

Abstract
Context
The value of assessing various emerging lipid-related markers for prediction of first cardiovascular events is debated.
Objective
To determine whether adding information on apolipoprotein B and apolipoprotein A-I, lipoprotein(a), or lipoprotein-associated phospholipase A2 to total cholesterol and high-density lipoprotein cholesterol (HDL-C) improves cardiovascular disease (CVD) risk prediction.
Design, Setting, and Participants
Individual records were available for 165 544 participants without baseline CVD in 37 prospective cohorts (calendar years of recruitment: 1968–2007) with up to 15 126 incident fatal or nonfatal CVD outcomes (10 132 CHD and 4994 stroke outcomes) during a median follow-up of 10.4 years (interquartile range, 7.6–14 years).
Main Outcome Measures
Discrimination of CVD outcomes and reclassification of participants across predicted 10-year risk categories of low (<10%), intermediate (10%–<20%), and high (≥20%) risk.
Results
The addition of information on various lipid-related markers to total cholesterol, HDL-C, and other conventional risk factors yielded improvement in the model’s discrimination: C-index change, 0.0006 (95% CI, 0.0002–0.0009) for the combination of apolipoprotein B and A-I; 0.0016 (95% CI, 0.0009–0.0023) for lipoprotein(a); and 0.0018 (95% CI, 0.0010–0.0026) for lipoprotein-associated phospholipase A2 mass. Net reclassification improvements were less than 1% with the addition of each of these markers to risk scores containing conventional risk factors. We estimated that for 100 000 adults aged 40 years or older, 15 436 would be initially classified at intermediate risk using conventional risk factors alone. Additional testing with a combination of apolipoprotein B and A-I would reclassify 1.1%; lipoprotein(a), 4.1%; and lipoprotein-associated phospholipase A2 mass, 2.7% of people to a 20% or higher predicted CVD risk category and, therefore, in need of statin treatment under Adult Treatment Panel III guidelines.
Conclusion
In a study of individuals without known CVD, the addition of information on the combination of apolipoprotein B and A-I, lipoprotein(a), or lipoprotein-associated phospholipase A2 mass to risk scores containing total cholesterol and HDL-C led to slight improvement in CVD prediction.
Routinely used risk prediction scores for cardiovascular disease (CVD) contain information on total cholesterol and high-density lipoprotein cholesterol (HDL-C) and several other conventional risk factors.1,2 There is considerable interest in whether CVD prediction can be improved by assessment of various additional lipid-related markers either to replace, or supplement, traditional cholesterol measurements in these scores.3
Proposals to replace information on total cholesterol and HDL-C with single parameters, such as the total cholesterol:HDL-C ratio or non–HDL-C (ie, total cholesterol - HDL-C),4,5 have been motivated by a desire for greater simplicity and a belief that these parameters better reflect the underlying atherosclerotic process. For example, non–HDL-C reflects the cholesterol content of several proatherogenic lipoprotein subfractions (very low-density lipoprotein, intermediate-density lipoprotein, and chylomicron remnants) in addition to low-density lipoprotein cholesterol.
Similar considerations apply to proposals to replace information on total cholesterol and HDL-C with apolipoprotein B and apolipoprotein A-I.6–9 Because apolipoprotein B and A-I are the principal surface proteins found on proatherogenic lipoproteins and HDL, respectively, they might be more strongly related to CVD risk than is the cholesterol contained in these lipoproteins. However, perhaps partly due to inconclusive epidemiological evidence, there are conflicting guidelines about the relevance of apolipoprotein B and A-I to CVD prediction.1,6–11
There is also debate about the value of supplementing conventional risk factors with targeted assessment of lipoprotein(a). In 2010, the European Atherosclerosis Society Consensus Panel recommended lipoprotein(a) measurement to augment risk assessment in people at intermediate (10%–<20%) or high (≥20%) predicted 10-year CVD risk.12 However, the 2010 American College of Cardiology Foundation/ American Heart Association Task Force on Practice Guidelines did not support this recommendation.6–8 Similar uncertainties apply to the incremental predictive value of assessing circulating concentrations of lipoprotein–associated phospholipase A2.8
Complementing previous reports from this collaboration,13–15 the current analysis has 2 objectives. First, to determine whether replacing information on total cholesterol and HDL-C with various lipid parameters improves prediction of first-onset CVD outcomes. Second, to determine whether additional information on apolipoprotein B and A-I, lipoprotein(a), or lipoprotein-associated phospholipase A2 to prognostic models containing information on total cholesterol, HDL-C, and other conventional risk factors improves CVD risk prediction.
METHODS
Study Design
Details of this collaboration have been published.16 Eligible prospective studies had information for each participant on total cholesterol, HDL-C, age, sex, smoking status, diabetes, and blood pressure; assayed triglyceride, apolipoprotein B and A-I, lipoprotein(a), or lipoprotein-associated phospholipase A2 mass or activity; had not selected participants on the basis of having had previous CVD (defined in each study at the initial examination); recorded cause-specific mortality, vascular morbidity (nonfatal myocardial infarction or stroke), or both during follow-up using well-defined criteria; and recorded more than 1 year of follow-up. Because information on directly measured LDL-C, adiposity measures, family history of CVD, and socioeconomic factors was available only in subsets of the participants, these variables were not included in the main analysis. eTables 1–4 and eAppendix 1 provide study details, including assay methods, acronyms, and references (available at http://www.jama.com). Data from the Apolipoprotein Related Mortality Risk Study (AMORIS) could not be incorporated into these current analyses because it did not measure baseline levels of HDL-C, blood pressure, smoking status, body mass index, or diabetes (eTable 5).17 In registering fatal outcomes, all contributing studies in this analysis used International Classification of Disease coding to at least 3 digits and ascertainment was based on death certificates, with 29 studies also involving review of medical records, autopsy findings, and other supplementary sources. Studies used definitions of myocardial infarction based on World Health Organization or similar criteria and of stroke based on clinical and brain imaging features. The study was approved by the Cambridgeshire ethics review committee.
Statistical Analysis
Because recent risk scores have tended to combine coronary heart disease (CHD) and stroke outcomes due to the existence of shared risk factors and treatments,18 the primary outcome used herein was first-onset CVD, defined as fatal or nonfatal CHD event or any stroke. We compared prognostic models that replaced information on total cholesterol and HDL-C with various nontraditional lipid parameters that have been previously proposed, including the total cholesterol: HDL-C ratio (which is mathematically equivalent to the non–HDL-C:HDL-C ratio); the HDL-C: total cholesterol ratio; non–HDL-C; apolipoprotein B and A-I; apolipoprotein B: A-I ratio; apolipoprotein A-I:B ratio; total cholesterol and apolipoprotein A-I; apolipoprotein B and HDL-C, and loge transformations of ratios.
We also evaluated supplementing risk scores containing total cholesterol and HDL-C with triglyceride, apolipoprotein B, apolipoprotein A-I, lipoprotein(a), and lipoprotein-associated phospholipase A2 mass or activity. Lipoprotein(a) was modeled nonlinearly by including linear and quadratic terms of log-transformed lipoprotein(a). Because of differences in the mean and standard deviation of concentrations of lipoprotein-associated phospholipase A2 recorded across studies using different assay methods (eTables 3 and 4), values were standardized within each study. Cox proportional hazards modeling allowed for separate baseline hazards by study (and, when appropriate, by trial group) and sex but estimated common coefficients (loge hazard ratios) across studies. We censored deaths from non-CVD causes. Prognostic models were compared using measures of risk discrimination and reclassification.19–21 We extended our previous methods19 to a 2-stage approach allowing examination of between-study heterogeneity, calculating the C index and the D measure, and their changes, within each study separately before pooling results. Studies were weighted by numbers of CVD outcomes (eAppendix 2). Between-study heterogeneity in the risk discrimination measures and their changes was quantified by the I2 statistic.22 The proportional hazards assumption was satisfied. For participants in studies with at least 10 years of follow-up, we constructed reclassification tables using data from studies that had recorded both fatal and nonfatal CVD outcomes to examine movement of participants between 3 predicted 10-year CVD risk categories (<10%, 10%–<20%, and ≥20%) upon addition of lipid-related markers to conventional risk factors8 and summarized these using the net reclassification improvement.20
Our clinical modeling involved 3 key assumptions. First, we assumed the use of sequential screening, ie, initial screening with conventional risk factors alone followed by additional measurement of further lipid-related markers in people at 10% to less than 20% predicted 10-year CVD risk. Second, we assumed statin allocation would reduce CVD risk by 20% in people without a history of CVD (including in people at <20% predicted 10-year risk). This estimate was derived from relative risk reductions observed with statins in a meta-analysis of randomized trials (eAppendix 2).8,23 Third, we assumed a policy of statin allocation per Adult Treatment Panel III guidelines,24 that is, people at 20% or more of predicted CVD risk plus others, such as people with diabetes irrespective of their predicted 10-year risk. Analyses were performed using Stata statistical software version 11.0 (StataCorp), 2-sided P values, and 95% CIs.
RESULTS
Individual records were available for 165 544 participants without baseline CVD in 37 prospective cohorts (calendar years of recruitment, 1968–2007) with up to 15 126 incident fatal and nonfatal CVD outcomes (10132 CHD and 4994 stroke events) recorded during median follow-up of 10.4 years (interquartile range [IQR], 7.6–14 years). The Table describes the baseline characteristics of participants and presents adjusted hazard ratios for CVD with baseline levels of risk factors (supplemented by eTables 1–3, available at http://www.jama.com).
Replacement of Cholesterol With Other Lipid-Related Markers
Replacing total cholesterol and HDL-C with information on various lipid-related markers did not improve risk discrimination or reclassification (Figure 1 and eTable 6). For example, replacement of information on total cholesterol and HDL-C with apolipoprotein B and A-I significantly worsened risk discrimination (C-index change: −0.0028; P <.001) and risk classification (net reclassification improvement: −1.08%; P =.01). No improvement in risk discrimination was observed in subgroups defined by baseline age, sex, elevated triglyceride, history of diabetes, and other conventional risk factors (eg, lipids, blood pressure, smoking status, metabolic syndrome), use of lipid- or blood pressure–lowering medications at entry, fasting status, type of assay, predicted 10-year CVD risk, and study design (eFigure 1). In separate analyses of CHD and stroke as individual outcomes, replacement of information on total cholesterol and HDL-C with various lipid-related markers did not improve risk discrimination (eFigure 2).
Figure 1. Changes in Cardiovascular Disease Risk Discrimination and Reclassification When Replacing Cholesterol Markers With Lipid-Related Markers.
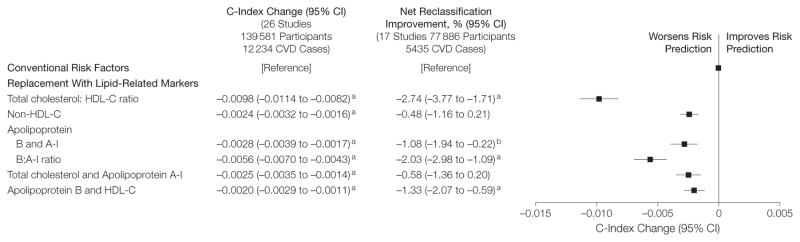
The model analyzed patients with conventional risk factors of age, systolic blood pressure, smoking status, history of diabetes, and total and high-density lipoprotein cholesterol (HDL-C), each of which were included as individual linear terms. The models were stratified by sex. Overall, the C-index for a model containing conventional cardiovascular disease (CVD) risk factors was 0.7244 (95% CI, 0.7200–0.7289). The net reclassification improvement analysis was calculated only for participants in studies that had at least 10 years of follow-up.
aP<.001 for comparison against the model containing conventional risk factors.
bP<.05 for comparison against the model containing conventional risk factors.
Addition of Lipid-Related Markers
Prognostic models for CVD that added lipid-related markers to models containing total cholesterol and HDL-C and other conventional risk factors changed the C index by the amounts shown in Figure 2 and eTable 7, available at http://www.jama.com. However, none of these lipid-related markers significantly improved CVD risk classification. Again, broadly similar results to those observed overall for the lipid-related markers were found in clinically relevant subgroups, including participants who reported using lipid-lowering medications at entry. Although there was tentative evidence of effect-modification in some groups (eFigures 3–6), cautious interpretation is required given the multiplicity of comparisons made. First, apolipoprotein A-I and B, as well as lipoprotein(a), could improve CVD prediction more in individuals with higher total cholesterol or in people initially classified at 10% to less than 20% predicted 10-year risk (P <.001 and P =.02, respectively; eFigures 3 and 4). Second, the addition of apolipoprotein B and A-I could preferentially improve CVD risk discrimination in men (P =.01), participants using blood pressure–lowering medications at entry (P =.005), and individuals with lower HDL-C (P =.022; eFigure 3). Third, the addition of apolipoprotein B and A-I significantly improved risk discrimination for CHD (C-index increase of 0.0010; P <.001) but not for stroke (C-index increase of −0.0002; P =.30). By contrast, addition of lipoprotein(a) or lipoprotein-associated phospholipase A2 mass provided improvements for CHD that were similar to those for stroke (eFigure 7).
Figure 2. Changes in Cardiovascular Disease Risk Discrimination and Classification After Adding Lipid-Related Markers.

The model containing conventional risk factors include age, systolic blood pressure, smoking status, history of diabetes, total and high-density lipoprotein cholesterol (HDL-C), each included as individual linear terms. Models were stratified by sex.
aNet reclassification improvement was calculated only for participants in studies with at least 10 years of follow-up. Change in C-index adding lipoprotein(a) greater than 30 mg/dL was 0.0001 (95% CI, −0.0001 to 0.0003).
bTriglyceride values were log-transformed.
cP<.05 for comparison against model containing conventional risk factors.
dP<.001 for comparison against model containing conventional risk factors.
eLipoprotein(a) was modeled nonlinearly by including linear and quadratic terms of log-transformed lipoprotein(a).
Similar results to those described above were observed in analyses that used the D measure (eFigures 8 and 9), or that were restricted to studies with at least 10 years of follow-up (eFigure 10). Levels of lipid-related markers contributed relatively little to heterogeneity in the study-specific C index, which was mostly due to differing age ranges across cohorts (eFigures 11–15). We could not reliably evaluate the effect of joint assessment of apolipoprotein B and A-I, lipoprotein(a), and lipoprotein-associated phospholipase A2 because only about 10% of the participants in this analysis had concomitant information on all these parameters.
Clinical Modeling
We modeled a population of 100 000 adults aged 40 years or older with similar age structure as the European standard population and an age- and sex-specific incidence of CVD as in the current study; 15 436 people would be initially classified at 10% to less than 20% 10-year predicted CVD risk using conventional risk factors alone, of whom 13 622 would remain after excluding those recommended for statin treatment by Adult Treatment Panel III guidelines (such as people with diabetes irrespective of their predicted 10-year risk24 (Figure 3 and eAppendix 2). For these 13 622 people, assessment of lipoprotein(a) would reclassify 555 people (4.1%) to 20% or greater predicted risk, 86 of whom would be expected to have a CVD event within 10 years; assessment of lipoprotein-associated phospholipase A2 mass would reclassify 365 people (2.7%), 72 of whom would be expected to have a CVD event within 10 years; and assessment of the combination of apolipoprotein B or A-I would reclassify 154 people (1.1%), 16 of whom would be expected to have a CVD event within 10 years (eFigure 16). Assuming statin allocation per the Adult Treatment Panel III guidelines,24 such targeted assessment could help prevent about 17 (ie, 0.20 × 86) extra CVD outcomes over 10 years for those additionally tested for lipoprotein(a), 14 (0.20× 72) extra CVD outcomes over 10 years for those tested for lipoprotein-associated phospholipase A2 mass, or 3 (0.20 × 16) extra CVD outcomes over 10 years for those tested for a combination of apolipoprotein B or A-I. In other words, such targeted assessment of individuals at intermediate CVD risk could help prevent 1 extra CVD outcome over 10 years for every 801 assessed for lipoprotein(a) (ie, 13 622/17), 973 assessed for lipoprotein-associated phospholipase A2 mass (13 622/14), and 4541 assessed for the combination of apolipoprotein B and A-I (13 622/3). Under these circumstances, statins would be newly allocated to about 33 of 801 people (4.1%) assessed for lipoprotein(a), 26 of 973 people (2.7%) assessed for lipoprotein-associated phospholipase A2 mass, or 50 of 4541 (1.1%) assessed for the combination of apolipoprotein B and A-I. Alternatively, assuming use of the more selective statin allocation policies in Canada9 or the United Kingdom, then the numbers needed to screen listed above should each be multiplied by 0.6.
Figure 3. Modeling of Reclassification per 100 000 People Initially Screened With Conventional Risk Factors and Then Additional Targeted Assessment of Lipid-Related Markers.
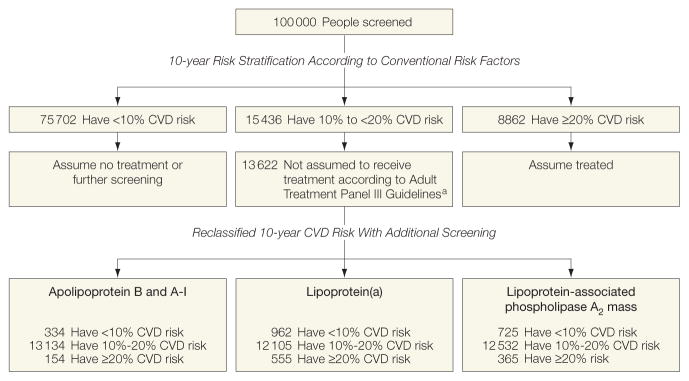
Conventional risk factors were age, smoking status, systolic blood pressure, history of diabetes, total and high-density lipoprotein cholesterol (stratified by sex).
aFollowing Adult Treatment Panel III (ATP-III) guidelines, this model assumes that people who should receive statins are those at a 20% or higher predicted 10-year cardiovascular disease (CVD) risk and other people (eg, those with diabetes) who merit statins irrespective of predicted 10-year CVD risk. People reporting statin use at baseline were also assumed to merit statin allocation.
COMMENT
In contrast with some existing guidelines,1,6,7,9 the current analysis has shown that replacement of information on total cholesterol and HDL-C with various lipid parameters does not improve CVD prediction. For example, none of the following measures were superior to total cholesterol and HDL-C when they replaced traditional cholesterol measurements in risk prediction scores: the total cholesterol:HDL-C ratio; non–HDL-C; the linear combination of apolipoprotein B and A-I; or the apolipoprotein B:A-I ratio. Furthermore, replacement of total cholesterol and HDL-C with apolipoprotein B and A-I actually significantly worsened risk discrimination. These findings applied to clinically relevant subpopulations, including people with diabetes and people with elevated triglyceride levels.
With regards to the value of adding information on various emerging lipid-related markers to risk scores already containing total cholesterol, HDL-C, and other conventional risk factors, we observed slight potential for improvement in CVD prediction. This conclusion was suggested by the following analyses. First, we showed that each of the lipid-related markers studied herein slightly increased CVD prediction when using measures (eg, the C index and D measure) that are independent of clinical risk categories. Second, we found that none of these markers significantly improved reclassification of participants across the clinical risk cutoff levels that are currently used to inform treatment decisions. Third, we modeled a scenario assuming targeted lipid-related marker assessment in people judged as being at intermediate risk (10%– <20% 10-year predicted CVD risk) after initial screening by conventional risk factors alone. If such targeted measurement were to be coupled with allocation of statins per US Adult Treatment Panel III guidelines,24 then our data suggest that it could help prevent 1 extra CVD outcome over 10 years for approximately every 4500 people additionally screened with a combination of apolipoprotein B and A-I, or about 800 people screened with lipoprotein(a), or about 1000 people screened with lipoprotein-associated phospholipase A2 mass.
The generalizability of our findings has been enhanced by inclusion of data from 165 000 participants in 15 countries and by the general lack of heterogeneity in the results. To enhance validity, we have restricted analysis to prospective studies with extended follow-up. For example, although some large retrospective case-control studies have reported stronger associations of apolipoprotein B and A-I with CHD than those observed herein, it remains uncertain to what extent this difference might be explained by factors such as changes in lipid levels observed in the hours after the onset of infarction in case-control studies of acute myocardial infarction.25,26 In contrast with literature-based reviews,27 our access to individual participant data has enabled time-to-event analysis, analysis of clinically relevant subgroups, and consistent comparison across studies. To estimate incremental improvement in CVD prediction, we have studied only people with complete information on conventional risk factors. Our findings are consistent with a separate and complementary analyses of the evidence from randomized trials of patients treated with statins.2,28
This study has potential limitations. Our analysis does not, of course, address etiological and therapeutic questions being explored in randomized trials. Reclassification analyses are intrinsically sensitive to choice of follow-up interval and clinical risk categories. Somewhat greater clinical impact than suggested by our analysis would be estimated if we had used less conservative modeling assumptions (eg, use of more effective statin regimens23 and longer time horizons) or alternative disease outcomes (such as an exclusive focus on CHD rather than on CHD plus stroke). Conversely, our clinical models could have overestimated potential benefits of assessing lipid-related markers because not all people eligible for statins will receive them or be willing, adherent, or able to take them.31 Although we did not find that our results varied importantly by assay methods used, further study of this issue is needed, perhaps particularly for lipid-related markers for which measurements have only recently been standardised.12,32 Furthermore, large studies are needed to assess whether concurrent assesment of lipoprotein(a) concentration and apolipoprotein(a) isoform size confers greater improvement in CVD prediction than lipoprotein(a) alone (such assessment was not possible in the current study because it lacked concomitant data on such isoforms). This study had a limited ability to study lipid-related markers in combination with one another and to investigate populations not of European descent.
In summary, in a study of individuals without known cardiovascular disease, replacing information on total cholesterol and HDL-C with apolipoprotein B and apolipoprotein A-I worsened CVD prediction. Furthermore, addition of the combination apolipoprotein B and A-I, lipoprotein(a), or lipoprotein-associated phospholipase A2 to risk scores containing total cholesterol and HDL-C provided slight improvement in CVD prediction. The clinical benefits of using any of these biomarkers remains to be established.
Supplementary Material
Table.
Summary of Available Data and Hazard Ratios for Cardiovascular Disease With Measured Baseline Levels of Risk Factors
| Studies With Information on Apolipoproteins (26 Studies, 139 581 Participants, 12 234 CVD Cases)
|
Studies With Information on Lipoprotein(a) (24 Studies, 133 502 Participants, 12 639 CVD Cases)
|
Studies With Information on Lipoprotein-Associated Phospholipase A
2 (11 Studies, 32075 Participants, 6150 CVD Cases) |
||||
|---|---|---|---|---|---|---|
| Mean (SD) or No. (No. of CVD Cases) | Hazard Ratioa (95% CI) | Mean (SD) or No. (No. of CVD Cases) | Hazard Ratioa (95% CI) | Mean (SD) or No. (No. of CVD Cases) | Hazard Ratioa (95% CI) | |
| Conventional risk factors | ||||||
| Age at survey, y | 56.42 (8.41) | 1.87 (1.73–2.02) | 56.86 (8.38) | 1.81 (1.69–1.93) | 63.78 (7.52) | 1.62 (1.43–1.83) |
|
| ||||||
| Sex | ||||||
| Men | 68 520 (7734) | NAb | 64 402 (7910) | NAb | 15 814 (3583) | NAb |
|
| ||||||
| Women | 71 061 (4500) | NAb | 69 100 (4729) | NAb | 16 261 (2567) | NAb |
|
| ||||||
| Current smoking | ||||||
| No | 102 261 (7137) | 1.0 [Reference] | 97 949 (7483) | 1.0 [Reference] | 21 972 (3677) | 1.0 [Reference] |
|
| ||||||
| Yes | 37 320 (5097) | 1.79 (1.66–1.94) | 35 553 (5156) | 1.87 (1.73–2.02) | 10 103 (2473) | 1.63 (1.38–1.91) |
|
| ||||||
| History of diabetes | ||||||
| No | 131 610 (10 722) | 1.0 [Reference] | 126 328 (11 103) | 1.0 [Reference] | 29 904 (5534) | 1.0 [Reference] |
|
| ||||||
| Yes | 7971 (1512) | 2.04 (1.76–2.35) | 7174 (1536) | 2.05 (1.77–2.38) | 2171 (616) | 1.76 (1.57–1.98) |
|
| ||||||
| Systolic blood pressure, mm Hg | 135.19 (18.38) | 1.31 (1.26–1.37) | 134.17 (18.15) | 1.34 (1.29–1.38) | 138.88 (21.04) | 1.29 (1.24–1.35) |
|
| ||||||
| Traditional lipids, mg/dL | ||||||
| Total cholesterol | 226 (42.5) | 1.22 (1.17–1.27) | 229 (42.1) | 1.19 (1.15–1.24) | 225 (41.7) | 1.13 (1.05–1.22) |
|
| ||||||
| HDL-C | 51.4 (14.7) | 0.83 (0.78–0.87) | 50.6 (14.7) | 0.82 (0.77–0.88) | 52.5 (12.7) | 0.85 (0.77–0.94) |
|
| ||||||
| Triglyceridec | 115 (80–168)d | 1.19 (1.15–1.23) | 115 (80–168)d | 1.18 (1.14–1.22) | 97 (80–142)d | 1.11 (1.05–1.16) |
|
| ||||||
| Lipid-related markers | ||||||
| Non–HDL-C, mg/dL | 175 (43.6) | 1.27 (1.22–1.33) | 178 (43.2) | 1.25 (1.19–1.31) | 173 (42.9) | 1.18 (1.10–1.27) |
|
| ||||||
| Total-C:HDL-C ratioc | 4.4 (3.5–5.4)d | 1.32 (1.24–1.39) | 4.4 (3.5–5.5)d | 1.31 (1.23–1.39) | 4.4 (3.6–5.4)d | 1.20 (1.09–1.32) |
|
| ||||||
| Apolipoprotein B, mg/dL | 110 (29) | 1.24 (1.19–1.29) | ||||
|
| ||||||
| Apolipoprotein A-I, mg/dL | 146 (32) | 0.87 (0.84–0.90) | ||||
|
| ||||||
| Apolipoprotein B:A-I ratioc | 0.7 (0.6–0.9)d | 1.30 (1.24–1.36) | ||||
|
| ||||||
| Lipoprotein(a), mg/dLc | 10.9 (4.4–28.0)d | 1.13 (1.09–1.18) | ||||
|
| ||||||
| Lipoprotein-associated phospholipase A2 | ||||||
| Activity | NAe | 1.12 (1.04–1.20) | ||||
|
| ||||||
| Mass | NAe | 1.15 (1.09–1.21) | ||||
Abbreviations: CVD, cardiovascular disease; HDL-C, high-density lipoprotein cholesterol; NA, not applicable.
SI conversion factors: To convert total cholesterol and HDL-C from mg/dL to mmol/L, multiply by 0.0259; triglycerides from mg/dL to mmol/L, multiply by 0.0113; lipoprotein(a) from mg/dL to mmol/L, multiply by 0.357; and apolipoprotein B and A-I from mg/dL to g/L, multiply by 0.01.
Hazard ratio (95% confidence interval) per 1 SD higher age, systolic blood pressure, measured biomarker level or compared to relevant reference category. Hazard ratios were adjusted for age, smoking status, systolic blood pressure, and history of diabetes, where appropriate.
Models were stratified by sex.
Variables were loge transformed.
Median and interquartile range.
Concentrations of lipoptotein–associated phospholipase A2 were standardized to a mean (SD) of 0 (1) within each study due to different assays yielding different absolute levels.
Acknowledgments
Funding/Support: The study was funded by grants from the British Heart Foundation; UK Medical Research Council; and UK National Institute of Health Research, Cambridge Biomedical Research Centre.
Role of the Sponsor: None of these organizations were involved in the design and conduct of the study; collection, management, analysis, and interpretation of the data; and preparation, review, or approval of the manuscript.
Authors/Writing Group: Emanuele Di Angelantonio, MD, Pei Gao, PhD, Lisa Pennells, PhD, Stephen Kaptoge, PhD, Department of Public Health and Primary Care, University of Cambridge, Cambridge, England; Muriel Caslake, PhD, Institute of Cardiovascular and Medical Sciences, University of Glasgow, Glasgow, Scotland; Alexander Thompson, PhD, Adam S. Butterworth, PhD, Nadeem Sarwar, PhD, David Wormser, PhD, and Danish Saleheen, MD, Department of Public Health and Primary Care, University of Cambridge; Christie M. Ballantyne, MD, Department of Medicine, Baylor College of Medicine, Houston, Texas; Bruce M. Psaty, MD, Departments of Medicine, Epidemiology, and Health Services, University of Washington, Seattle; Johan Sundström, MD, Uppsala Clinical Research Center, Uppsala University, Uppsala, Sweden; Paul M Ridker, MD, Division of Preventive Medicine, Brigham and Women’s Hospital, Boston, Massachusetts; Dorothea Nagel, PhD, Klinikum der Universität München, LMU, München, Germany; Richard F. Gillum, MD, Center for Disease Control and Prevention, Washington, DC; Ian Ford, PhD, Robertson Centre for Biostatistics, University of Glasgow; Pierre Ducimetiere, PhD, INSERM, France; Stefan Kiechl, MD, Department of Neurology, Medical University Innsbruck, Innsbruck, Austria; Wolfgang Koenig, MD, Department of Internal Medicine II-Cardiology, University of Ulm Medical Center, Ulm, Germany; Robin P. F. Dullaart, MD, University Hospital Groningen, University Medical Center Groningen, Groningen, the Netherlands; Gerd Assmann, MD, Assmann-Stiftung fur Pravention, Germany; Ralph B. D’Agostino, Sr, PhD, Department of Mathematics and Statistics, Boston University, Boston; Gilles R. Dagenais, MD, Départment de medicine, Institut universitaire de cardiologie et pneumologie de Québec, Québec, Canada; Jackie A. Cooper, MSc, Centre for Cardiovascular Genetics, University College London, England; Daan Kromhout, PhD, Division of Human Nutrition, Wageningen University, Wageningen, the Netherlands; Altan Onat, MD, Institute for Experimental Medical Research, Istanbul University, Istanbul, Turkey; Robert W. Tipping, MS, Merck Research Laboratories, Philadelphia, Pennsylvania; Agustín Gómez-de-la-Cámara, MD, Unidad de investigation, Hospital 12 de Octubre, Madrid, Spain; Annika Rosengren, MD, Department of Medicine, Sahlgrenska Academy, University of Gothenburg, Göteborg, Sweden; Susan E. Sutherland, PhD, Medical University of South Carolina; John Gallacher, PhD, Department of Epidemiology, Cardiff University, Cardiff, Wales; F. Gerry R. Fowkes, FRCPE, Wolfson Unit, Public Health Sciences, University of Edinburgh, Edinburgh; Edoardo Casiglia, MD, Department of Clinical and Experimental Medicine, University of Padova, Padova, Italy; Albert Hofman, MD, Department of Epidemiology, Erasmus Medical Center, Rotterdam, the Netherlands; Veikko Salomaa, MD, Department of Epidemiology, National Institute for Health and Welfare, Helsinki, Finland; Elizabeth Barrett-Connor, MD, Department of Family and Preventive Medicine, Division of Epidemiology, University of California, San Diego; Robert Clarke, MD, Clinical Trials Service Unit, University of Oxford, Oxford; Eric Brunner, PhD, Department of Epidemiology and Public Health, University College London; J. Wouter Jukema, MD, Department of Cardiology, Leiden University Medical Center, Leiden, the Netherlands; Leon A. Simons, MD, Lipid Research Department, University of New South Wales, Darlinghurst, Australia; Manjinder Sandhu, PhD, Department of Public Health and Primary Care; Nicholas J. Wareham, FRCP, MRC Epidemiology Unit, University of Cambridge; Kay-Tee Khaw, FMedSci, Department of Public Health and Primary Care, University of Cambridge; Jussi Kauhanen, MD, University of Eastern Finland, Kuopio, Finland; Jukka T. Salonen, MD, Metabolic Analytical Services Inc and University of Helsinki, Helsinki; William J. Howard, MD, MedStar Health Research Institute, Washington Hospital Center, Washington, DC; Børge G. Nordestgaard, MD, Department of Clinical Biochemistry, University of Copenhagen, Copenhagen, Denmark; Angela M. Wood, PhD, and Simon G. Thompson, FMedSci, Department of Public Health and Primary Care, University of Cambridge; S. Matthijs Boekholdt, MD, Academic Medical Center, Amsterdam, the Netherlands; Naveed Sattar, FRCP, and Chris Packard, PhD, Institute of Cardiovascular and Medical Sciences, University of Glasgow; Vilmundur Gudnason, MD, Icelandic Heart Association and University of Iceland, Reykjavik, Iceland; and John Danesh, FRCP, Department of Public Health and Primary Care, University of Cambridge.
Author Contributions: Dr Di Angelantonio had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. Dr Di Angelantonio, Dr Gao, Dr Pennells, and Dr Kaptoge contributed equally to the study.
Study concept and design: Di Angelantonio, Gao, Pennells, Kaptoge, A. Thompson, Sarwar, Gillum, Kiechl, Hofman, Sandhu, Khaw, Kauhanen, Sattar, Packard, Danesh.
Acquisition of data: Di Angelantonio, Kaptoge, A. Thompson, Sarwar, Wormser, Ballantyne, Psaty, Ridker, Nagel, Gillum, Ford, Ducimetiere, Kiechl, Koenig, Assmann, D’Agostino, Dagenais, Cooper, Kromhout, Tipping, Gómez-de-la-Cámara, Rosengren, Sutherland, Gallacher, Fowkes, Casiglia, Salomaa, Barrett-Connor, Clarke, Brunner, Jukema, Simons, Sandhu, Wareham, Khaw, Kauhanen, Salonen, Howard, Nordestgaard, Wood, Boekholdt, Sattar, Packard, Gudnason, Danesh.
Analysis and interpretation of data: Di Angelantonio, Gao, Pennells, Kaptoge, Caslake, A. Thompson, Butterworth, Sarwar, Wormser, Saleheen, Sundström, Ridker, Dullaart, Onat, Sutherland, Salomaa, Simons, Nordestgaard, Wood, S. G. Thompson, Sattar, Packard, Danesh.
Drafting of the manuscript: Di Angelantonio, Pennells, Kaptoge, A. Thompson, Butterworth, Sarwar, Kromhout, Casiglia, Salomaa, Wood, Packard, Danesh.
Critical revision of the manuscript for important intellectual content: Di Angelantonio, Gao, Pennells, Kaptoge, Caslake, Thompson, Butterworth, Saleheen, Ballantyne, Psaty, Ridker, Sundström, Nagel, Gillum, Ford, Ducimetiere, Kiechl, Koenig, Dullaart, Assmann, D’Agostino, Dagenais, Cooper, Kromhout, Onat, Tipping, Gómez-de-la-Cámara, Rosengren, Sutherland, Gallacher, Fowkes, Casiglia, Salomaa, Barrett-Connor, Clarke, Kauhanen, Salonen, Howard, Nordestgaard, S. G. Thompson, Boekholdt, Sattar, Gudnason, Danesh.
Statistical analysis: Gao, Pennells, Kaptoge, Wormser, Saleheen, Gillum, D’Agostino, Sutherland, Brunner, Jukema, Simons, Sandhu, Wareham, Khaw, Wood, S. G. Thompson.
Obtained funding: Caslake, A. Thompson, Psaty, Koenig, Dullaart, Kromhout, Rosengren, Fowkes, Casiglia, Hofman, Salomaa, Barrett-Connor, Brunner, Jukema, Wareham, Khaw, Kauhanen, Salonen, Nordestgaard, S. G. Thompson, Packard, Gudnason, Danesh.
Administrative, technical, or material support:
Di Angelantonio, Caslake, Ridker, Ford, Kiechl, Koenig, Dullaart, Assmann, Dagenais, Kromhout, Onat, Tipping, Sutherland, Salomaa, Barrett-Connor, Jukema, Simons, Khaw, Packard, Danesh.
Study supervision: Di Angelantonio, Pennells, Kaptoge, Caslake, Wormser, Gómez-de-la-Cámara, Gallacher, Fowkes, Casiglia, Hofman, Salomaa, Brunner, Jukema, Simons, Kauhanen, Wood, S. G. Thompson, Sattar, Danesh.
Conflict of Interest Disclosures: All authors have completed and submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Dr Di Angelantonio reported that he has received research funding from the British Heart Foundation and European Union; compensation for consultancy and lecturers from Lead-up Medical Network, Merck Sharp and Dohme, and John Wiley & Sons; support for travel or accommodations from Pfizer; and royalties from Elsevier (France). Dr Gao reported that she has received research funding from the British Heart Foundation and the Medical Research Council. Dr Pennells reported that she has received research funding from the Medical Research Council. Dr Kaptoge reported that he has received research funding from British Heart Foundation, Medical Research Council, UK National Institute of Health Research, and Cambridge Biomedical Research Centre. Dr Caslake reported that she has received research funding from Roche and Merck Sharp and Dohme and support for travel and accommodations from Randox Laboratories and Denka Seiken. Dr A. Thompson reported that he has received research funding from the British Heart Foundation and Medical Research Council; has been an employee of Roche Products Ltd since September 2011; and has received compensation from GlaxoSmithKline for giving lectures. Dr Sarwar reported that he has been an employee of Pfizer Inc since April 2011. Dr Wormser reported that he has received research funding from the British Heart Foundation and Medical Research Council. Dr Saleheen reported that he has received research funding from the National Institute of Health and Wellcome Trust. Dr Ballantyne reported that she has received research funding from Abbott, Amarin, AstraZeneca, Bristol-Myers Squibb, GlaxoSmithKline, Gentech, Kowa, Merck, Novartis, Roche, Sanofi-Synthelabo, Takeda, National Institutes of Health, American Diabetes Association, and American Heart Association and compensation for consultancy and for lectures from Abbott, Adnexus, Amarin, Amylin, AstraZeneca, Bristol-Myers Squibb, Esperion, Genetech, GlaxoSmithKline, Idera Pharma, Kowa, Merck, Novartis, Omthera, Pfizer, Resverlogix, Roche, Sanofi-Synthelabo, and Takeda. Dr Psaty reported that he serves on a data safety and monitoring board for a clinical trial of a device funded by the manufacturer Zoll LieCor and on the Steering Committee for the Yale Open-Data Project funded by Medtronic. Dr Ridker reported that he is listed as a coinventor on patents held by the Brigham and Women’s Hospital that relate to the use of inflammatory biomarkers in cardiovascular disease and diabetes that have been licensed to AstraZeneca and Seimens; received research funding from AstraZeneca and Novartis; and compensation for consultancy from Merck, Vascular Biogenics, ISIS Inc, and Genzyme. Dr Kiechl reported that he has received research funding from the Fonds zur Förderung der wissenschaftlichen Forschung. Dr Koenig reported that he has received research funding from the Else Kröner Fresenius Foundation, Vifor Company, European Union, Federal Ministry of Education and Research and Boehringer Ingelheim; and compensation for consultancy and lecturers from Roche, Cerenis, Anthera, BioInvet, Novartis, AstraZeneca, Merck Sharp and Dohme, Roche, and Boehringer Ingelheim. Dr Assmann reported that he has received compensation for consultancy and lecturers from Merck Sharp and Dohme, and Residual Risk Reduction Initiative and has served on board for the Residual Risk Reduction Initiative. Dr Dagenais reported that he has received research funding from the National Heart, Lung, and Blood Institute, National Institute of Diabetes and Digestive and Kidney Diseases, Canadian Institutes Health Research, and Heart and Stroke of Canada and compensation for expert testimony and lecturers from Abbott and Boehringer Ingelheim. Dr Kromhout reported that he has received research funding from the Netherlands Prevention Foundation. Mr Tipping reported that he is an employee of Merck. Dr Rosengren reported that she has received research funding from the Swedish Research Council and Swedish Heart and Lung Foundation. Dr Barrett-Connor reported that she has received research funding from the National Institutes of Health. Dr Clarke reported that he has received research funding from the British Heart Foundation. Dr Brunner reported that he has received research funding from the Medical Research Council and British Heart Foundation. Dr Jukema reported that he has received research funding from the European Union. Dr Sandhu reported that he has received research funding from the British Heart Foundation. Dr Wareham reported that he has received research funding from the Medical Research Council. Dr Khaw reported that she has received research funding from the UK Medical Research Council and Cancer Research. Dr Howard reported that he has received compensation for consultancy and lecturers from Merck, Abbott, and GlaxoSmithKline. Dr S. G. Thompson reported that he has received research funding from the British Heart Foundation. Dr Sattar reported that he has received research funding from Pfizer and has served on the advisory board for Niaspan for Merck Sharp and Dohme. Dr Packard reported that he has received compensation for consultancy and lecturers from AstraZeneca, Roche, and Merck Sharp and Dohme. Dr Danesh reported that he has received research funding from the British Heart Foundation, BUPA Foundation, Cambridge Biomedical Research Centre, Denka, diaDexus, European Union, European Research Council, Evelyn Trust, Fogarty International Centre, GlaxoSmithKline, Medical Research Council, Merck Sharp and Dohme, National Heart, Lung, and Blood Institute, National Institute of Neurological Disorders and Stroke, National Institute for Health Research, Novartis, Pfizer, Roche, Wellcome Trust, and UK Biobank and has served on advisory boards for Merck, Pfizer, and Novartis. All other members of the writing committee declare that they have no conflicts of interest.
Investigators of Participating Studies: AFTCAPS: R. W. Tipping; ARIC: C. M. Ballantyne, R. Hoogeveen, S. S. Virani, C. Ndumele, and V. Nambi; BRUN: J. Willeit, P. Willeit, A. Mayr, P. Santer, and S. Kiechl; CaPS: J. Gallacher, J. W. G. Yarnell, and Y. Ben-Shlomo; CASTEL: E. Casiglia and V. Tikhonoff; CHARL: S. E. Sutherland, P. J. Nietert, J. E. Keil, and D. L. Bachman; CHS: B. M. Psaty, M. Cushman, R. P. Tracy, and N. Jenny, see http://www.chs-nhlbi.org for acknowledgments; COPEN: B. G. Nordestgaard, A. Tybjærg-Hansen, R. Frikke-Schmidt, M. Benn, and P. R. Kamstrup; DRECE: A. Gómez de la Cámara, J. A. Gutiérrez-Fuentes, J. A. Gómez Gerique, and M. A. Rubio Herrera; DUBBO: L. A. Simons, Y. Friedlander, and J. McCallum; EAS: F.G.R. Fowkes, J. F. Price, A. J. Lee, and J. Bolton; EPICNOR: M. Sandhu, N. J. Wareham, and K-T Khaw; FINRISK92: K. Harald, P. R. Jousilahti, E. Vartiainen, and V. Salomaa; FRAMOFF: R. B. D’Agostino, Sr, P. A. Wolf, R. S. Vasan, and E. J. Benjamin; GRIPS: P. Cremer and D. Nagel; KIHD: J. Kauhanen, J. T. Salonen, K. Nyyssönen, and T-P Tuomainen; MOGERAUG1: W. Koenig, C. Meisinger, A. Döring, and W. Mraz; MOSWEGOT: A. Rosengren, L. Wilhelmsen, and G. Lappas; NHANES III: R. F. Gillum and J. Kwagyan; NPHS II: J. A. Cooper, and K. A. Bauer; PREVEND: R. P. F. Dullaart, S. J. L. Bakker, R. T. Gansevoort, P. van der Harst, and H. L. Hillege; PRIME: P. Ducimetiere, P. Amouyel, D. Arveiler, A. Evans, and J. Ferrières; PROCAM: H. Schulte and G. Assmann; PROSPER: J. W. Jukema, D. J. Stott, R. G. J. Westendorp, and B. M. Buckley; QUEBEC: B. Cantin, B. Lamarche, J-P Després, and G. R. Dagenais; RANCHO: E. Barrett-Connor, D. L. Wingard, and L. B. Daniels; REYK: V. Gudnason, T. Aspelund, G. Sigurdsson, B. Thorsson, and G. Eiriksdottir; ROTT: J. C. M. Witteman, I. Kardys, A. Dehghan, M. A. Ikram, and A. Hofman; SHS: W. J. Howard, B. V. Howard, Y. Zhang, L. Best, and J. Umans; TARFS: A. Onat and G. Can; ULSAM: J. Sundström, L. Lind, V. Giedraitis, and E. Ingelsson; WHITE1: M. Marmot, R. Clarke, R. Collins, and A. Fletcher; WHITE2: E. Brunner, M. Shipley, and M. Kivimaki; WHS: P. M Ridker, J. Buring, N. Rifai, and N. Cook; WOSCOPS: I. Ford, and M. Robertson; and ZUTE: E. J. M. Feskens, J. M. Geleijnse, and D, Kromhout.
Data Management Team: M. Walker, S. Watson.
Coordinating Centre: M. Alexander, A. S. Butterworth, E. Di Angelantonio, O. H. Franco, P. Gao, R. Gobin, J. M. Gregson, P. Haycock, S. Kaptoge, L. Pennells, D. Saleheen, J. Sanderson, N. Sarwar, A. Thompson, S. G. Thompson, M. Walker, S. Watson, A. M. Wood, D. Wormser, X. Zhao, and J. Danesh (principal investigator).
Footnotes
Online-Only Material: The 7 eTables, 16 eFigures, 2 eAppendixes, and Author Audio Interview are available at http://www.jama.com.
References
- 1.Perk J, De Backer G, Gohlke H, et al. European Guidelines on cardiovascular disease prevention in clinical practice (version 2012): The Fifth Joint Task Force of the European Society of Cardiology and Other Societies on Cardiovascular Disease Prevention in Clinical Practice (constituted by representatives of nine societies and by invited experts) [published online May 2, 2012] Eur Heart J. doi: 10.1093/eurheartj/ehs092. [DOI] [PubMed] [Google Scholar]
- 2.Parish S, Offer A, Clarke R, et al. Lipids and Lipoproteins and Risk of Different Vascular Events in the MRC/BHF Heart Protection Study. Circulation. 2012;125(20):2469–2478. doi: 10.1161/CIRCULATIONAHA.111.073684. [DOI] [PubMed] [Google Scholar]
- 3.Arsenault BJ, Boekholdt SM, Kastelein JJP. Lipid parameters for measuring risk of cardiovascular disease. Nat Rev Cardiol. 2011;8(4):197–206. doi: 10.1038/nrcardio.2010.223. [DOI] [PubMed] [Google Scholar]
- 4.Ridker PM, Rifai N, Cook NR, et al. Non-HDL cholesterol, apolipoproteins A-I and B100, standard lipid measures, lipid ratios, and CRP as risk factors for cardiovascular disease in women. JAMA. 2005;294 (3):326–333. doi: 10.1001/jama.294.3.326. [DOI] [PubMed] [Google Scholar]
- 5.Arsenault BJ, Rana JS, Stroes ESG, et al. Beyond low-density lipoprotein cholesterol. J Am Coll Cardiol. 2009;55(1):35–41. doi: 10.1016/j.jacc.2009.07.057. [DOI] [PubMed] [Google Scholar]
- 6.Contois JH, McConnell JP, Sethi AA, et al. AACC Lipoproteins and Vascular Diseases Division Working Group on Best Practices. Apolipoprotein B and cardiovascular disease risk. Clin Chem. 2009;55(3):407–419. doi: 10.1373/clinchem.2008.118356. [DOI] [PubMed] [Google Scholar]
- 7.Brunzell JD, Davidson M, Furberg CD, et al. American Diabetes Association; American College of Cardiology Foundation. Lipoprotein management in patients with cardiometabolic risk. Diabetes Care. 2008;31(4):811–822. doi: 10.2337/dc08-9018. [DOI] [PubMed] [Google Scholar]
- 8.Greenland P, Alpert JS, Beller GA, et al. American College of Cardiology Foundation; American Heart Association. 2010 ACCF/AHA guideline for assessment of cardiovascular risk in asymptomatic adults. J Am Coll Cardiol. 2010;56(25):e50–e103. doi: 10.1016/j.jacc.2010.09.001. [DOI] [PubMed] [Google Scholar]
- 9.Genest J, McPherson R, Frohlich J, et al. 2009 Canadian Cardiovascular Society/Canadian guidelines for the diagnosis and treatment of dyslipidemia and prevention of cardiovascular disease in the adult—2009 recommendations. Can J Cardiol. 2009;25(10):567–579. doi: 10.1016/s0828-282x(09)70715-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Myers GL, Christenson RH, Cushman M, et al. NACB LMPG Committee Members. National Academy of Clinical Biochemistry Laboratory Medicine Practice guidelines. Clin Chem. 2009;55(2):378–384. doi: 10.1373/clinchem.2008.115899. [DOI] [PubMed] [Google Scholar]
- 11.Reiner Z, Catapano AL, De Backer G, et al. European Association for Cardiovascular Prevention & Rehabilitation; ESC Committee for Practice Guidelines (CPG) 2008–2010 and 2010–2012 Committees. ESC/EAS Guidelines for the management of dyslipidaemias. Eur Heart J. 2011;32(14):1769–1818. doi: 10.1093/eurheartj/ehr158. [DOI] [PubMed] [Google Scholar]
- 12.Nordestgaard BG, Chapman MJ, Ray K, et al. European Atherosclerosis Society Consensus Panel. Lipoprotein(a) as a cardiovascular risk factor: current status. Eur Heart J. 2010;31(23):2844–2853. doi: 10.1093/eurheartj/ehq386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Emerging Risk Factors Collaboration. Lipoprotein (a) concentration and the risk of coronary heart disease, stroke, and nonvascular mortality. JAMA. 2009;302(4):412–423. doi: 10.1001/jama.2009.1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Emerging Risk Factors Collaboration. Major lipids, apolipoproteins, and risk of vascular disease. JAMA. 2009;302(18):1993–2000. doi: 10.1001/jama.2009.1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lp-PLA2 Studies Collaboration. Lipoprotein-associated phospholipase A2 and risk of coronary disease, stroke, and mortality. Lancet. 2010;375(9725):1536–1544. doi: 10.1016/S0140-6736(10)60319-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Danesh J, Erqou S, Walker M, et al. Emerging Risk Factors Collaboration. Analysis of individual data on lipid, inflammatory and other markers in over 1.1 million participants in 104 prospective studies of cardiovascular diseases. Eur J Epidemiol. 2007;22(12):839–869. doi: 10.1007/s10654-007-9165-7. [DOI] [PubMed] [Google Scholar]
- 17.Walldius G, Jungner I, Holme I, Aastveit AH, Kolar W, Steiner E. High apolipoprotein B, low apolipoprotein A-I, and improvement in the prediction of fatal myocardial infarction (AMORIS study) Lancet. 2001;358(9298):2026–2033. doi: 10.1016/S0140-6736(01)07098-2. [DOI] [PubMed] [Google Scholar]
- 18.D’Agostino RB, Sr, Vasan RS, Pencina MJ, et al. General cardiovascular risk profile for use in primary care: the Framingham Heart Study. Circulation. 2008;117(6):743–753. doi: 10.1161/CIRCULATIONAHA.107.699579. [DOI] [PubMed] [Google Scholar]
- 19.Fibrinogen Studies Collaboration. Measures to assess the prognostic ability of the stratified Cox proportional hazards model. Stat Med. 2009;28(3):389–411. doi: 10.1002/sim.3378. [DOI] [PubMed] [Google Scholar]
- 20.Pencina MJ, D’Agostino RB, Sr, D’Agostino RB, Jr, Vasan RS. Evaluating the added predictive ability of a new marker: from area under the ROC curve to reclassification and beyond. Stat Med. 2008;27(2):157–172. doi: 10.1002/sim.2929. [DOI] [PubMed] [Google Scholar]
- 21.Steyerberg EW, Vickers AJ, Cook NR, et al. Assessing the performance of prediction models: a framework for traditional and novel measures. Epidemiology. 2010;21(1):128–138. doi: 10.1097/EDE.0b013e3181c30fb2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Higgins JP, Thompson SG. Quantifying heterogeneity in a meta-analysis. Stat Med. 2002;21 (11):1539–1558. doi: 10.1002/sim.1186. [DOI] [PubMed] [Google Scholar]
- 23.Baigent C, Blackwell L, Emberson J, et al. Cholesterol Treatment Trialists’ (CTT) Collaboration. Efficacy and safety of more intensive lowering of LDL cholesterol. Lancet. 2010;376(9753):1670–1681. doi: 10.1016/S0140-6736(10)61350-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults. Executive summary of the third report of The National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) JAMA. 2001;285(19):2486–2497. doi: 10.1001/jama.285.19.2486. [DOI] [PubMed] [Google Scholar]
- 25.McQueen MJ, Hawken S, Wang X, et al. INTERHEART study investigators. Lipids, lipoproteins, and apolipoproteins as risk markers of myocardial infarction in 52 countries (the INTERHEART study): a case-control study. Lancet. 2008;372(9634):224–233. doi: 10.1016/S0140-6736(08)61076-4. [DOI] [PubMed] [Google Scholar]
- 26.Parish S, Peto R, Palmer A, et al. International Studies of Infarct Survival Collaborators. The joint effects of apolipoprotein B, apolipoprotein A1, LDL cholesterol, and HDL cholesterol on risk. Eur Heart J. 2009;30(17):2137–2146. doi: 10.1093/eurheartj/ehp221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sniderman AD, Williams K, Contois JH, et al. A meta-analysis of low-density lipoprotein cholesterol, non–high-density lipoprotein cholesterol, and apolipoprotein B as markers of cardiovascular risk. Circ Cardiovasc Qual Outcomes. 2011;4(3):337–345. doi: 10.1161/CIRCOUTCOMES.110.959247. [DOI] [PubMed] [Google Scholar]
- 28.Boekholdt SM, Arsenault BJ, Mora S, et al. Association of LDL cholesterol, non–HDL cholesterol, and apolipoprotein B levels with risk of cardiovascular events among patients treated with statins: a meta-analysis. JAMA. 2012;307(12):1302–1309. doi: 10.1001/jama.2012.366. [DOI] [PubMed] [Google Scholar]
- 29.ClinicalTrials.gov. [Accessed April 10, 2012];Treatment of HDL to Reduce the Incidence of Vascular Events HPS2-THRIE [NCT00461360] http://clinicaltrials.gov/ct/show/NCT00461630.
- 30.White H, Held C, Stewart R, et al. Study design and rationale for the clinical outcomes of the STABILITY Trial (STabilization of Atherosclerotic plaque By Initiation of darapLadIb TherapY) comparing darapladib versus placebo in patients with coronary heart disease. Am Heart J. 2010;160(4):655–661. doi: 10.1016/j.ahj.2010.07.006. [DOI] [PubMed] [Google Scholar]
- 31.Maningat P, Breslow JL. Needed: pragmatic clinical trials for statin-intolerant patients. N Engl J Med. 2011;365(24):2250–2251. doi: 10.1056/NEJMp1112023. [DOI] [PubMed] [Google Scholar]
- 32.Kamstrup PR, Tybjaerg-Hansen A, Steffensen R, Nordestgaard BG. Genetically elevated lipoprotein(a) and increased risk of myocardial infarction. JAMA. 2009;301(22):2331–2339. doi: 10.1001/jama.2009.801. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.