

Abstract
Some studies of animal models of middle cerebral artery occlusion indicate that inflammation plays a key role in the pathogenesis of cerebral ischemia and secondary damage. Flurbiprofen has been suggested to alleviate cerebral ischemia/reperfusion injury in both focal and global cerebral ischemia models, but the mechanisms underlying the protective action are still incompletely understood. In this study we want to investigate the protective effect of flurbiprofen after transient middle cerebral artery occlusion (MCAO) in rats and the role of the NF-κB signaling pathway on this neuroprotective effect. Male Wistar rats were subjected to transient middle cerebral artery occlusion for 2 h, followed by 24 h reperfusion. Flurbiprofen was administrated via tail-vein injection at the onset of reperfusion. HE staining and Immunohistochemistry were carried out to detect the morphological changes in ischemic penumbra cortex. The expression of inflammatory cytokines genes (IL-1β, IL-6 and TNF-α) and the levels of p-NF-κB (p65) in ischemic penumbra cortex were measured by RT-PCR and western blot. Administration of flurbiprofen at the doses of 5 mg/kg and 10 mg/kg significantly attenuated cerebral ischemia/reperfusion injury, as shown by a reduction in the morphological changes and inhibition of pro-inflammatory cytokine expression in ischemic penumbra cortex. Moreover, our findings further demonstrated that the inhibition of NF-κB activity was involved in the neuroprotective effect of flurbiprofen on inflammatory responses. Flurbiprofen protects against cerebral injury by reducing expression of inflammatory cytokines genes and this effect may be partly due to the inhibition of NF-κB signaling pathway.
Keywords: Flurbiprofen, focal cerebral ischemia, inflammation, nuclear factor-κB
Introduction
A growing number of recent experiments have showed that excessive inflammatory response plays a key role in the pathogenesis of cerebral ischemia and secondary damage [1,4]. Especially, some studies revealed that focal cerebral ischemia triggers inflammation response around the ischemic area and subsequent reperfusion can exacerbates it. Following permanent as well as transient middle cerebral artery occlusion (MCAO) in rodents, acute inflammatory reactions that start within hours and persist for days after the insult are currently being thought to contribute to the neuronal death. In various animal models of MCAO, increased neutrophil accumulation has been founded in the microvessels and ischemic cerebral cortex [5,7]. It has been reported that adhesion molecules, cytokines (IL-6, IL-1, TNF-α, and TGF-β), and chemokines, which are produced immediately after the onset of cerebral ischemia contribute to irreversible brain injuries [7,8]. Inhibition of neutrophil adhesion and neutrophil function in experimental models of stroke have all been shown to have reductions in infarct volumes and improved outcomes [7]. Moreover, several studies have suggested that TNF-α null mice [8], iNOS knock-out mice [9] or COX-2 deficient mice [10,11] have alleviated brain damage and were more resistant to cerebral ischemia.
Nuclear factor-κB (NF-κB) is one of the most important transcription factors activated after cerebral ischemia. NF-κB is involved in inflammatory responses that potentiate ischemic injury [12] activating many genes involved in the pathogenesis of cerebral ischemia, such as iNOS, IL-1β, TNF-α, ICAM-1, COX-2, and IL-6. Recent studies in cerebral ischemia models indicate that NF-κB’s actions are largely detrimental and animals treated with pharmacological NF-κB inhibition would experience anti-inflammatory effects [13]. Many studies also suggested that inhibiting NF-κB activation or knocking out p50 subunit of NF-κB is protection and develops smaller infarct volume [14].
Many studies demonstrate that NSAIDS may provide neuroprotective effect in cerebral ischemic conditions [15,16]. Flurbiprofen axetil (FA) which dissolved in lipid microsphere may promote aggregation of flurbiprofen granular at inflammatory lesion sites and absorption in a short time, both factors which help to effectively target therapy. Flurbiprofen has been found to be highly efficacious in neuroprotection in neuro-degeneration [17] from both histological and behavioral standpoints, and it significantly improved neurologic deficit score and mean brain infarct volume percentage under cerebral ischemic conditions, but the underlying molecular mechanisms remain poorly understood. Our previous study demonstrated that flurbiprofen inhibites apoptosis in the ischemic cerebral parenchyma in rats through the activation of Akt/GSK-3β pathway [18].
In this study we want to investigate whether the neuroprotective effect of flurbiprofen was associated with inhibition of inflammatory responses and NF-κB signaling pathway.
Materials and methods
Experimental animals
Transient middle cerebral artery occlusion (MCAO) was induced in male Wistar rats (240-300 g) provided by the Institute of Medical Experimental Animals, Shandong University. Animals were housed in a room at a controlled temperature of 25 ± 2°C and a relative humidity of 50 ± 10% with alternating 12 h light and dark cycles and free access to food and water. All rats were acclimatized in our animal facility for at least 1 week prior to experiments. Stressful stimuli were avoided. All surgical procedures were performed following institutional approval in accordance with Shandong University Guide for the Care and Use of Laboratory Animals. Rats were randomly dividedinto five groups: sham group, ischemia/reperfusion (I/R) group, I/R + 5 mg/kg, I/R + 10 mg/kg dose of flurbiprofen axetil groups (the dosages were used in the previous study by Salzberg-Brenhouse [19]) and 1 mL/kg vehicle group (lipo-microballoons in which flurbiprofen was dissolved). Flurbiprofen axetil and vehicle were administered via tail-vein injection at the onset of reperfusion.
Focal cerebral ischemia
Transient focal cerebral ischemia was produced by the MCAO procedure as described previously [20]. After 120 minutes of MCAO, reperfusion was initiated by careful withdrawing the suture. The sham-operated rats received all surgical procedures but without the suture inserted. During and after the surgery, rectal temperature was controlled with a homeothermic blanket and kept at 37 ± 0.5°C. After 24 h of reperfusion, the rats were anesthetized and then decapitated. The brains were immediately frozen in liquid nitrogen for the following studies.
Histopathology and immunohistochemistry
The brain was fixed by transcardial perfusion with a buffered 4% paraformaldehyde solution and paraffin-embedded. The ischemic penumbra cortex was sectioned at a thickness of 4 μm. Regular HE staining was performed for morphological observation.
Immunohistochemistry was performed using the Histostain-plus bulk kit (Zymed laboratories, CA, USA). Briefly, ischemic penumbra sections were incubated in 3% hydrogen dioxide solution for 20 minutes, rinsed twice with PBS and the Antigen was retrieved with microwave irradiation, then the sections were incubated with the appropriate primary antibody overnight at 4°C. On the following day, ischemic cerebral penumbra sections were rinsed 3 times with PBS, incubated with anti-rabbit immunoglobulin G secondary antibody coupled with HRP. The bound antiserum was visualized by incubating with DAB kit (Zymed laboratories, CA, USA). Immunostained cells were analyzed under bright-field microscopy.
Cerebral myeloperoxidase assay
Myeloperoxidase (MPO) is one of the distinct indicators for the tissue infiltration of neutrophilic granulocytes following cerebral ischemia. 10% ischemic penumbra tissue homogenate was prepared to analyze MPO activity by spectrophotometry with O-dianisidine dihydrochloride and hydrogen peroxide at 460 nm [21]. The procedures were according to the description of the kit.
RNA extraction and RT-PCR analysis
Total cellular RNA was extracted with Trizol reagent (Invitrogen Corporation, Carlsland, CA) according to the manufacturer’s recommended procedures. Briefly, first-strand DNA was synthesized from 1 μg of total RNA using a Reaction Ready TM first strand cDNA synthesis kit (SABioscience Corporation, Frederick, MD). After incubation at 70°C for 3 min and cooling down to 37°C for 10 min, RT cocktail was added to the annealing mixture and further incubated at 37°C for 60 min. Two milliliter of 1:2 diluted cDNA was subjected to real-time quantitative PCR using Bio-Rad iCycler iQ Multicolor Real-Time PCR Detection System (Bio-Rad Life Science Research, Hercules, CA). PCR was performed in a 25 mL volume using SYBR Green ER qPCR Super Mix for iCycler (Invitrogen Corporation, Carlsland, CA). All primers were purchased from Sangon Biotech (Shanghai) Corporation. All PCR assays were performed in triplicate. The following primer pairs were used: for TNF-α: CTCTTCAAGGGACAAGGCTG (forward) and TCACAGAGCAATGACTCCAAAG (reverse); IL-1β: ATTACCACTTGTTGGCTTA (forward) and TGTGATGTTCCCATTAGAC (reverse); IL-6: GAGAGCATTGGAAGTTGGGG (forward) and CTTCCAGCCAGTTGCCTTCT (reverse).
Western blot
Western blotting was performed as described previously [22]. Tissues (n = 5, in each group) corresponding to the ischemic penumbra cortex were minced into fragments, homogenized in lysis buffer (50 mmol/L Tris-HCL pH 6.8, 150 mmol/L NaCl, 5 mmol/L EDTA, 0.5% sodium deoxycholate, 0.5% NP-40 and protease inhibitor cocktail) and spun down (2000 g, 5 min, 4°C). The protein concentrations were determined using a BCA Protein Assay reagent kit (Pierce, Rockford, IL). The lysates were separated by 12% SDS-PAGE and then transferred to a nitrocellulose membrane (Bio-Rad, Hercules, CA). After blocking with solution of 5% skim milk powder in 1×TBS for one hour, the membranes were incubated with primary antibodies of p-NF-κB (p65), NF-κB (p65) (Cell Signaling, Beverly, MA, USA) overnight at 4°C, respectively. After washing three times with TBS-T for 5 min, the blot was incubated with horseradish peroxidase-labeled second antibodies of anti-mouse IgG or anti-rabbit IgG (1: 6000 dilution, Protein Tech Group, Chicago, IL, USA), respectively. The blot was again washed three times with TBS-T before being exposed to the Super Signal West Dura Extended Duration substrate (Pierce Bio technology, Rockford, IL). An enhanced chemiluminescent (ECL) detection system was used according to the manufacturer’s protocol, and immunoblots were exposed to autoradiography film (Hyperfilm-ECL, Amersham Pharmacia Biotech). To document the loading controls, the membrane was reprobed with a primary antibody against housekeeping protein GAPDH.
Statistical analysis
All analyses were performed using SPSS v11.0 (SPSS Inc, Chicago, IL). Data were expressed as means ± SEM. Student’s t-test and one-way analysis of variance test were used for statistical analyses of the data. Differences were considered statistically significant for values of P < 0.05.
Results
Histological observation of cerebral ischemic damage
HE staining was performed to observe the morphological changes, as shown in Figure 1. In the sham group, the ischemic penumbra cortex tissues remain intact and normal cell organelles, neurons kept arranged well and the nuclei were centered with clear staining and the cytoplasm was abundant. In contrast, in ischemia/reperfusion and vehicle groups, the penumbra cortex tissues were damaged severely in the infarct region. Neurons were markedly degenerated and necrotic, and their arrangement was disordered. However, after administration of flurbiprofen, the extent of damage was significantly diminished, and the number of normal neurons was markedly increased as well.
Figure 1.
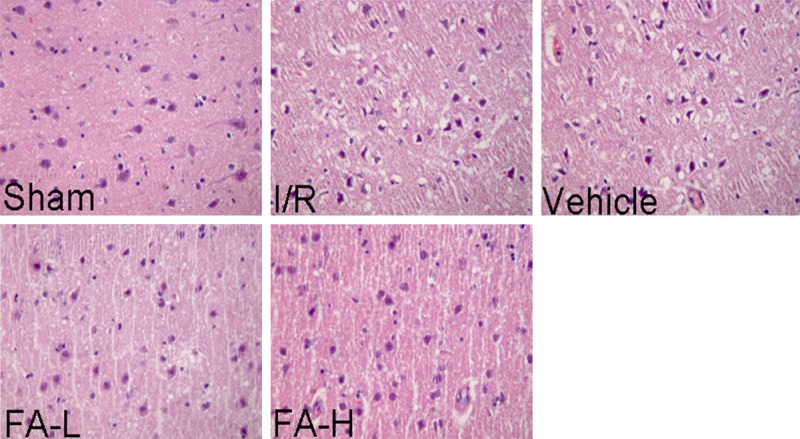
Representative image of histological assay of the ischemic penumbra tissues of rats. Ischemic penumbra sections obtained from injured cerebral hemispheres were stained with haematoxylin and eosin, observed using Leica microscope. In ischemia/reperfusion and vehicle groups, the ischemic penumbra tissues were damaged seriously. Neurons were degenerated and necrotic, and their arrangement was disordered. However, flurbiprofen can obviously decrease the extent of damage induced by ischemia/reperfusion insult. Magnification, 400脳.
Effects of flurbiprofen on neutrophils infiltration to ischemic penumbra cortex
As shown in Figure 3D, MPO activity was greatly increased in ischemia/reperfusion and vehicle groups compared with the sham group (P < 0.05). However, treatment with flurbiprofen at doses of 5 mg/kg and 10 mg/kg significantly relieved the increase of MPO activity induced by MCAO.
Figure 3.
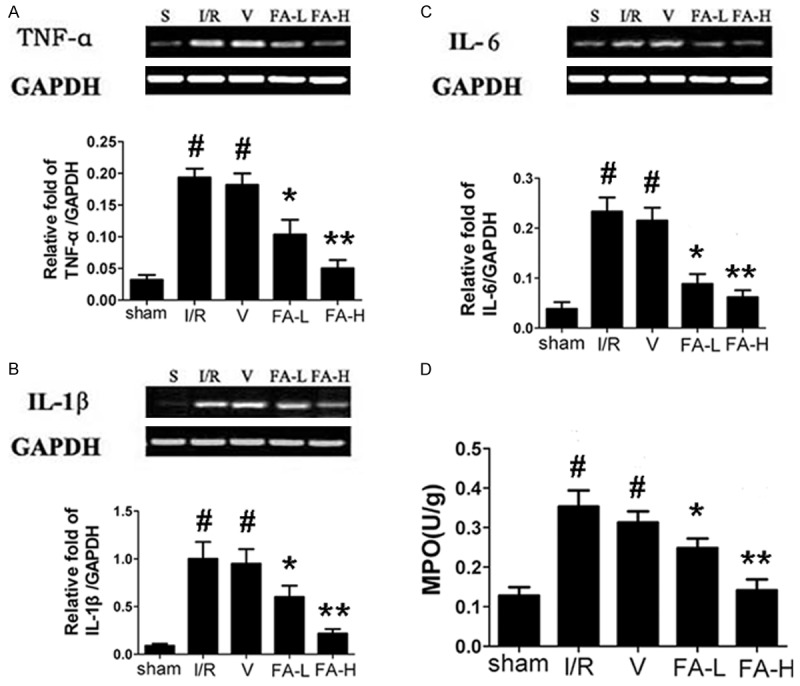
Effect of flurbiprofen on the mRNA expression of TNF-α, IL-6 and IL-1β in the ischemic penumbra of rats. Flurbiprofen was administered at the beginning of reperfusion at the dose of 5 mg/kg and 10 mg/kg. In ischemia/reperfusion and vehicle groups, the mRNA expression of these cytokines was significantly enhanced compared with sham group. Whereas, flurbiprofen can significantly reduce the mRNA expression of these cytokines induced by ischemia/reperfusion insult. The mRNA expression was determined by RT-PCR analysis and bands were quantified by densitometry, normalized to GAPDH levels (A-C). (D) Ischemic penumbra tissue homogenates were prepared to analyze MPO activity by spectrophotometry. Treatment with flurbiprofen at doses of 5 mg/kg and 10 mg/kg significantly relieved the increase of MPO activity induced by ischemia/reperfusion insult. All data were represented as means ± S.D. For each independent experiment, n = 5. #P < 0.05 vs. sham group, *P < 0.05 vs. ischemia/reperfusion and vehicle group, **P < 0.05 vs. FA-L group.
Effects of flurbiprofen on inflammatory cytokine gene expression
After 24 hours of reperfusion, the mRNA levels of the inflammatory cytokines TNF-α, IL-6 and IL-1β showed significantly increased expression in ischemia/reperfusion and vehicle groups compared with the sham group (P < 0.05). But the increase of the cytokines expression caused by MCAO was lower after administration of flurbiprofen at doses of 5 mg/kg and 10 mg/kg (P < 0.05) (Figure 3A-C).
Effect of flurbiprofen on NF-κB (p65) phosphorylation in ischemic penumbra cortex
The expression of NF-κB (p65) in ischemic penumbra cortex was detected by immunohistochemistry assay and western blot assay. As shown in Figure 2 and Figure 4, NF-κB (p65) was predominantly located in the nucleus in ischemia/reperfusion and vehicle groups and flurbiprofen can significantly inhibit its translocation from cytoplasm into the nucleus (P < 0.05). Furthermore, through western blot assay of nuclear extract, the present study showed that the level of p-NF-κB (p65) in ischemia/reperfusion and vehicle groups was markedly higher than that in the sham and flurbiprofen groups (P < 0.05). No significant differences in the level of p-NF-κB (p65) were found between 5 mg/kg and 10 mg/kg groups (P > 0.05). Taken together, these findings suggested that NF-κB (p65) signaling pathway is most likely involved in the neuroprotective effect of flurbiprofen against cerebral ischemic injury.
Figure 2.
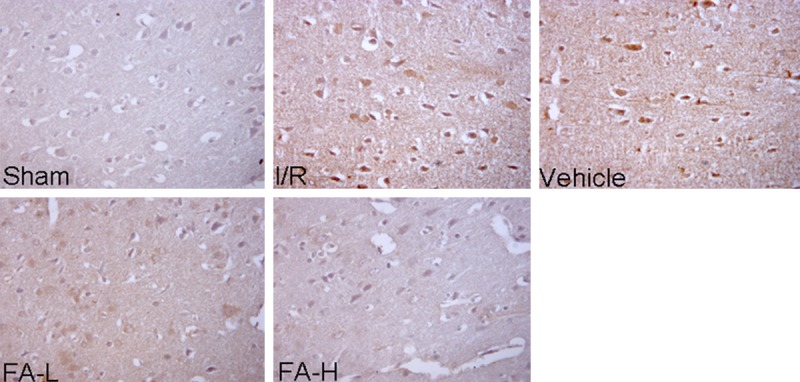
Representative image of immunohistological assay for NF-κB (p65) in the ischemic penumbra tissues of rats. Flurbiprofen inhibits translocation of NF-κB into the nucleus induced by ischemia/reperfusion. In ischemia/reperfusion and vehicle groups, the expression of NF-κB (p65) (brown staining) in ischemic penumbra was detected by immunohistological assay after 24 hours of reperfusion. NF-κB (p65) predominantly located in the nucleus, whereas flurbiprofen can significantly inhibit its translocation from cytoplasm into the nucleus. Magnification, 400×.
Figure 4.
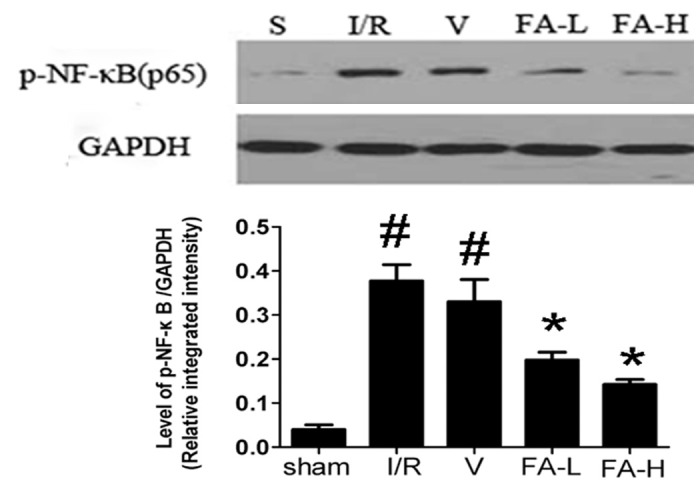
Effect of flurbiprofen on the level of p-NF-κB (p65) in the ischemic penumbra of rats. Western blot showed that the level of p-NF-κB (p65) in nuclear extract of ischemia/reperfusion and vehicle groups was markedly higher compared with sham and flurbiprofen groups. Flurbiprofen can obviously reduce the level of p-NF-κB (p65) in nuclear extract. All data were represented as means ± S.D. For each independent experiment, n = 5. #P < 0.05 vs. sham group, *P < 0.05 vs. ischemia/reperfusion and vehicle group.
Discussion
Stroke is accompanied by a strong inflammatory response that exacerbates injury [23] and is known that postischemic inflammation plays a pivotal role in the delayed progression of brain damage [10]. Microglial activation is detected 2 hours after the onset of ischemia and elevated numbers of neutrophils within brain parenchyma are found by 6 hours of reperfusion [24]. Therapies that prevent the leukocyte infiltration during the acute phase [25] and anti-inflammatory drugs like minocycline and indomethacin are known to induce neuroprotection after ischemia [26,27]. It has been reported that flurbiprofen could provide neuroprotective effect in neuro-degeneration and cerebral ischemic conditions; however, the mechanisms still remain poorly elucidated. Our previous study demonstrated that flurbiprofen protected rat brains against cerebral ischemia/reperfusion injury through inhibition of apoptosis [18]. The major finding of the present study is that flurbiprofen at doses of either 5 mg/kg or 10 mg/kg could inhibit the inflammatory response in the ischemic penumbra of rats exposed to cerebral ischemia/reperfusion by limiting the increase of MPO activity and cytokines gene expression (Figure 3D). Moreover, our findings further revealed that the inhibition of NF-κB was involved in the neuroprotective effect of flurbiprofen against cerebral ischemia/reperfusion (Figure 4).
Some studies suggest that NSAIDS may be neural protective in cerebral ischemic conditions [15,16]. As we all known, animal models over-expressing human COX-2 are more susceptible to injury mediated by cerebral ischemia[28], while COX-2 deficient mice are more resistant [10]. In animal models of stroke, ibuprofen reduced neuronal injury and improved cerebral blood flow and neurological outcome in global ischemia [29] and decreased infarct size in focal ischemia [30]. The COX inhibitor flurbiprofen, which utilizes a lipid microsphere drug delivery system, may facilitate effective target therapy. In addition, the lipid microsphere easily permeates cell membrane and greatly promotes the absorption of the drug and results in shortened onset time. Since it is widely recognized that neutrophil infiltration plays a central role in cerebral ischemia/reperfusion injury and inhibition of neutrophil infiltration and activation during cerebral ischemia/reperfusion phase has become a target of neuroprotection, we directly examined the neutrophil accumulation in the ischemic brain using MPO staining analysis. The present study showed that flurbiprofen might prevent ischemia/reperfusion-induced brain injury and inflammation partially due to inhibition of the increased of MPO activity (Figure 3D).
Accumulating evidence indicates that inflammatory cytokines, TNF-α, IL-1β and IL-6, were found to obviously increase upon ischemia/reperfusion insult. Administration of TNF-α during ischemic brain insult has been shown to augment the injury, as evidenced by increased tissue damage and neurological deficits [25]. As we all known, NF-κB serves as a key transcription factor for a variety of genes involved in stroke inflammatory responses, including TNF-α, IL-1β and IL-8 [31]. NF-κB activation was associated with the phosphorylation of IkBα and the nuclear translocation of p65 [32,33], thus animals exempted from NF-κB activation or deficient in NF-κB were less susceptible to brain ischemia/reperfusion injury [34]. Phosphorylation of p65 at the serine 536 in the transactivation domain enhances the transcriptional activity of NF-κB [35]. Previous studies also suggested that following focal ischemia NF-kB activation in neurons mediates infiltration of neutrophils into the brain parenchyma, and therapeutic paradigms that prevent NF-κB activation induce neuroprotection [36]. In the present study, the levels of mRNAs of TNF-α, IL-1β and IL-6 in the ischemic penumbra was significantly increased in response to ischemic injury, which can be substantially inhibited by flurbiprofen (Figure 3A-C). Furthermore, we found that NF-κB (p65) was predominantly located in the nucleus upon ischemia/reperfusion insult and flurbiprofen treatment can significantly inhibit its translocation from cytoplasm into the nucleus. On the other hand, our data also showed that the level of p-NF-κB (p65) in ischemia/reperfusion group was markedly higher than that in the sham and flurbiprofen groups by western blot assay of nuclear extract (Figure 4). Taken together, these findings revealed that NF-κB (p65) is involved in the neuroprotection of flurbiprofen against ischemic injury.
Several studies have demonstrated that NSAIDs such as sulindac, sodium salicylate, ibuprofen, and flurbiprofen induce anti-inflammatory and antiproliferative effects independent of cyclooxygenase activity and prostaglandinsynthesis inhibition and indicated that NSAIDs may modulate several signaling pathways including p38 kinase, ERK, and PKC [37,38]. Although detailed molecular mechanism(s) that account for flurbiprofen induced neuroprotection in cerebral ischemia injury requires further studies, the current study suggests tischemia-reperfusion injury by reducing inflammatory activity and this effect may be partly due to the inhibition of NF-κB signaling pathway.
In conclusions, our results demonstrate that flurbiprofen protects against cerebral injury by reducing expression of inflammatory cytokines genes and relieving the increase of MPO activity and these effects may be partly due to the inhibition of NF-κB signaling pathway.
Acknowledgements
This work was supported by Shandong Province Natural Science Foundation (ZR2010HM082; ZR2009CM098) and Shandong Province Science and Technology Plan Project (2010GWZ20246).
Disclosure of conflict of interest
None.
References
- 1.Bowen KK, Naylor M, Vemuganti R. Prevention of inflammation is a mechanism of preconditioning-induced neuroprotection against focal cerebral ischemia. Neurochem Int. 2006;49:127–135. doi: 10.1016/j.neuint.2006.02.011. [DOI] [PubMed] [Google Scholar]
- 2.Collino M, Aragno M, Mastrocola R, Gallicchio M, Rosa AC, Dianzani C, Danni O, Thiemermann C, Fantozzi R. Modulation of the oxidative stress and inflammatory response by PPAR-gamma agonists in the hippocampus of rats exposed to cerebral ischemia/reperfusion. Eur J Pharmacol. 2006;530:70–80. doi: 10.1016/j.ejphar.2005.11.049. [DOI] [PubMed] [Google Scholar]
- 3.Chamorro A, Hallenbeck J. The harms and benefits of inflammatory and immune responses in vascular disease. Stroke. 2006;37:291–293. doi: 10.1161/01.STR.0000200561.69611.f8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lin Z, Zhu D, Yan Y, Yu B. Herbal formula FBD extracts prevented brain injury and inflammation induced by cerebral ischemia-reperfusion. J Ethnopharmacol. 2008;118:140–147. doi: 10.1016/j.jep.2008.03.023. [DOI] [PubMed] [Google Scholar]
- 5.del Zoppo GJ, Schmid-Schonbein GW, Mori E, Copeland BR, Chang CM. Polymorphonuclear leukocytes occlude capillaries following middle cerebral artery occlusion and reperfusion in baboons. Stroke. 1991;22:1276–1283. doi: 10.1161/01.str.22.10.1276. [DOI] [PubMed] [Google Scholar]
- 6.Dereski MO, Chopp M, Knight RA, Rodolosi LC, Garcia JH. The heterogeneous temporal evolution of focal ischemic neuronal damage in the rat. Acta Neuropathol. 1993;85:327–333. doi: 10.1007/BF00227730. [DOI] [PubMed] [Google Scholar]
- 7.Huang J, Upadhyay UM, Tamargo RJ. Inflammation in stroke and focal cerebral ischemia. Surg Neurol. 2006;66:232–245. doi: 10.1016/j.surneu.2005.12.028. [DOI] [PubMed] [Google Scholar]
- 8.Adibhatla RM, Hatcher JF. Secretory phospholipase A2 IIA is up-regulated by TNF-alpha and IL-1alpha/beta after transient focal cerebral ischemia in rat. Brain Res. 2007;1134:199–205. doi: 10.1016/j.brainres.2006.11.080. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 9.Iadecola C, Zhang F, Casey R, Nagayama M, Ross ME. Delayed reduction of ischemic brain injury and neurological deficits in mice lacking the inducible nitric oxide synthase gene. J Neurosci. 1997;17:9157–9164. doi: 10.1523/JNEUROSCI.17-23-09157.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Iadecola C, Niwa K, Nogawa S, Zhao X, Nagayama M, Araki E, Morham S, Ross ME. Reduced susceptibility to ischemic brain injury and N-methyl-D-aspartate-mediated neurotoxicity in cyclooxygenase-2-deficient mice. Proc Natl Acad Sci U S A. 2001;98:1294–1299. doi: 10.1073/pnas.98.3.1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scali C, Giovannini MG, Prosperi C, Bellucci A, Pepeu G, Casamenti F. The selective cyclooxygenase-2 inhibitor rofecoxib suppresses brain inflammation and protects cholinergic neurons from excitotoxic degeneration in vivo. Neuroscience. 2003;117:909–919. doi: 10.1016/s0306-4522(02)00839-4. [DOI] [PubMed] [Google Scholar]
- 12.Seegers H, Grillon E, Trioullier Y, Vath A, Verna JM, Blum D. Nuclear factor-kappa B activation in permanent intraluminal focal cerebral ischemia in the rat. Neurosci Lett. 2000;288:241–245. doi: 10.1016/s0304-3940(00)01245-3. [DOI] [PubMed] [Google Scholar]
- 13.Phillips JB, Williams AJ, Adams J, Elliott PJ, Tortella FC. Proteasome inhibitor PS519 reduces infarction and attenuates leukocyte infiltration in a rat model of focal cerebral ischemia. Stroke. 2000;31:1686–1693. doi: 10.1161/01.str.31.7.1686. [DOI] [PubMed] [Google Scholar]
- 14.Yi JH, Park SW, Kapadia R, Vemuganti R. Role of transcription factors in mediating post-ischemic cerebral inflammation and brain damage. Neurochem Int. 2007;50:1014–1027. doi: 10.1016/j.neuint.2007.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McGeer PL, Schulzer M, McGeer EG. Arthritis and anti-inflammatory agents as possible protective factors for Alzheimer’s disease: a review of 17 epidemiologic studies. Neurology. 1996;47:425–432. doi: 10.1212/wnl.47.2.425. [DOI] [PubMed] [Google Scholar]
- 16.Wang C, Liu JL, Sang HF, Lu Y, Dong HL, Xiong LZ. Therapeutic time window of flurbiprofen axetil’s neuroprotective effect in a rat model of transient focal cerebral ischemia. Chin Med J (Engl) 2008;121:2572–2577. [PubMed] [Google Scholar]
- 17.Candelario-Jalil E, Gonzalez-Falcon A, Garcia-Cabrera M, Alvarez D, Al-Dalain S, Martinez G, León OS, Springer JE. Assessment of the relative contribution of COX-1 and COX-2 isoforms to ischemia-induced oxidative damage and neurodegeneration following transient global cerebral ischemia. J Neurochem. 2003;86:545–555. doi: 10.1046/j.1471-4159.2003.01812.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sun B, Chen L, Wei X, Xiang Y, Liu X, Zhang X. The Akt/GSK-3beta pathway mediates flurbiprofen-induced neuroprotection against focal cerebral ischemia/reperfusion injury in rats. Biochem Biophys Res Commun. 2011 Jun 17;409:808–813. doi: 10.1016/j.bbrc.2011.05.095. [DOI] [PubMed] [Google Scholar]
- 19.Salzberg-Brenhouse HC, Chen EY, Emerich DF, Baldwin S, Hogeland K, Ranelli S, Lafreniere D, Perdomo B, Novak L, Kladis T, Fu K, Basile AS, Kordower JH, Bartus RT. Inhibitors of cyclooxygenase-2, but not cyclooxygenase-1 provide structural and functional protection against quinolinic acid-induced neurodegeneration. J Pharmacol Exp Ther. 2003;306:218–228. doi: 10.1124/jpet.103.049700. [DOI] [PubMed] [Google Scholar]
- 20.Wei X, Liu H, Sun X, Fu F, Zhang X, Wang J, An J, Ding H. Hydroxysafflor yellow A protects rat brains against ischemia-reperfusion injury by antioxidant action. Neurosci Lett. 2005;386:58–62. doi: 10.1016/j.neulet.2005.05.069. [DOI] [PubMed] [Google Scholar]
- 21.Schierwagen C, Bylund-Fellenius AC, Lundberg C. Improved method for quantification of tissue PMN accumulation measured by myeloperoxidase activity. J Pharmacol Methods. 1990;23:179–186. doi: 10.1016/0160-5402(90)90061-o. [DOI] [PubMed] [Google Scholar]
- 22.Chen L, Zhang Y, Sun X, Li H, LeSage G, Javer A, Zhang X, Wei X, Jiang Y, Yin D. Synthetic resveratrol aliphatic acid inhibits TLR2-mediated apoptosis and an involvement of Akt/GSK3beta pathway. Bioorg Med Chem. 2009;17:4378–4382. doi: 10.1016/j.bmc.2009.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zheng Z, Yenari MA. Post-ischemic inflammation: molecular mechanisms and therapeutic implications. Neurol Res. 2004;26:884–892. doi: 10.1179/016164104X2357. [DOI] [PubMed] [Google Scholar]
- 24.Barone FC, Arvin B, White RF, Miller A, Webb CL, Willette RN, Lysko PG, Feuerstein GZ. Tumor necrosis factor-alpha. A mediator of focal ischemic brain injury. Stroke. 1997;28:1233–1244. doi: 10.1161/01.str.28.6.1233. [DOI] [PubMed] [Google Scholar]
- 25.Yenari MA, Liu J, Zheng Z, Vexler ZS, Lee JE, Giffard RG. Antiapoptotic and anti-inflammatory mechanisms of heat-shock protein protection. Ann N Y Acad Sci. 2005;1053:74–83. doi: 10.1196/annals.1344.007. [DOI] [PubMed] [Google Scholar]
- 26.Yrjanheikki J, Tikka T, Keinanen R, Goldsteins G, Chan PH, Koistinaho J. A tetracycline derivative, minocycline, reduces inflammation and protects against focal cerebral ischemia with a wide therapeutic window. Proc Natl Acad Sci U S A. 1999;96:13496–13500. doi: 10.1073/pnas.96.23.13496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Monje ML, Toda H, Palmer TD. Inflammatory blockade restores adult hippocampal neurogenesis. Science. 2003;302:1760–1765. doi: 10.1126/science.1088417. [DOI] [PubMed] [Google Scholar]
- 28.Dore S, Otsuka T, Mito T, Sugo N, Hand T, Wu L, Hurn PD, Traystman RJ, Andreasson K. Neuronal overexpression of cyclooxygenase-2 increases cerebral infarction. Ann Neurol. 2003;54:155–162. doi: 10.1002/ana.10612. [DOI] [PubMed] [Google Scholar]
- 29.Patel PM, Drummond JC, Sano T, Cole DJ, Kalkman CJ, Yaksh TL. Effect of ibuprofen on regional eicosanoid production and neuronal injury after forebrain ischemia in rats. Brain Res. 1993;614:315–324. doi: 10.1016/0006-8993(93)91050-3. [DOI] [PubMed] [Google Scholar]
- 30.Antezana DF, Clatterbuck RE, Alkayed NJ, Murphy SJ, Anderson LG, Frazier J, Hurn PD, Traystman RJ, Tamargo RJ. High-dose ibuprofen for reduction of striatal infarcts during middle cerebral artery occlusion in rats. J Neurosurg. 2003;98:860–866. doi: 10.3171/jns.2003.98.4.0860. [DOI] [PubMed] [Google Scholar]
- 31.Cechetto DF. Role of nuclear factor kappa B in neuropathological mechanisms. Prog Brain Res. 2001;132:391–404. doi: 10.1016/S0079-6123(01)32090-3. [DOI] [PubMed] [Google Scholar]
- 32.Pahl HL. Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene. 1999;18:6853–6866. doi: 10.1038/sj.onc.1203239. [DOI] [PubMed] [Google Scholar]
- 33.Waddick KG, Uckun FM. Innovative treatment programs against cancer: II. Nuclear factor-kappaB (NF-kappaB) as a molecular target. Biochem Pharmacol. 1999;57:9–17. doi: 10.1016/s0006-2952(98)00224-x. [DOI] [PubMed] [Google Scholar]
- 34.Schneider A, Martin-Villalba A, Weih F, Vogel J, Wirth T, Schwaninger M. NF-kappaB is activated and promotes cell death in focal cerebral ischemia. Nat Med. 1999;5:554–559. doi: 10.1038/8432. [DOI] [PubMed] [Google Scholar]
- 35.Takeshima E, Tomimori K, Kawakami H, Ishikawa C, Sawada S, Tomita M, Senba M, Kinjo F, Mimuro H, Sasakawa C, Fujita J, Mori N. NF-kappaB activation by Helicobacter pylori requires Akt-mediated phosphorylation of p65. BMC Microbiol. 2009;9:36. doi: 10.1186/1471-2180-9-36. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 36.Williams AJ, Hale SL, Moffett JR, Dave JR, Elliott PJ, Adams J, Tortella FC. Delayed treatment with MLN519 reduces infarction and associated neurologic deficit caused by focal ischemic brain injury in rats via antiinflammatory mechanisms involving nuclear factor-kappaB activation, gliosis, and leukocyte infiltration. J Cereb Blood Flow Metab. 2003;23:75–87. doi: 10.1097/01.WCB.0000039285.37737.C2. [DOI] [PubMed] [Google Scholar]
- 37.Tegeder I, Pfeilschifter J, Geisslinger G. Cyclooxygenase-independent actions of cyclooxygenase inhibitors. FASEB J. 2001;15:2057–2072. doi: 10.1096/fj.01-0390rev. [DOI] [PubMed] [Google Scholar]
- 38.Yoon JB, Kim SJ, Hwang SG, Chang S, Kang SS, Chun JS. Non-steroidal anti-inflammatory drugs inhibit nitric oxide-induced apoptosis and dedifferentiation of articular chondrocytes independent of cyclooxygenase activity. J Biol Chem. 2003;278:15319–15325. doi: 10.1074/jbc.M212520200. [DOI] [PubMed] [Google Scholar]