is protein or nucleic acid. The capture moiety most commonly used in recent reports is the antibody (Scheme 4).44, 47, 48 The antibody-based biobarcode assay has been successfully developed for the detection of several cancer-specific biomarkers and viral proteins.44, 45, 47–50 Antibodies are useful recognition elements because they can be used to detect many important protein biomarkers, they are widely available and they can be efficiently conjugated to AuNPs and a variety of solid phases. However, there are limitations to antibody-based detection. Compared to DNA biomarkers, proteins are less stable and more sensitive to thermal and chemical degradation. Also, in order to pull down the target biomarker in a sandwich format, antibodies must be selected to bind two distinct epitopes of the protein target. Usually a monoclonal antibody is functionalized to the solid phase, while a monoclonal or polyclonal antibody is used on the biobarcode probe,51 and for many protein targets, two distinct antibodies are not available for this format.
The type of capture moiety used for DNA targets is a short sequence of DNA that binds to the target by base-pair recognition (Scheme 5). A second DNA sequence conjugated to the solid phase is complementary to a different region of the DNA target and is used to separate the biobarcode probe from the analyte solution. The DNA capture-based biobarcode assay has been demonstrated to detect a synthetic anthrax gene,46 and could be developed to detect a range of biologically relevant nucleic acid targets such as genomic DNA, mRNA, microRNA and small interfering RNA. Unlike antibodies, synthetic DNA can be made to complement virtually any known sequence; therefore, DNA capture moieties do not suffer from issues of availability. Nucleic acid targets, however, are often double-stranded or form tertiary structures, so competition with the natural complement for base-pairing with target becomes an issue. Additionally, nucleic acid biomarkers are generally much longer than the targeted sequence and the overhanging ends of the nucleic acid can interfere with the performance of the assay.
DNA tag sequences used for the biobarcode assay must be unique, otherwise the risk of cross reactivity and false positives is high. An NCBI BLAST search can be performed to determine if there are any off targets for the biobarcode sequence that may be present in a biological assay (http://blast.ncbi.nlm.nih.gov/Blast.cgi).51 The ideal length of the DNA tags for the microarray detection method is 25 – 30 bases. Universal DNA tag sequences, with corresponding microarray chips and probes, have been developed for this assay.51 If using qPCR to amplify and detect the tags, longer sequences are recommended (45 – 80+ bases) for efficient PCR. DNA tags can be made from ssDNA or dsDNA. For ssDNA, the tags are anchored at one end to the gold surface and can be released by DTT prior to detection, but this method requires the DNA to be separated from the DTT, due to its interference with downstream detection methods. On the other hand, dsDNA is anchored to the gold surface by one end of ssDNA complementary to the strand that will be detected. The DNA tag is then annealed to the anchored DNA strand during probe synthesis and released from the anchored strand by heating prior to tag detection. However, we have observed that releasing complementary DNA from AuNPs by heating can be inefficient at lower temperatures (65 – 80 °C), and DNA tag release at higher temperatures (80 – 95 °C) tends to disrupt the gold-thiol interaction and release the anchored DNA from the gold surface. The release of this thiol-modified anchor DNA has the potential to interfere with the detection of the DNA tags by forming dsDNA with the DNA tags and introducing background noise to the assay.
Detection of the biobarcodes after release can be performed using qPCR, microarrays, or fluorescence. qPCR is the most established method for nucleic acid detection and has been the method of choice for the detection of biobarcode tags in the most recent reports. qPCR allows the most sensitive quantitative analysis of the number of biobarcode DNA tags released in the assay, achieving limits of detection several orders of magnitude lower than ELISA-based methods.44, 47, 48 The Mirkin group developed another technique for biobarcode detection called the scanometric method, in which tags are hybridized to a microarray chip and are detected using DNA-functionalized AuNPs and silver and hydroquinone for signal enhancement.45, 52 The scanometric method has allowed the biobarcode assay to be performed entirely on a chip, independent of expensive and cumbersome thermocyclers or fluorometers, while still obtaining limits of detection in the high attomolar range.49 The method is highly sensitive, reaching limits of detection in the low attomolar range. Disadvantages of the scanometric method include the time required to perform the assay and the dependence on specialized materials and equipment.51 Another method to detect the biobarcode tags is to use fluorescent dyes. The DNA tags can be modified with fluorescent dyes during synthesis or the dyes can be incorporated into dsDNA after tag release. Although fluorescence detection is the simplest and fastest method, it is not as commonly used because, in practice, it is not sensitive enough to detect concentrations of DNA tags below the high nanomolar range.
Synthesis
The synthesis of both antibody-based and DNA capture-based biobarcode AuNPs is well established.44, 51 Biobarcode probes are most commonly synthesized on 15 nm or 30 nm AuNPs. During synthesis, 15 nm AuNPs are generally more stable than 30 nm AuNPs because of their higher surface area-to-volume ratio, but they have four times less surface area for probes to bind. For a typical synthesis reaction, the thiol-modified DNA biobarcode tag is added in 50 – 100-fold excess of the recognition element concentration for 15 nm AuNPs and 200 – 300-fold excess for 30 nm AuNPs in order to achieve a 1:1 recognition element to AuNP ratio with multiple DNA tags.
Biobarcode AuNPs have less synthetic constraints than hAuNPs with regard to steric issues. Since detection of DNA tags is generally performed after they are released into solution, signal is independent of the structure and function of the DNA tags on the AuNP surface. Therefore, overcrowding of DNA tags on the AuNP surface does not influence detection of the DNA tags as it does with hAuNPs, which require surface coverage that does not impede the opening of the hairpin structure upon binding the target.
For a more thorough description of the design and synthesis of the biobarcode assay components, the Mirkin group has published an in-depth protocol on the subject.51
Characterization
Careful characterization of the components on the biobarcode assay is necessary for proper analysis of successful design and synthesis of the probes. The components that are most critical to characterize are the number of biobarcode tags on the probe, the number of capture moieties on the probe and the number of capture moieties on the solid phase. Generally, the number of DNA tags or antibodies attached is quantified by subtracting the amount measured in the supernatant after the functionalization step from the total amount added. Generally, absorbance at 260 nm is used to quantify DNA, and absorbance at 280 nm is used to quantify antibodies.44, 51 Issues arise, however, because the supernatant absorbance method is not trivial. For example, not all the supernatant can be removed from the AuNPs without some loss of AuNPs. Additionally, the method does not account for DNA or antibody loss from binding to the tube walls, pipette tips, etc. To account for miscalculations that may result from these issues, a fluorescence-based method has also been described to quantify the number of DNA tags on the AuNP. The fluorescence method requires fluorophore-labeled oligonucleotides to be used as surrogates for quantification.36, 47 This method, however, is not ideal because the number of DNA tags on the probes used in the biobarcode assay are not directly measured. Alternatively, if using DNA tags designed for qPCR, the tags could be released form the AuNPs and quantified against a standard curve by qPCR. Characterization of the other components of the assay, such as the functionalized solid phase or the capture antibody on the AuNP, is rarely reported in the literature. To validate the effectiveness of the antibody-functionalized solid phase, our group uses a sandwich-ELISA type validation with quantum dot-conjugated primary antibodies that can be visualized by fluorescence microscopy. Although this method is not completely quantitative, it has practical relevance to the functionality of the assay. Additionally, to validate that the AuNPs were functionalized with antibodies, we have performed quartz crystal microbalance studies where antibody and DNA-functionalized AuNPs are flowed over an antigen-coated quartz crystal. An increase in mass compared to a DNA-only-functionalized AuNP negative control indicates that the antibody on the AuNP specifically binds the antigens on the quartz crystal (Figure 3).44
Figure 3.

Characterization of antibody-based biobarcode AuNPs by monitoring mass change of an antigen-coated quartz crystal during the addition of antibody and DNA-functionalized AuNPs (black) or DNA-only-functionalized AuNPs (gray). (A) Mass change of quartz crystals while flowing infected cells, PBS wash, BSA block solution, a second PBS wash and biobarcode AuNPs over the quartz crystal. (B) Normalized mass change after the addition of each biobarcode AuNP. Antibody and DNA-functionalized AuNPs but not DNA-only-functionalized AuNPs bind the antigen-coated quartz crystal. Reprinted with permission from Ref. 44.
Outlook
The biobarcode AuNP assay is a unique alternative to the nucleic acid-antibody conjugates used in IPCR assays. It has been shown to reach low attomolar limits of detection,45 which is comparable to conventional IPCR or qPCR assays.53 It has received relatively little attention most likely due to the fact that it has not proven to perform reliably in complex biological matrices. As with all immuno-based detection assays (ELISA, Western blot, etc.) and DNA-based detection assays (PCR, microarray, etc.), nonspecific interactions of antibodies or DNA sequences with nontarget molecules introduces noise into the assay and can result in a relatively high number of false positives.54, 55 Consequently, most biobarcode assays are carried out in simple biological matrices and assay conditions (see Table 1).
Another issue with the biobarcode AuNP assays is the discrepancy between the number of reported barcodes per particle and the actual signal amplification, which is only rarely discussed in the literature.44, 47 Using the fluorescence-based surrogate DNA tag quantification method, Chen et al. determined that their AuNP probes were functionalized with about 80 DNA tags per particle. Using this value and the limit of their DNA detection method, they obtained a theoretical limit of detection in the low attomolar range. However, the actual limit of detection was determined to be about two orders of magnitude higher.47 Our laboratories observed a similar trend when our assay was compared to ELISA.44 The source of this discrepancy may be that the number of detectable DNA tags per AuNP is much less than the calculated value, or that the AuNP probes bind target biomarkers less efficiently than standard ELISA reagents. Proper characterization of all the components of the assay, including the target-binding efficiencies of the capture moiety and functionalized solid phase, may reveal the source of this discrepancy.
In general, a standard for proper characterization of the components is lacking in the field of DNA biobarcode tag AuNPs. The number of DNA tags per AuNP is generally reported, based on the absorbance at 260 nm in the supernatant before and after conjugation, but methods are rarely described for quantifying the number of antibodies attached. Standard techniques for characterizing the AuNP probes and functionalized solid phases need to be established and employed in the field.
Although using biobarcode AuNPs for detection has not been shown to be significantly more reliable or sensitive than IPCR, it offers several advantages. One major advantage of the biobarcode assay is that the use of multiple DNA tags per capture moiety provides an intrinsic amplification of signal prior to the detection of the tags. Additionally, the synthesis of the biobarcode probes is simple and standardized,51 and can easily be adapted into a format for multiplexed detection of more than one target per assay.50 Another potential advantage of the biobarcode assay is that the DNA tag sequence could be tailored to encode functions beyond those of a label for signal amplification. For example, the tags could function as catalytic nucleic acids to perform logic gate operations for a variety of applications.56 One potential application of functional nucleic acid tags could be to reduce background signal noise that results from the nonspecific interactions of the capture moieties with nontarget molecules.
Aptamer and DNAzyme-Functionalized Gold
Two classical functions of nucleic acids, complementary base-pairing and information storage, are the basis for hairpin DNA and biobarcode-based sensors, respectively, as described in the previous two sections. The potential for nucleic acids to bind non-nucleic acid substrates and/or catalyze substrate turnover was discovered by the systematic evolution of ligands by exponential enrichment (SELEX) process and has been exploited to develop a third type of sensor. SELEX is an in vitro, artificial selection process that has been used extensively for generating short nucleic acid sequences with a range of desirable functions from large libraries of random DNA sequences.12, 13, 56, 57 Two major classes of synthetic, functional nucleic acid-based molecules have advanced from the SELEX process: aptamers and aptazymes. Aptamers have resulted from a selection process that exploits the affinity of nucleic acid sequences for a specific target and have been developed to recognize ions, small molecules, proteins and whole cells.58 Aptazymes, or DNAzymes and ribozymes, are selected for their catalytic activities in the presence of specific cofactors. They have also been developed to detect a wide range of targets including nucleotides, metal ions, carbohydrates, small metabolites, a variety of proteins and specific cell types.59 Aptamers and DNAzymes are becoming popular alternatives to their protein counterparts, antibodies and enzymes, respectively, because they can be generated quickly, inexpensively and in large scale against almost any target of interest. Additionally, aptamers and DNAzyme are extremely stable in a variety of conditions and can be renatured after exposure to cycles of extreme temperatures.12 Most aptamers and DNAzymes, however, have been developed for proof-of-principle studies and have limited impact toward clinically relevant applications or processes. Moreover, most DNAzymes fail to function efficiently in complex biological samples.
The detection readout mechanisms of aptamers and DNAzymes are diverse and include self-cleavage, fluorescence, colorimetric dye turnover and electron transfer.58 Gold surfaces can physically enhance the detection of these readout mechanisms. Furthermore, AuNPs provide a means for transportation and concentration of aptamers and DNAzymes in biological sensing applications. Therefore, there has been increasing interest in the use of gold surfaces as a platform for aptamer and DNAzyme assays, and many configurations have been developed that take advantage of the unique properties of gold surfaces.
Design & Applications
AuNPs and gold electrodes have been used as platforms in three main formats for aptamer and DNAzyme assays: controlled AuNP aggregation, DNA capture DNAzyme AuNPs and electrochemistry. To design an effective aptamer or DNAzyme assay, the assay configuration and functional nucleic acid sequence must be considered together. The design constraints for aptamer and DNAzyme probes are specific to each format that is used.
Aptamer- and DNAzyme-dependent controlled AuNP aggregation is one popular format. The extremely high extinction coefficient of AuNPs and the plasmon shift associated with aggregation causes sensitive, colorimetric change that can be visualized by the human eye and can be quantified with a simple absorbance reading. Functional nucleic acids have been used to both induce60, 61 and disrupt AuNP aggregation (Scheme 6).5, 62–64 One method to induce AuNP aggregation is by using aptamer-functionalized AuNPs that target specific molecules and bridge multiple AuNPs upon recognition. Virtually any aptamer or set of aptamers could be used for this type of assay, as long as they recognize at least two moieties on the same target (Scheme 6A), similar to the previously discussed complementary DNA capture biobarcode AuNP assay for detecting specific DNA sequences. Medley et al. developed a sensitive AuNP-based, aptamer-induced aggregation assay that selectively targets T-cell lymphoblastic leukemia or B-cell lymphoma cancer cells using cell-type specific aptamers. Absorbance at 650 nm, indicative of AuNP aggregation, was used to detect and quantify the cancer cells (Figure 4). A limit of detection of 90 or 1000 cancer cells was achieved using absorbance spectroscopy or visual detection, respectively.61 Disruption of AuNP aggregation, on the other hand, has been carried out using DNAzymes that cleave oligonucleotide bridges between AuNPs in the presence of biologically relevant DNAzyme cofactors (Scheme 6B). Wang et al. used a self-cleaving DNAzyme to detect adenosine triphosphate (ATP). The ratio of absorbance at 522 nm (free AuNPs) and 700 nm (aggregated AuNPs) was used to quantify the amount of ATP present in a sample (Figure 5). A limit of detection of 600 nM or 2 μM ATP was achieved using absorbance spectroscopy or visual detection, respectively.63
Scheme 6.

Aptamer and DNAzyme-dependent controlled AuNP aggregation for biomarker detection. (A) Aptamer-functionalized AuNPs bind target biomarker inducing the formation of AuNP aggregates. (B) Networks of DNA-functionalized AuNPs held together by complementary DNAzyme strands are disrupted upon binding DNAzyme cofactor biomarkers. The state of aggregation is detected by absorbance spectrophotometry or dynamic light scattering (DLS).
Figure 4.

Aptamer-induced AuNP aggregation in the presence of target cells detected by increased absorbance at 650 nm. (A) Schematic representation of the assay. (B) Absorbance spectra using various samples and controls. Strong absorbance at 650 nm demonstrates specific recognition of target cells. Reprinted with permission from Ref. 61.
Figure 5.

Self-cleaving DNAzyme-induced disruption of AuNP aggregation in the presence of ATP based detected by increased absorbance at 522 nm. (A) Shift in absorbance spectra with increasing concentration of ATP (right to left: 0, 0.6, 2.2, 4.4, 17.7, 44.2, 88.5, 132.7 M ATP). (B) Absorbance ratio (A522/A700) plotted against increasing concentrations of ATP. Quantifying ATP concentration from the absorbance ratio is possible because of their linear relationship. Reprinted with permission from Ref. 63.
The DNA capture DNAzyme assay format takes advantage of the AuNP as a platform to localize and facilitate the transport of multiple DNAzymes by complementary binding to a nucleic acid target. This is similar to the biobarcode assay, except that the signal does not come from DNA tags but from the catalytic turnover of organic dyes by the DNAzymes. The peroxidase-mimicking DNAzyme and the “8–17” self-cleaving DNAzyme have been conjugated to AuNPs for the development of biosensor applications.56, 65–67 In this assay format, a solid phase functionalized with a capture moiety is used to isolate the target prior to the addition of the DNAzyme-functionalized AuNP (Scheme 7). Both Fu et al. and Niazov et al. use peroxidase-mimicking DNAzymes to detect nucleic acid biomarkers. Fu et al. used a magnetic microparticle solid phase and detected synthetic Chlamydia DNA down to 50 fM.66 Using a gold chip solid phase, Niazov et al. detected synthetic DNA down to 100 pM.67 Another DNAzyme assay has been developed that takes advantage of the quenching properties of AuNPs for target-induced fluorescence emission.67 Upon binding to the Pb(II) ion, self-cleavage of the fluorophore-modified “8–17” DNAzyme allows the fluorophore to be released from the AuNP. The limit of Pb(II) detection using this format was determined to be 5 nM.
Scheme 7.

DNA capture DNAzyme assay for nucleic acid detection. (A) Capture DNA-functionalized magnetic microparticles (MMPs) or (B) gold chips hybridize to the target nucleic acid biomarker to separate it from the analyte solution. DNAzyme and DNA tag-functionalized AuNPs hybridize to the target nucleic acid. The DNAzyme substrate is added and the product is detected by absorbance spectrophotometry or luminescence.
Many aptamers and DNAzymes modified with redox reporters have been functionalized to gold electrodes to make electrochemical biosensors.68 The properties of aptamers and DNAzymes must be carefully considered to develop functional electron transfer agents. One characteristic of aptamers and DNAzymes is that they undergo substantial structural changes upon target binding or catalysis. Functional nucleic acids associated with redox reagents, such as methylene blue or [Ru(NH3)6]3+, have been designed to take advantage of these structural changes. Upon binding a target biomarker, the redox reagents approach the surface of the electrode and transfer electrons (Scheme 8). Using this approach, Xiao et al. used a methylene blue-modified “8–17” DNAzyme to detect Pb(II). While bound to its nucleic acid substrate, the double-stranded “8–17” DNAzyme is rigid and the methylene blue is held away from the electrode surface. Upon binding the Pb(II) cofactor, the substrate strand is cleaved and released and the DNAzyme becomes flexible, allowing the methylene blue redox reagent to approach the electrode surface and transfer electrons. With this format, Pb(II) was detected at 300 nM concentration.69 Another characteristic of aptamers and DNAzymes is that they are highly negatively charged and can be saturated with a relatively high concentration of redox reagents. Upon binding positively charged proteins, the redox reagents are displaced and electron transfer can be detected. Using this technique, Li et al. use an anti-thrombin aptamer conjugated to AuNPs on a gold electrode to detect the positively charged thrombin protein in serum. Upon binding thrombin, the [Ru(NH3)6]3+ redox reporter was displaced and electron transfer was detected at concentrations of thrombin as low as 1 pM.70
Scheme 8.

Electrochemistry with aptamer- and DNAzyme-functionalized electrodes for biomarker detection. (A) Aptamer-functionalized electrodes are saturated with a redox reagent. Upon binding the target biomarker, the redox is displaced from the aptamer inducing electron transfer with the electrode surface. (B) Methylene blue-modified DNAzymes functionalized to electrodes bind the biomarker cofactor and cleave the complementary strand, allowing the methylene blue on the flexible single-stranded DNAzyme to transfer electrons with the electrode surface. Electron transfer is detected using voltammetry.
Synthesis
Synthesis considerations of aptamer- and DNAzyme-functionalized gold surfaces are similar in many respects to other nucleic acid-functionalized AuNP probes. The nucleic acids are conjugated to the surface via the gold-thiol interaction. Loading optimization is important as overcrowding on the surface could inhibit the ability for aptamers to bind a target or DNAzymes to catalyze substrate turnover. As with other methods for functionalizing AuNPs to gold surfaces, a second, nonfunctional ligand could be mixed with the functional nucleic acid to prevent overcrowding. For example, as described above, a 3′ thiol-modified oligothymine can be used to laterally space the aptamers and DNAzymes to prevent structural constraints that would result from overcrowding.22 Additionally, since most aptamers and DNAzymes that are used to functionalize gold surfaces are selected in an environment free of gold, the nucleotide structure may be different on the gold surface. For example, adenine, cytosine and guanine nucleobases have a higher affinity for the gold surface than thymine and are generally excluded in the last ~10 bases of the oligonucleotide.71 If the structure of the aptamer or DNAzyme is dependent on adenine, cytosine and guanine bases near the 5′ end of the DNA molecule, the structure and/or function of the nucleic acid may be compromised.
A substantial amount of research has been devoted to establishing synthesis parameters for proper electron transfer from aptamers and DNAzymes to an electrode surface. White et al. demonstrated that a higher aptamer packing density on the surface of the electrode increases the signal that results from electron transfer but decreases the overall gain of the sensor. The group also found that thicker self-assembled monolayers on the electrode surface increased the gain (Figure 6) but decreased the resulting current through the electrode.68 Moreover, the length of redox reagent-modified ssDNA can be optimized to maximize signal in the target recognition state. Uzawa et al. have recently reported that the rate of electron transfer from the redox reagent on the end of a ssDNA bound to an electrode scales inversely with the length of the DNA molecule and is independent of the packing density of the DNA molecule.72 These data indicates that by optimizing the length of the functional nucleic acid and the thickness of the self-assembled monolayer on the electrode, sensor performance can be greatly increased.
Figure 6.

The effect of three thicknesses of self-assembled monolayers (SAMs) on signal gain. The signal gain from the two-, three- and six-carbon alkane thiol SAMs (C2, C3 and C6) plotted against the concentration of the target (cocaine). Signal gain is highest with the C6 SAM. Reprinted with permission from Ref. 68.
Characterization
The same standards of characterization for nucleic acid-functionalized gold surfaces discussed earlier remain appropriate for aptamer- and DNAzyme-functionalized gold surfaces. Absorbance at 260 nm before and after functionalization should be used to quantify the number of functional nucleic acids per AuNP, or in the case of electrochemical sensors, the probe density on the electrode surface. For aptamer-functionalized AuNPs, an assay equivalent to a sandwich ELISA could be used, using the functionalized AuNP as a capture antibody analog, to quantify the functional nucleic acids attached. The results of the ELISA would indicate the number of functional probes on the gold surface. For DNAzyme-functionalized gold surfaces, the number of functional probes could be calculated by comparing the enzymatic activity of the DNAzymes on the gold surface to a standard dilution series of DNAzymes free in solution. The surface density of functional DNA on electrochemical sensors is most commonly quantified by measuring the maximum peak current at the saturating concentration of redox reagent, and then solving for the DNA surface density based on the resulting charge. This method along with many other commonly used methods for quantifying surface coverage of DNA on electrodes is not described in this feature article as they are covered in great detail elsewhere.73
To validate the practical application of the aptamer- or DNAzyme-functionalized AuNP assays, a direct comparison could be made to their antibody- or enzyme-based analogs. This can only be accomplished if an analogous antibody or enzyme is available. To demonstrate the efficacy of an aptamer designed to recognize human vascular endothelial growth factor, Drolet et al. used a sandwich ELISA to directly compare the aptamer to a monoclonal antibody raised against the same target.74 The group reported that the performance of the aptamer was equivalent to the monoclonal antibody in sensitivity and specificity, and they concluded that it was an effective alternative to the monoclonal antibody for the ELISA format they used. This is a rare example of a comparative characterization to demonstrate the validity of an aptamer-based assay. Similar studies could be performed with the antibody and enzyme counterparts for the aptamer and DNAzyme-based AuNP and electrochemistry assays being developed.
Outlook
The development of biological sensors from SELEX-designed functional nucleic acids holds great promise for the future of the field of molecular detection and diagnostics. A recent report demonstrated that an aptamer selected for its affinity for epidermal growth factor receptor (EGFR) could induce selective, receptor-mediated endocytosis of AuNPs into cell types that express EGFR but not into cells that do not express EGFR.75 The group established a precedent for selective AuNP delivery into endosomes and provided a means for selective drug delivery or in vivo imaging of specific cell types. Additionally, the integration of unnatural bases into random oligonucleotides is being investigated.76 In addition to the four natural DNA nucleobases, incorporation of the unnatural nucleobases would substantially increase the size, diversity and functional properties of the SELEX library and would open the window to a plethora of targets for aptamer and DNAzyme-based sensors.
Aptamer and DNAzyme-based sensors are popular formats for the detection of biologically relevant molecules because of their simplicity, stability and sensitivity. Although the controlled AuNP aggregation format has become popular because of its simple design, it suffers from low sensitivity with limits of detection of small molecules or ions in the high nM to high μM range5, 60, 62–64 or down to 90 cells.61 AuNP assays that rely on DNAzyme dye turnover, conversely, increase the effective concentration of the probes and provide a more sensitive readout. This format, compared to controlled aggregation, has been shown to detect more biologically relevant molecules, such as DNA and telomerase, and has reached limits of detection of DNA targets or ions from low fM to low nM.65–67 However, because nucleic acid-functionalized AuNPs are sensitive to irreversible aggregation in the presence of many nontarget molecules or ions, false positives or false negatives can result if the composition of the assay buffer is not precisely controlled. Consequently, many of the studies using the nucleic acid-functionalized AuNPs for detection have limited use in biological applications where the complexity of the biological milieu can be confounding. The electrochemistry format sidesteps the aggregation problems, except in the cases in which AuNPs are used to amplify signal,77, 78 and also provides a modular platform that are easily functionalized to detect many different targets for a host of applications. Because electrochemical platforms are able to detect binding-induced conformational changes, they can be made reagentless and inexpensively. They can also enable real-time detection in complex biological samples.79 Electrochemistry, however, has not significantly improved the sensitivity of biomarker detection, reaching limits of detection of proteins and ions in the low pM to high nM range.69, 70, 77, 78
As described above, an effective aptamer and DNAzyme surface coverage is critical to the success or failure of the probe as a sensor and should be optimized for each assay.67, 68 Yet compared to other assays involving nucleic acid-functionalized AuNPs, aptamer- and DNAzyme-functionalized AuNPs are generally poorly characterized and the surface coverage of aptamers or DNAzymes per AuNP is not commonly reported. The techniques used to characterize the functional components of the assay should be employed and the results should be reported as standard practice. Only with such rigor can independent comparisons between assay systems be made.
Conclusion
The unique properties of gold allow it to be used as an effective platform for the development of sensors for biomarker detection. The platform concentrates biomarkers at the sensor surface and provides a functional interface for nucleic acid reporter molecule. This design allows the sensors to be used in a variety of conditions and expands the applications for which they can be used. The design flexibility of nucleic acid-functionalized gold probes also allows them to be easily integrated into a variety of device formats. Moreover, the diversity and sensitivity of detection readout permits the probes use in low resource settings where access to specialized laboratory equipment is not available. Because of the ease of preparation, probes can be developed for multiple targets, which would allow multiplexed detection of variety of biologically relevant disease biomarkers.
Progress in the field of nucleic acid-functionalized gold biosensor development is dependent on standardized design, synthesis and characterization practices and effectively reported methods. With a robust standard for probe development in place, the challenges of increasing the sensitivity and specificity of the detection assays in complex biological matrices could be more effectively addressed by the field.
Acknowledgments
D.W.W. and F.R.H. acknowledge the NIH (R21- EB009238) and DTRA (HDTRA-1-10-0067). Additionally, a portion of this effort was funded by a grant from the Bill & Melinda Gates Foundation through the Global Health Grand Challenges in Global Health initiative. N.M.A. acknowledges support from the NSF Graduate Research Fellowship (DGE 0909667). We would like to thank M.F. Richards for critical comments regarding this manuscript.
Contributor Information
Nicholas M. Adams, Email: n.adams@vanderbilt.edu, VU Station B 351822, Department of Chemistry, Vanderbilt University, Nashville, TN 37235, USA. VU Station B 351631, Department of Biomedical Engineering, Vanderbilt University, Nashville, TN 37235, USA
Stephen R. Jackson, Email: s.jackson@vanderbilt.edu, VU Station B 351822, Department of Chemistry, Vanderbilt University, Nashville, TN 37235, USA
Frederick R. Haselton, Email: rick.haselton@vanderbilt.edu, VU Station B 351631, Department of Biomedical Engineering, Vanderbilt University, Nashville, TN 37235, USA
David W. Wright, Email: david.wright@vanderbilt.edu, VU Station B 351822, Department of Chemistry, Vanderbilt University, Nashville, TN 37235, USA.
References
- 1.White KA, Rosi NL. Nanomedicine. 2008;3:543–553. doi: 10.2217/17435889.3.4.543. [DOI] [PubMed] [Google Scholar]
- 2.Wilson R. Chemical Society Reviews. 2008;37:2028–2045. doi: 10.1039/b712179m. [DOI] [PubMed] [Google Scholar]
- 3.Boisselier E, Astruc D. Chemical Society Reviews. 2009;38:1759–1782. doi: 10.1039/b806051g. [DOI] [PubMed] [Google Scholar]
- 4.Moyano DF, Rotello VM. Langmuir. 2011;27:10376–10385. doi: 10.1021/la2004535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu J, Lu Y. Journal of the American Chemical Society. 2003;125:6642–6643. doi: 10.1021/ja034775u. [DOI] [PubMed] [Google Scholar]
- 6.Lee JS, Han MS, Mirkin CA. Angewandte Chemie. 2007;119:4171–4174. [Google Scholar]
- 7.Dasary SSR, Singh AK, Senapati D, Yu H, Ray PC. Journal of the American Chemical Society. 2009;131:13806–13812. doi: 10.1021/ja905134d. [DOI] [PubMed] [Google Scholar]
- 8.Hao RZ, Song HB, Zuo GM, Yang RF, Wei HP, Wang DB, Cui ZQ, Zhang Z, Cheng ZX, Zhang XE. Biosensors and Bioelectronics. 2011;26:3398–3404. doi: 10.1016/j.bios.2011.01.010. [DOI] [PubMed] [Google Scholar]
- 9.Sperling RA, Rivera Gil P, Zhang F, Zanella M, Parak WJ. Chemical Society Reviews. 2008;37:1896–1908. doi: 10.1039/b712170a. [DOI] [PubMed] [Google Scholar]
- 10.El-Sayed MA. Acc Chem Res. 2001;34:257–264. doi: 10.1021/ar960016n. [DOI] [PubMed] [Google Scholar]
- 11.Myroshnychenko V, Rodriguez-Fernandez J, Pastoriza-Santos I, Funston AM, Novo C, Mulvaney P, Liz-Marzan LM, Garcia dAFJ. Chem Soc Rev. 2008;37:1792–1805. doi: 10.1039/b711486a. [DOI] [PubMed] [Google Scholar]
- 12.Liu J, Cao Z, Lu Y. Chemical Reviews. 2009;109:1948–1998. doi: 10.1021/cr030183i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wilson DS, Szostak JW. Annu Rev Biochem. 1999;68:611–647. doi: 10.1146/annurev.biochem.68.1.611. [DOI] [PubMed] [Google Scholar]
- 14.Alivisatos AP, Johnsson KP, Peng X, Wilson TE, Loweth CJ, Bruchez MP, Jr, Schultz PG. Nature (London) 1996;382:609–611. doi: 10.1038/382609a0. [DOI] [PubMed] [Google Scholar]
- 15.Mirkin CA, Letsinger RL, Mucic RC, Storhoff JJ. Nature (London) 1996;382:607–609. doi: 10.1038/382607a0. [DOI] [PubMed] [Google Scholar]
- 16.Wang K, Tang Z, Yang CJ, Kim Y, Fang X, Li W, Wu Y, Medley CD, Cao Z, Li J, Colon P, Lin H, Tan W. Angew Chem, Int Ed. 2009;48:856–870. doi: 10.1002/anie.200800370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marras SAE, Kramer FR, Tyagi S. Nucleic Acids Research. 2002;30:e122. doi: 10.1093/nar/gnf121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fan C, Wang S, Hong JW, Bazan GC, Plaxco KW, Heeger AJ. Proc Natl Acad Sci U S A. 2003;100:6297–6301. doi: 10.1073/pnas.1132025100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dubertret B, Calame M, Libchaber AJ. Nat Biotechnol. 2001;19:365–370. doi: 10.1038/86762. [DOI] [PubMed] [Google Scholar]
- 20.Maxwell DJ, Taylor JR, Nie S. J Am Chem Soc. 2002;124:9606–9612. doi: 10.1021/ja025814p. [DOI] [PubMed] [Google Scholar]
- 21.Elghanian R, Storhoff JJ, Mucic RC, Letsinger RL, Mirkin CA. Science (Washington, D C) 1997;277:1078–1080. doi: 10.1126/science.277.5329.1078. [DOI] [PubMed] [Google Scholar]
- 22.Song S, Liang Z, Zhang J, Wang L, Li G, Fan C. Angew Chem, Int Ed. 2009;48:8670–8674. S8670/1–S8670/6. doi: 10.1002/anie.200901887. [DOI] [PubMed] [Google Scholar]
- 23.Jayagopal A, Halfpenny KC, Perez JW, Wright DW. J Am Chem Soc. 2010;132:9789–9796. doi: 10.1021/ja102585v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harry SR, Hicks DJ, Amiri KI, Wright DW. Chem Commun (Cambridge, U K) 2010;46:5557–5559. doi: 10.1039/c001969k. [DOI] [PubMed] [Google Scholar]
- 25.Strohsahl CM, Miller BL, Krauss TD. Nat Protoc. 2007;2:2105–2110. doi: 10.1038/nprot.2007.301. [DOI] [PubMed] [Google Scholar]
- 26.Du H, Strohsahl CM, Camera J, Miller BL, Krauss TD. J Am Chem Soc. 2005;127:7932–7940. doi: 10.1021/ja042482a. [DOI] [PubMed] [Google Scholar]
- 27.Cederquist KB, Keating CD. Langmuir. 2010;26:18273–18280. doi: 10.1021/la1031703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Perez JW, Haselton FR, Wright DW. Analyst (Cambridge, U K) 2009;134:1548–1553. doi: 10.1039/b904191e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lai RY, Lagally ET, Lee SH, Soh HT, Plaxco KW, Heeger AJ. Proc Natl Acad Sci U S A. 2006;103:4017–4021. doi: 10.1073/pnas.0511325103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fan C, Plaxco KW, Heeger AJ. Proc Natl Acad Sci U S A. 2003;100:9134–9137. doi: 10.1073/pnas.1633515100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Turkevich J, Stevenson PC, Hillier J. Discuss Faraday Soc. 1951;(11):55–75. [Google Scholar]
- 32.Frens G. Kolloid-Z Z Polym. 1972;250:736–41. [Google Scholar]
- 33.Frens G. Nature (London), Phys Sci. 1973;241:20–2. [Google Scholar]
- 34.Hill HD, Millstone JE, Banholzer MJ, Mirkin CA. ACS Nano. 2009;3:418–424. doi: 10.1021/nn800726e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Park SJ, Taton TA, Mirkin CA. Science. 2002;295:1503–6. doi: 10.1126/science.1067003. [DOI] [PubMed] [Google Scholar]
- 36.Demers LM, Mirkin CA, Mucic RC, Reynolds RA, Letsinger RL, Elghanian R, Viswanadham G. Analytical Chemistry. 2000;72:5535–5541. doi: 10.1021/ac0006627. [DOI] [PubMed] [Google Scholar]
- 37.Liu X, Tan W. Anal Chem. 1999;71:5054–5059. doi: 10.1021/ac990561c. [DOI] [PubMed] [Google Scholar]
- 38.Yao G, Tan W. Anal Biochem. 2004;331:216–223. doi: 10.1016/j.ab.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 39.Tyagi S, Kramer FR. Nat Biotechnol. 1996;14:303–8. doi: 10.1038/nbt0396-303. [DOI] [PubMed] [Google Scholar]
- 40.Bonnet G, Tyagi S, Libchaber A, Kramer FR. Proc Natl Acad Sci U S A. 1999;96:6171–6176. doi: 10.1073/pnas.96.11.6171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang H, Zhao Q, Li XF, Le XC. Analyst. 2007;132:724–737. doi: 10.1039/b704256f. [DOI] [PubMed] [Google Scholar]
- 42.Sano T, Smith C, Cantor C. Science. 1992;258:120–122. doi: 10.1126/science.1439758. [DOI] [PubMed] [Google Scholar]
- 43.Adler M, Wacker R, Niemeyer CM. Analyst. 2008;133:702–718. doi: 10.1039/b718587c. [DOI] [PubMed] [Google Scholar]
- 44.Perez JW, Vargis EA, Russ PK, Haselton FR, Wright DW. Analytical Biochemistry. 2011;410:141–148. doi: 10.1016/j.ab.2010.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nam JM, Thaxton CS, Mirkin CA. Science. 2003;301:1884–1886. doi: 10.1126/science.1088755. [DOI] [PubMed] [Google Scholar]
- 46.Nam JM, Stoeva SI, Mirkin CA. Journal of the American Chemical Society. 2004;126:5932–5933. doi: 10.1021/ja049384+. [DOI] [PubMed] [Google Scholar]
- 47.Chen L, Wei H, Guo Y, Cui Z, Zhang Z, Zhang XE. Journal of Immunological Methods. 2009;346:64–70. doi: 10.1016/j.jim.2009.05.007. [DOI] [PubMed] [Google Scholar]
- 48.Yin H-q, Jia M-x, Shi L-j, Yang S, Zhang L-y, Zhang Q-m, Wang S-q, Li G, Zhang J-g. Journal of Virological Methods. In Press, Uncorrected Proof. [Google Scholar]
- 49.Goluch ED, Nam JM, Georganopoulou DG, Chiesl TN, Shaikh KA, Ryu KS, Barron AE, Mirkin CA, Liu C. Lab on a Chip. 2006;6:1293–1299. doi: 10.1039/b606294f. [DOI] [PubMed] [Google Scholar]
- 50.Stoeva SI, Lee JS, Smith JE, Rosen ST, Mirkin CA. Journal of the American Chemical Society. 2006;128:8378–8379. doi: 10.1021/ja0613106. [DOI] [PubMed] [Google Scholar]
- 51.Hill HD, Mirkin CA. Nat Protocols. 2006;1:324–336. doi: 10.1038/nprot.2006.51. [DOI] [PubMed] [Google Scholar]
- 52.Taton TA, Mirkin CA, Letsinger RL. Science. 2000;289:1757–1760. doi: 10.1126/science.289.5485.1757. [DOI] [PubMed] [Google Scholar]
- 53.Malou N, Raoult D. Trends in Microbiology. 2011;19:295–302. doi: 10.1016/j.tim.2011.03.004. [DOI] [PubMed] [Google Scholar]
- 54.Kricka LJ. Clin Chem. 2000;46:1037–1038. [PubMed] [Google Scholar]
- 55.Draghici S, Khatri P, Eklund AC, Szallasi Z. Trends in Genetics. 2006;22:101–109. doi: 10.1016/j.tig.2005.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Willner I, Shlyahovsky B, Zayats M, Willner B. Chemical Society Reviews. 2008;37:1153–1165. doi: 10.1039/b718428j. [DOI] [PubMed] [Google Scholar]
- 57.Famulok M, Hartig JS, Mayer G. Chem Rev (Washington, DC, U S) 2007;107:3715–3743. doi: 10.1021/cr0306743. [DOI] [PubMed] [Google Scholar]
- 58.Mairal T, Cengiz Özalp V, Lozano Sánchez P, Mir M, Katakis I, O’Sullivan C. Analytical and Bioanalytical Chemistry. 2008;390:989–1007. doi: 10.1007/s00216-007-1346-4. [DOI] [PubMed] [Google Scholar]
- 59.Muller S, Strohbach D, Wolf J. IEE Proceedings - Nanobiotechnology. 2006;153:31–40. doi: 10.1049/ip-nbt:20050047. [DOI] [PubMed] [Google Scholar]
- 60.Liu J, Lu Y. Chemical Communications. 2007:4872–4874. doi: 10.1039/b712421j. [DOI] [PubMed] [Google Scholar]
- 61.Medley CD, Smith JE, Tang Z, Wu Y, Bamrungsap S, Tan W. Analytical Chemistry. 2008;80:1067–1072. doi: 10.1021/ac702037y. [DOI] [PubMed] [Google Scholar]
- 62.Liu J, Lu Y. Analytical Chemistry. 2004;76:1627–1632. doi: 10.1021/ac0351769. [DOI] [PubMed] [Google Scholar]
- 63.Wang J, Wang L, Liu X, Liang Z, Song S, Li W, Li G, Fan C. Advanced Materials. 2007;19:3943–3946. [Google Scholar]
- 64.Miao X, Ling L, Shuai X. Chemical Communications. 2011;47:4192–4194. doi: 10.1039/c0cc05344a. [DOI] [PubMed] [Google Scholar]
- 65.Niazov T, Pavlov V, Xiao Y, Gill R, Willner I. Nano Letters. 2004;4:1683–1687. [Google Scholar]
- 66.Fu R, Li T, Park HG. Chemical Communications. 2009:5838–5840. doi: 10.1039/b907762f. [DOI] [PubMed] [Google Scholar]
- 67.Kim JH, Han SH, Chung BH. Biosensors and Bioelectronics. 2011;26:2125–2129. doi: 10.1016/j.bios.2010.09.018. [DOI] [PubMed] [Google Scholar]
- 68.White RJ, Phares N, Lubin AA, Xiao Y, Plaxco KW. Langmuir. 2008;24:10513–10518. doi: 10.1021/la800801v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Xiao Y, Rowe AA, Plaxco KW. Journal of the American Chemical Society. 2006;129:262–263. doi: 10.1021/ja067278x. [DOI] [PubMed] [Google Scholar]
- 70.Li L, Zhao H, Chen Z, Mu X, Guo L. Analytical and Bioanalytical Chemistry. 2010;398:563–570. doi: 10.1007/s00216-010-3922-2. [DOI] [PubMed] [Google Scholar]
- 71.Storhoff JJ, Elghanian R, Mirkin CA, Letsinger RL. Langmuir. 2002;18:6666–6670. [Google Scholar]
- 72.Uzawa T, Cheng RR, White RJ, Makarov DE, Plaxco KW. Journal of the American Chemical Society. 2010;132:16120–16126. doi: 10.1021/ja106345d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gorodetsky AA, Buzzeo MC, Barton JK. Bioconjugate Chemistry. 2008;19:2285–2296. doi: 10.1021/bc8003149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Drolet DW, Moon-McDermott L, Romig TS. Nat Biotech. 1996;14:1021–1025. doi: 10.1038/nbt0896-1021. [DOI] [PubMed] [Google Scholar]
- 75.Li N, Larson T, Nguyen HH, Sokolov KV, Ellington AD. Chemical Communications. 2010;46:392–394. doi: 10.1039/b920865h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Keefe AD, Cload ST. Current Opinion in Chemical Biology. 2008;12:448–456. doi: 10.1016/j.cbpa.2008.06.028. [DOI] [PubMed] [Google Scholar]
- 77.He P, Shen L, Cao Y, Li D. Analytical Chemistry. 2007;79:8024–8029. doi: 10.1021/ac070772e. [DOI] [PubMed] [Google Scholar]
- 78.Shen L, Chen Z, Li Y, He S, Xie S, Xu X, Liang Z, Meng X, Li Q, Zhu Z, Li M, Le XC, Shao Y. Analytical Chemistry. 2008;80:6323–6328. doi: 10.1021/ac800601y. [DOI] [PubMed] [Google Scholar]
- 79.Vallee-Belisle A, Plaxco KW. Curr Opin Struct Biol. 2010;20:518–526. doi: 10.1016/j.sbi.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]