

Abstract
Objective:
To determine whether oral quinacrine increases survival in sporadic Creutzfeldt-Jakob disease (sCJD).
Methods:
This NIH/National Institute on Aging–funded, double-blinded, placebo-controlled, stratified randomization treatment trial was conducted at the University of California, San Francisco from February 2005 through May 2009 (ClinicalTrials.gov, NCT00183092). Subjects were randomized (50:50) to quinacrine (300 mg daily) or placebo with inpatient evaluations at baseline, and planned for months 2, 6, and 12. Subjects returning for their month-2 visit were offered open-label quinacrine. The primary outcome was survival from randomization to month 2.
Results:
Of 425 patients referred, 69 subjects enrolled, 54 subjects were randomized to active drug or placebo, and 51 subjects with sCJD were included in survival analyses. Survival for the randomized portion of the trial (first 2 months) showed no significant difference between the 2 groups (log-rank statistic, p = 0.43; Cox proportional relative hazard = 1.43, quinacrine compared with placebo, 95% confidence interval = 0.58, 3.53). The quinacrine-treated group, however, declined less on 2 of 3 functional scales, the modified Rankin and Clinical Dementia Rating, than the placebo group during the first 2 months.
Conclusion:
This interventional study provides Class I evidence that oral quinacrine at 300 mg per day does not improve 2-month survival of patients with sCJD, compared with placebo. Importantly, this study shows that double-blinded, placebo-controlled, randomized treatment trials are possible in prion disease. Furthermore, the quantitative data collected on the course of sCJD will be useful for future trials.
Classification of evidence:
This study provides Class I evidence that quinacrine does not improve survival for people with sCJD when given orally at a dose of 300 mg per day for 2 months.
Sporadic Creutzfeldt-Jakob disease (sCJD), the most common form of human prion disease, is a rapidly progressive, uniformly fatal condition. Numerous drugs have been tried and have failed in animal models of prion disease.1 Only one double-blinded, randomized controlled trial, with a primary outcome of cognitive function, has been conducted in sCJD; in that study, flupirtine showed mild benefits in cognition, but no survival benefit.2 The antimalarial drug quinacrine and antipsychotic chlorpromazine were shown to eliminate prions in vitro,3,4 but chlorpromazine likely has a higher toxicity risk at the expected therapeutic dose compared with quinacrine.3 Because quinacrine was used safely for decades to treat cerebral malaria and is known to have excellent CNS penetration,5–8 we offered a compassionate quinacrine protocol to patients with sCJD referred to our center over 34 months. We found that those who chose quinacrine survived significantly longer than those who did not choose quinacrine (see supplementary data on the Neurology® Web site at www.neurology.org). From this experience and case reports, we concluded that quinacrine was also well tolerated and easy to monitor in sCJD, with minimal toxicity.7,9 Encouraged by preliminary observations, we conducted a placebo-controlled, double-blinded treatment study to determine the efficacy of quinacrine on survival in CJD.
METHODS
Standard protocol approvals, registrations, and patient consents.
Study activities were reviewed and approved by our institutional review board and monitored by a data safety monitoring board (DSMB). Written informed consent was obtained from each subject or legally authorized representative. This study is registered at ClinicalTrials.gov, number NCT00183092.
Design.
This study, funded by the NIH/National Institute on Aging, was a phase IIb, single-center, randomized, double-blinded, placebo-controlled study on the effect of oral quinacrine (300 mg per day) on survival in sCJD. The primary research question was: Is there Class I evidence that oral quinacrine at a dose of 300 mg per day for 2 months extends survival in sCJD? The study was conducted at the University of California, San Francisco (UCSF) between February 2005 and May 2009.
Subject selection.
Inclusion and exclusion criteria are shown in table e-1. If eligibility of a subject was in question, at least 3 neurologists reviewed the case and made a consensus decision. Study recruitment and the detailed diagnostic process are discussed in the supplemental data.
Study drug initiation.
Randomization to treatment group (placebo or quinacrine, 50:50) was performed by the pharmacist in variable block sizes (2, 4, and/or 6) and stratified by Barthel Index scores (≤30 or >30) (see supplemental data for details on study drug assignment). After appropriate baseline laboratory safety testing (e-Methods), subjects received a loading dose of study drug: five 200-mg doses, administered orally every 6 hours and supplemented with 1 g of sodium bicarbonate to prevent gastrointestinal upset. After completing the loading dose, subjects began the study drug at 100 mg, 3 times daily. After monitoring for 24 hours, they were discharged home on study drug.
Study monitoring and safety.
Telephone follow-up calls with caregivers occurred every 2 weeks for the first 2 months, during which study personnel collected modified Barthel Index,10 Rankin Scale,11 Clinical Dementia Rating (CDR) scale,12 and Neuropsychiatric Inventory (NPI) scores,13 and screened for quinacrine toxicity or side effects. Specimen collection kits (urine and blood) for safety monitoring were shipped to caregivers monthly, for remote safety monitoring (table e-2). Clinically significant adverse side effects, as judged by study physicians and the study pharmacist, resulted in a reduction or temporary discontinuation of study drug. All unexpected side effects, adverse effects (AEs), and serious AEs (SAEs) were reported to our institutional review board. The DSMB reviewed study safety and progress. A midstudy interim analysis was performed (see statistical analysis section and e-Methods).
Follow-up.
Subjects were reevaluated and baseline testing was repeated at UCSF at months 2, 6, and 12 when possible. Subjects returning to UCSF for their month-2 visit were given the option of switching to open-label quinacrine (see e-Methods for rationale). Subjects returning at month 2 who opted for open-label quinacrine were switched to oral quinacrine (at 100 mg, 3 times daily) without a loading dose and were monitored for 24 hours. Participants returning at month 2 could also choose to remain on study drug. Subjects not returning at month 2 stayed on the study drug.
After month 2, participants, including those who discontinued treatment, were followed on a monthly basis until death or until study cessation. Life-extending measures (e.g., intubation, feeding tubes) were recorded (as potential surrogate outcomes for death). Autopsies were coordinated by the UCSF Memory & Aging Center. Most brains were examined at UCSF and less frequently by the National Prion Disease Pathology Surveillance Center (NPDPSC; Cleveland, OH). The NPDPSC was provided brain samples from all autopsies and performed prion typing.
Outcome measures.
The primary outcome measure was survival time from randomization to month 2, when the randomized controlled portion of the trial ended. We also conducted secondary survival analyses of survival after the month-2 time point, including the nonrandomized portion of the study. For subjects who were still alive at last follow-up (month 12), date of last contact was used for survival analysis. Life-extending measures, such as feeding tubes and intubation, were recorded for all subjects as possible confounds in the survival analysis. Other secondary measures included scores on functional tests and rating scales (modified Barthel Index, Rankin, CDR, Mini-Mental State Examination [MMSE], and NPI), a quantifiable neurologic examination, neurocognitive testing (see e-Methods for tests), EEG, CSF “biomarkers,” and brain MRI.
Statistical analysis.
Sample size.
We intended to randomize 60 serial subjects with sCJD over 3 years (see e-Results for sample size calculation). Based on survival analysis from compassionate-use quinacrine (supplemental data), we estimated that 30 subjects with sCJD per group would yield an 80% power (α 0.05 level) to detect a doubling of mean survival (0.9 months) from randomization in the quinacrine group compared with the placebo group. Because preliminary data suggested that the modified Barthel score predicted survival independent of treatment, we stratified the randomization by block design based on this score.
Baseline demographic variables, various functional tests and rating scales (MMSE, CDR–Sum of Boxes [CDR-SB], modified Barthel Index, Rankin, and NPI), and neuropsychological testing were compared between treatment groups using independent sample t tests for continuous variables and Fisher exact test for noncontinuous variables (tables 1 and 2).
Table 1.
Baseline characteristics by treatment group
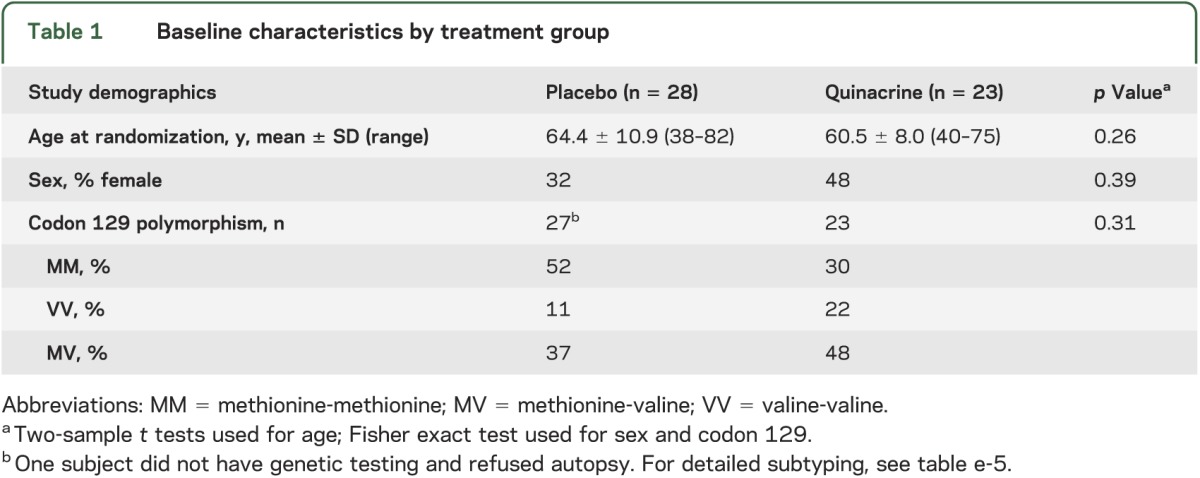
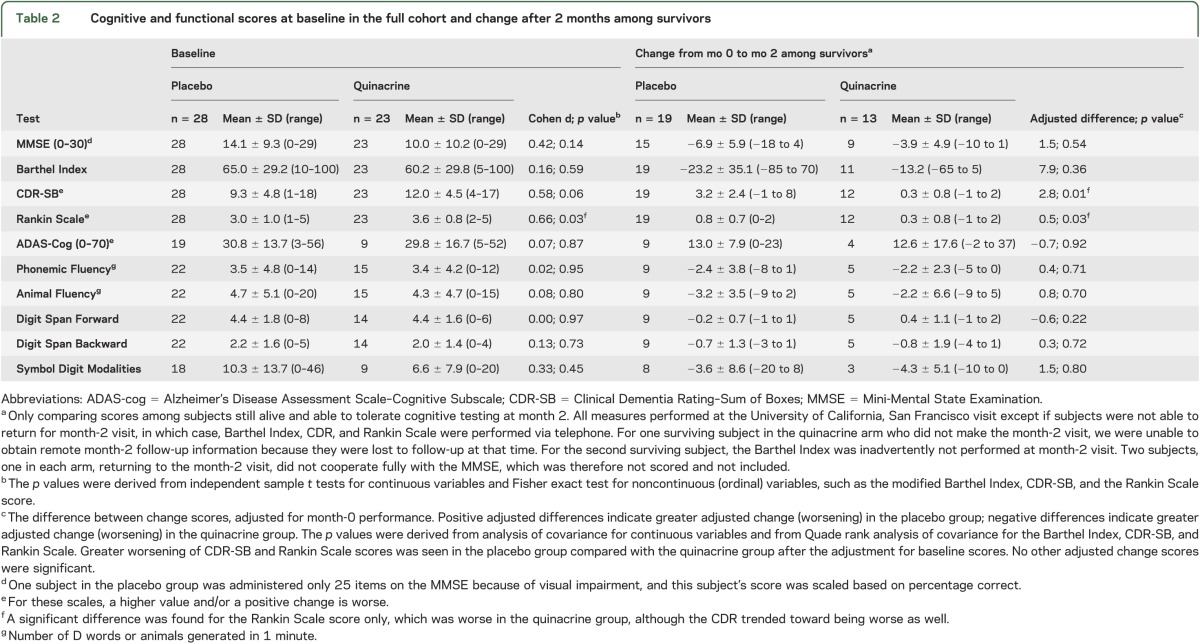
Table 2.
Cognitive and functional scores at baseline in the full cohort and change after 2 months among survivors
Primary outcome analyses of the randomized portion of the trial.
Survival during the first 2 months between treatment groups was analyzed using a log-rank test and associated Cox proportional hazards model, following the intention-to-treat principle. We planned an interim analysis of the survival data halfway through the study using the method of O'Brien and Fleming14 and an overall error rate of 0.05. These primary statistical analyses were performed using Stata.15
Secondary analysis of survival.
Because subjects returning to UCSF for their month-2 visit were able to choose whether or not to start open-label quinacrine, this eliminated true randomization from this point. We continued accumulating survival data. Survival from randomization to death or end of study was analyzed using a Cox proportional hazards model with a time-dependent treatment group variable.
Secondary outcome analysis.
Among the subjects who survived to month 2, we compared changes in the values of the MMSE, CDR-SB, modified Barthel Index, Rankin Scale, and neuropsychological test scores between the baseline and follow-up visits using parametric analysis of covariance for continuous variables and Quade nonparametric analysis of covariance for ordinal variables, adjusting for baseline performance as a covariate, and using PASW 17.0 for Windows (SPSS Inc., Chicago, IL) (table 2).
RESULTS
The study enrolled the first subject in April 2005 and stopped enrollment in January 2009. Subjects were formally followed through the study protocol through May 1, 2009 (6 surviving), although data on subject survival were collected through October 15, 2010. In total, 425 patients were referred to the study (figure 1, CONSORT study flow16,17; table e-3).
Figure 1. Quinacrine CJD treatment trial flowchart.

a Ineligible includes referred subjects who did not meet inclusion criteria (e.g., had an alternative diagnosis, were too advanced to participate [could not follow simple commands and swallow], did not live in the United States or Canada, had no caregiver, or did not fulfill other inclusion criteria). Many potential or probable sCJD referrals did not want to participate in research, did not respond to follow-up, died before evaluation, were unable to travel, or did not wish to prolong life. b Includes one subject who was given a clinical diagnosis of possible Sprue and did not wish to have a brain biopsy to confirm CJD. The subject was therefore not randomized. Autopsy later revealed a diagnosis of sCJD. c Three subjects, who were originally randomized to the quinacrine arm, were clinically diagnosed with probable sCJD, but genetic results revealed one fCJD (D178N) case, one case with 9-OPRI mutation, and one fCJD (V180I) case. d Although formal study follow-up was discontinued on May 1, 2009, some families remained in contact with the research team after that date. These numbers reflect the number of subjects that were still alive at last contact as of October 15, 2010. e Two subjects, one in each arm, were still alive as of the last follow-up, but were both receiving life-extending measures. CJD = Creutzfeldt-Jakob disease; fCJD = familial CJD; 9-OPRI = 9 octapeptide repeat insertion; sCJD = sporadic CJD; UCSF = University of California, San Francisco.
Subjects: Enrollment, demographics, and baseline characteristics.
Sixty-nine subjects consented for the study. Subjects came from across the United States, as well as Canada, with a plurality from California (figure e-1). Eighteen enrolled subjects were considered screen failures and not randomized (see figure 1, table e-4). Fifty-four subjects were randomized to start study drug, but because PRNP analysis later identified 3 subjects who carry PrP gene mutations, only 51 subjects with sCJD were included in the survival analysis: 28 in the placebo arm and 23 in the quinacrine arm. No significant differences were found between treatment groups in baseline characteristics, including codon 129 polymorphism, except that the Rankin Scale scores were worse in the quinacrine-treated group (tables 2 and e-5). The 2 groups did not differ significantly on any of the baseline cognitive measures (table 2). Nevertheless, because fewer subjects in the quinacrine treatment group were able to tolerate the full battery of neuropsychological tests at baseline and scored an average of 4 MMSE points lower than the placebo group, a baseline difference in cognitive functioning between the groups cannot be ruled out (see supplemental information).
Primary outcome measure.
Per protocol, a midterm survival and adverse event analysis was conducted and no significant survival difference was found. The DSMB recommended study continuation (see e-Results).
Primary analyses.
Thirteen of 23 quinacrine subjects (57%) and 19 of 28 placebo subjects (68%) survived to month 2. Figure 2A presents Kaplan-Meier survival curves for both groups for the 2-month randomized, controlled portion of the trial. There was no significant difference in survival between the groups (log-rank statistic, p = 0.43; Cox proportional relative hazard = 1.43, quinacrine compared with placebo, 95% confidence interval [CI] = 0.58, 3.53). The hazard ratio itself suggests a survival benefit of placebo over quinacrine, but does not achieve statistical significance; additionally, the wide CI includes values of the hazard ratio less than 1, indicating a survival benefit for quinacrine over placebo as well.
Figure 2. Kaplan-Meier survival analysis from baseline to month 2 and to death.

(A) Kaplan-Meier survival analysis from month 0 (baseline) to month 2 for 51 randomized subjects with sCJD (placebo, n = 28; quinacrine, n = 23). These differences were not statistically significant (log-rank statistic, p = 0.43; Cox proportional relative hazard = 1.43 quinacrine compared with placebo, 95% confidence interval = 0.58, 3.53). (B) Kaplan-Meier survival analysis from baseline to death or date of life-extending measures (censored) or to date of last contact (censored) for 51 randomized subjects with sCJD. Four groups were based on the treatment arm at randomization and the treatment arm chosen at month 2, to obtain the placebo-placebo group (n = 5); the placebo-quinacrine group (n = 14); the quinacrine-placebo group (n = 3); and the quinacrine-quinacrine group (n = 10). Because groups in this figure are not based solely on randomization, but include subject choice at month 2, we did not conduct a formal statistical test of differences in survival. Note that these curves might appear to suggest a greater benefit of quinacrine than the data indicate, as the subjects who went on open-label quinacrine were survivors (and most in good enough condition to return for their month-2 visit) from the placebo and quinacrine groups. sCJD = sporadic Creutzfeldt-Jakob disease.
Secondary survival analysis (after month 2).
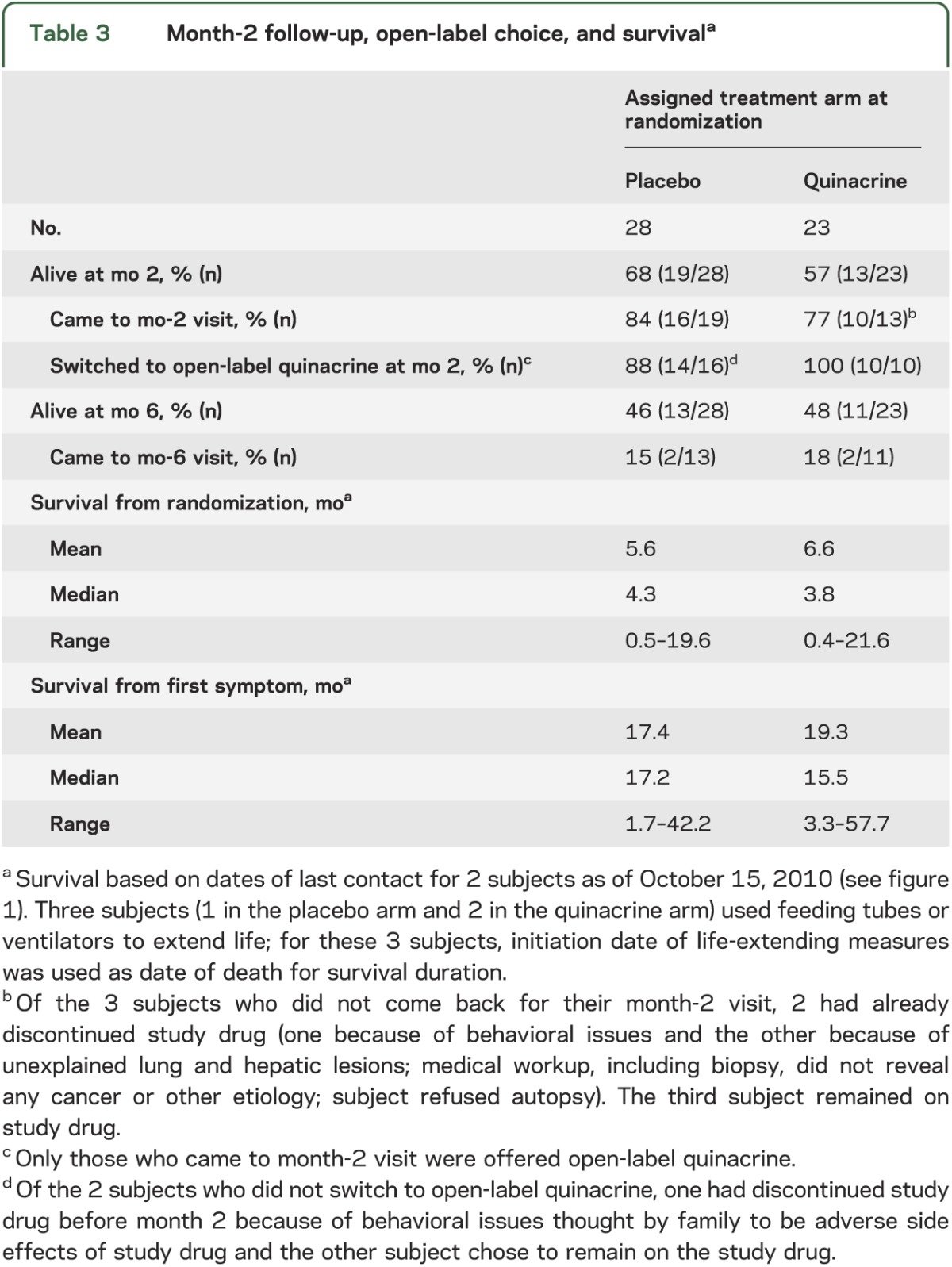
Twenty-six of the 32 surviving subjects (81%; 16/19 in the placebo group and 10/13 in the quinacrine group) returned to UCSF for their month-2 visits (table 3). The entire eligible quinacrine group and all but 2 (14/16) of the eligible placebo group opted to start open-label quinacrine. Because the study was no longer randomized after month 2, we assessed survival differences using time-dependent treatment choices, and censored the 3 subjects who chose life-extending measures. The survival times of subjects who chose quinacrine did not differ significantly from those who did not (Cox proportional hazards model with time-dependent treatment: relative hazard = 0.86; 95% CI = 0.44, 1.70; p = 0.67). Adjustment for baseline modified Barthel Index, MMSE or CDR scores, as well as sex, changed the treatment relative hazard only minimally, whereas adjustment for baseline Rankin Scale score reduced the relative hazard to 0.70 (95% CI = 0.36, 1.36). Thus, some of the uncorrected relative hazard for quinacrine was due to the baseline differences between the 2 groups; these results, however, do not indicate statistically significant increased survival for subjects who chose quinacrine compared with those who did not at month 2. To give an impression of the survival experience of the subjects, we divided subjects into 4 groups based on their treatment assignment before and after month 2 and plotted their Kaplan-Meier survival curves (figure 2B).
Table 3.
Month-2 follow-up, open-label choice, and survivala
Secondary outcome measures analysis.
In surviving patients at month 2, controlling for baseline performance, the quinacrine-treated group showed less decline than the placebo group on the CDR-SB and the modified Rankin Scale, but not the Barthel Index. For subjects able to undergo neurocognitive testing at month 2, controlling for baseline performance, there were no significant differences between groups in the change in scores between month 0 and month 2 on any of the cognitive tests (table 2; all p values >0.05).
Adverse events.
We examined AEs and SAEs in 2 timeframes: 1) through month 2, placebo arm vs quinacrine arm, and 2) after month 2, divided into those occurring within, vs outside of, 30 days of taking quinacrine. The number of AEs and the number of subjects with AEs through month 2 were similar between arms; the types of AEs differed, however. For example, elevated liver function tests were only seen in the quinacrine arm (table e-6). Through month 2, each arm had 3 SAEs, all different between arms (table e-7). Only one SAE, severe gastrointestinal distress, was assessed to be possibly due to quinacrine. Twenty-two percent of quinacrine arm and 18% of placebo arm subjects required dose reduction through month 2. After month 2, there were many more AEs and subjects with AEs within 30 days of taking quinacrine; more than half involved elevated liver function tests (12/27) or gastrointestinal distress (5/27). Of the 6 SAEs after month 2, 5 occurred within 30 days of taking quinacrine, but only one (behavioral change) was determined to be possibly or likely due to quinacrine. Overall, quinacrine was reasonably well tolerated.
DISCUSSION
This interventional study provides Class I evidence that oral quinacrine (300 mg per day) over a 2-month period did not prolong survival of subjects with sCJD. Time-dependent survival analysis corroborated findings of the randomized portion of the trial, showing no difference in survival based on whether subjects were receiving quinacrine or not. Although quinacrine has been shown to eliminate prions in vitro, the present findings do not support a favorable survival response in human prion disease, consistent with observational analyses of quinacrine in human prion disease18–20 and animal studies.21–23
The failure of quinacrine to extend survival in our study might be attributable to several reasons, including insufficient concentrations in the appropriate cellular compartment, inefficacy in vivo, as well as study design and limitations. Whereas our initial findings in cell culture3,24 and the long experience with quinacrine as antimalarial7,25,26 made it an exciting candidate for CJD treatment, our findings in humans are consistent with the disappointing results of several early animal studies.21,22,27 From our own and other studies,24,28–30 we concluded that a conformational change in the RML prion strain allowed it to replicate in the presence of high levels of quinacrine31 (see e-Discussion).
Quinacrine and some analogs have been studied by binding to recombinant human PrP (recHuPrP) (90–231) in the presence of SH-SY5Y cells.32 In these studies, the recHuPrP (90–231) was denatured at 53°C for 1 hour and then exposed to quinacrine or an analog. Quinacrine was more effective in rendering recHuPrP (90–231) susceptible to limited proteinase K digestion than most analogs, but was only marginally effective in protecting SH-SY5Y cells from the toxic effects of recHuPrP (90–231). Interestingly, one quinacrine analog, 6-chloro-2-methoxy-9-{[(1S,9aR)-(octahydro-2H-quinolizin-1-yl)methyl]amino}acridine (Q3), was quite effective in preventing the toxicity of recHuPrP (90–231).
It is possible that even though adequate total brain quinacrine concentrations were achieved,23,33 quinacrine levels in the extracellular compartment, where PrPSc resides on cell membranes, were too low.21 Although the anti-prion mechanism of quinacrine action in ScN2a cells is unknown, some investigators argue that quinacrine works through binding to PrPC.34 In other studies, we found that quinacrine inhibits PrPSc in dividing ScN2a cells but not in nondividing cells.23 Whether or not reductions in PrPSc levels in nondividing cells will be predictive of efficacy in vivo for putative prion therapeutics remains to be established.
It is notable that the vast majority of neurons in the adult CNS do not divide. Whether drug-resistant prions are more likely to emerge in nondividing cells is unknown, but conformational transformation in PrPSc followed by selection in cell culture is now well documented.31,35 Curiously, 2 subjects, both in the quinacrine arm, were clearly thought to have improved by their caregivers and study staff during the first 2 months of the study; both subjects also had slight improvement on scales and cognitive testing (see supplementary material). Because these and other subjects eventually declined after month 2, these improvements might have been due to the psychoactive side effects of quinacrine5,6,36 or due to a temporary reduction in prion load, as has been shown in mouse prion models treated with quinacrine. It appears that quinacrine, both in vitro and in vivo, might reduce PrPSc initially, but continuous treatment results in strain selection of PrPSc that is resistant to the effects of quinacrine.23
Although quinacrine did not prolong survival in this study, the quinacrine group showed less decline in 2 functional outcomes during the initial 2-month randomized phase of the study. The clinical significance of this is unclear as there were no differences in decline on a third functional scale or on any cognitive measures. Although we believe that prolonged survival is probably the most important first outcome for treating prion disease, preservation of or improvement in cognition or function might be viable targets in future CJD treatment trials. In fact, the flupirtine treatment trial in sCJD used cognition as the primary outcome.2
There were several limitations to this study. First, the study sample size was small although we enrolled a large number of subjects (n = 69) for such a rare disease. The final sample size of 51 subjects led to wide variability in estimates of the survival difference. Second, the study design, allowing subjects who came back to UCSF for their month-2 visits to switch to open-label quinacrine, restricted the true randomization period to the first 2 months. There also were significant limitations to the matching of groups after the month-2 time point. Although we adjusted for the fact that some subjects switched treatment arms at month 2 by using a time-dependent survival analysis, these data are influenced by many confounding factors. For example, only subjects who returned for their month-2 visit could elect to switch to open-label quinacrine. This group consisted of subjects who were able to travel, whereas most subjects who did not return probably were more impaired. Third, the treatment arms were not as matched as we anticipated; the quinacrine arm had significantly lower Rankin Scale scores. This difference was subsequently discovered to be due to a procedural error (see supplemental data for details). We do not believe that this significantly affected the study conclusions.
This represents the first report of a Class I treatment study of survival in sCJD. Although the outcome was negative, this study has shown that appropriate randomized controlled trials can be performed in rare, rapidly progressive, uniformly fatal neurodegenerative diseases. Methodologies are now in place for future trials. As with MRC PRION1,18 which was an observational trial of quinacrine in prion disease, we now have data quantifying the course of prion disease (manuscript in preparation), which will be essential for future prion trials.
As shown by baseline characteristics, our patients had significant cognitive and functional impairment at randomization. Earlier diagnosis of prion disease is probably necessary for potential treatments to have benefit and thus should be a goal of prion research along with finding new treatments. Given the rarity of prion disease, multinational trials, possibly with other study designs such as delayed start, should be considered in the future for faster enrollment and data collection.
Supplementary Material
ACKNOWLEDGMENT
The authors thank members of the DSMB (Wade Smith, MD, PhD [Chair]; Cheryl Jay, MD; James Rubenstein, MD; Dan Moore, PhD; and Timothy Davern, MD); staff of the UCSF CTSI CRC and Core Laboratory; Joan Hilton, PhD, for assistance with study randomization and midpoint analyses; referring physicians; the NPDPSC for assistance with PRNP genetic analyses, prion typing, and pathologic analyses; the Hillblom Foundation; the Sherman Fairchild Foundation; the Fight for Mike Homer Program; the Rainwater Charitable Foundation; the CJD Foundation for supporting the patients and families; and most importantly, the patients and their families.
GLOSSARY
- AE
adverse effect
- CDR
Clinical Dementia Rating
- CDR-SB
Clinical Dementia Rating–Sum of Boxes
- CI
confidence interval
- CJD
Creutzfeldt-Jakob disease
- DSMB
data safety monitoring board
- MMSE
Mini-Mental State Examination
- NPDPSC
National Prion Disease Pathology Surveillance Center
- NPI
Neuropsychiatric Inventory
- recHuPrP
recombinant human PrP
- SAE
serious adverse effect
- sCJD
sporadic Creutzfeldt-Jakob disease
- UCSF
University of California, San Francisco
Footnotes
Supplemental data at www.neurology.org
AUTHOR CONTRIBUTIONS
M.D. Geschwind: study concept and design, acquisition of data, analysis and interpretation, critical drafting and revision, study supervision. A.L. Kuo: acquisition of data, analysis and interpretation, critical drafting and revision, study supervision. K.S. Wong: acquisition of data, analysis. A. Haman: acquisition of data and analysis and interpretation, critical drafting and revision, study supervision. G. Devereux: acquisition of data, critical drafting and revision, study supervision. B.J. Raudabaugh, D.Y. Johnson, and C.C. Torres-Chae: acquisition of data, analysis. R. Finley: study concept and design, acquisition of data. P. Garcia: study concept and design, acquisition of data, analysis. J.N. Thai: acquisition of data, analysis. H.Q. Cheng: study concept and design, acquisition of data, critical drafting and revision. J.M. Neuhaus: study concept and design, analysis and interpretation, critical drafting and revision. S.A. Forner: acquisition of data, analysis and interpretation. J. Duncan: study concept and design, acquisition of data. K.L. Possin: analysis and interpretation, critical drafting and revision. S.J. DeArmond: study concept and design, acquisition of data. S.B. Prusiner: study concept and design, analysis and interpretation, critical drafting and revision. B.L. Miller: study concept and design, acquisition of data, analysis and interpretation, critical drafting and revision, study supervision.
STUDY FUNDING
Supported by NIH/National Institute on Aging (NIA) P01 AG02160, P0 AG010770, NIH/NIA K23 AG021989, R37 AG031220, and R01-AG031189. M.D.G. was funded by NIH/NIA K23 AG021989 and R01-AG031189.
DISCLOSURE
M. Geschwind receives grant support from the NIH/NIA, the Homer Family Fund, and has nothing to disclose related to this report. He has served as a consultant for Lundbeck Inc., MedaCorp, Gerson-Lehman Group, and The Council of Advisors, Guidepoint Global, and Neurophage. A. Kuo, K. Wong, A. Haman, G. Devereux, B. Raudabaugh, D. Johnson, and C. Torres-Chae report no disclosures. R. Finley has received funding from Novartis and the Speakers Bureau. P. Garcia received research grants from Medtronics, Inc., and UCB Pharma. J. Thai and H. Cheng report no disclosures. J. Neuhaus is funded by grants from the NIH as well as Forest Research Institute. S. Forner, J. Duncan, K. Possin, and S. DeArmond report no disclosures. S. Prusiner is funded by NIH grants (AG021601, AG02132, AG010770, AG031220). B. Miller receives grant support from the NIH/NIA and has nothing to disclose related to this report. Dr. Miller serves as a consultant for TauRx, Allon Therapeutics, and Siemens Medical Solutions. He has also received a research grant from Novartis. He is on the Board of Directors for the John Douglas French Foundation for Alzheimer's Research and for the Larry L. Hillblom Foundation. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Korth C, Peters PJ. Emerging pharmacotherapies for Creutzfeldt-Jakob disease. Arch Neurol 2006;63:497–501 [DOI] [PubMed] [Google Scholar]
- 2.Otto M, Cepek L, Ratzka P, et al. Efficacy of flupirtine on cognitive function in patients with CJD: a double-blind study. Neurology 2004;62:714–718 [DOI] [PubMed] [Google Scholar]
- 3.Korth C, May BC, Cohen FE, Prusiner SB. Acridine and phenothiazine derivatives as pharmacoptherapeutics for prion disease. Proc Natl Acad Sci USA 2001;98:9836–9841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Doh-Ura K, Iwaki T, Caughey B. Lysosomotropic agents and cysteine protease inhibitors inhibit scrapie-associated prion protein accumulation. J Virol 2000;74:4894–4897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Engel GL. Quinacrine effects on the central nervous system. JAMA 1966;197:505. [PubMed] [Google Scholar]
- 6.Gaskill HS, Fitz-Hugh T. Toxic psychosis following Atabrine. Bull US Army Med Dept 1945;86:63–69 [Google Scholar]
- 7.Wallace D. Antimalarial therapies. In: Wallace D, Hahn BH, editors. Dubois' Lupus Erythematosus, 5th ed. Baltimore: Williams & Wilkins; 1997:1117–1139 [Google Scholar]
- 8.Evans RL, Khalid S, Kinney JL. Antimalarial psychosis revisited. Arch Dermatol 1984;120:765–767 [PubMed] [Google Scholar]
- 9.Nakajima M, Yamada T, Kusuhara T, et al. Results of quinacrine administration to patients with Creutzfeldt-Jakob disease. Dement Geriatr Cogn Disord 2004;17:158–163 [DOI] [PubMed] [Google Scholar]
- 10.Shah S, Vanclay F, Cooper B. Improving the sensitivity of the Barthel Index for stroke rehabilitation. J Clin Epidemiol 1989;42:703–709 [DOI] [PubMed] [Google Scholar]
- 11.van Swieten JC, Koudstaal PJ, Visser MC, Schouten HJ, van Gijn J. Interobserver agreement for the assessment of handicap in stroke patients. Stroke 1988;19:604–607 [DOI] [PubMed] [Google Scholar]
- 12.Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules [see comments]. Neurology 1993;43:2412–2414 [DOI] [PubMed] [Google Scholar]
- 13.Cummings JL, Mega M, Gray K, Rosenberg-Thompson S, Carusi DA, Gornbein J. The Neuropsychiatric Inventory: comprehensive assessment of psychopathology in dementia. Neurology 1994;44:2308–2314 [DOI] [PubMed] [Google Scholar]
- 14.O'Brien PC, Fleming TR. A multiple testing procedure for clinical trials. Biometrics 1979;35:549–556 [PubMed] [Google Scholar]
- 15.Stata Statistical Software [computer program]. Version release 11.1. College Station, TX: Stata Corporation; 2010 [Google Scholar]
- 16.Altman DG, Schulz KF, Moher D, et al. The revised CONSORT statement for reporting randomized trials: explanation and elaboration. Ann Intern Med 2001;134:663–694 [DOI] [PubMed] [Google Scholar]
- 17.Moher D, Schultz KF, Altman DG. The CONSORT statement: revised recommendations for improving the quality of reports of parallel-group randomised trials. Lancet 2001;357:1191–1194 [PubMed] [Google Scholar]
- 18.Collinge J, Gorham M, Hudson F, et al. Safety and efficacy of quinacrine in human prion disease (PRION-1 study): a patient-preference trial. Lancet Neurol 2009;8:334–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Haik S, Brandel JP, Salomon D, et al. Compassionate use of quinacrine in Creutzfeldt-Jakob disease fails to show significant effects. Neurology 2004;63:2413–2415 [DOI] [PubMed] [Google Scholar]
- 20.Geschwind MD. Clinical trials for prion disease: difficult challenges, but hope for the future. Lancet Neurol 2009;8:304–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gayrard V, Picard-Hagen N, Viguie C, Laroute V, Andreoletti O, Toutain PL. A possible pharmacological explanation for quinacrine failure to treat prion diseases: pharmacokinetic investigations in an ovine model of scrapie. Br J Pharmacol 2005;144:386–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Collins SJ, Lewis V, Brazier M, Hill AF, Fletcher A, Masters CL. Quinacrine does not prolong survival in a murine Creutzfeldt-Jakob disease model. Ann Neurol 2002;52:503–506 [DOI] [PubMed] [Google Scholar]
- 23.Ghaemmaghami S, Ahn M, Lessard P, et al. Continuous quinacrine treatment results in the formation of drug-resistant prions. PLoS Pathog 2009;5:e1000673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.May BC, Fafarman AT, Hong SB, et al. Potent inhibition of scrapie prion replication in cultured cells by bis-acridines. Proc Natl Acad Sci USA 2003;100:3416–3421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rollo IM. Drugs used in the chemotherapy of malaria. In: Goodman LS, Gilman A, editors. The Pharmacological Basis of Therapeutics, 4th ed. London: Macmillan Company; 1970:1095–1124 [Google Scholar]
- 26.Van Dyke K, Lantz C, Szustkiewicz C. Quinacrine: mechanisms of antimalarial action. Science 1970;169:492–493 [PubMed] [Google Scholar]
- 27.Barret A, Tagliavini F, Forloni G, et al. Evaluation of quinacrine treatment for prion diseases. J Virol 2003;77:8462–8469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ryou C, Legname G, Peretz D, Craig JC, Baldwin MA, Prusiner SB. Differential inhibition of prion propagation by enantiomers of quinacrine. Lab Invest 2003;83:837–843 [DOI] [PubMed] [Google Scholar]
- 29.Huang Y, Okochi H, May BC, et al. Quinacrine is mainly metabolized to mono-desethyl quinacrine by CYP3A4/5 and its brain accumulation is limited by P-glycoprotein. Drug Metab Dispos 2006;34:1136–1144 [DOI] [PubMed] [Google Scholar]
- 30.Satoh K, Shirabe S, Eguchi K, et al. Toxicity of quinacrine can be reduced by co-administration of P-glycoprotein inhibitor in sporadic Creutzfeldt-Jakob disease. Cell Mol Neurobiol 2004;24:873–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ghaemmaghami S, Watts JC, Nguyen HO, Hayashi S, DeArmond SJ, Prusiner SB. Conformational transformation and selection of synthetic prion strains. J Mol Biol 2011;413:527–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Villa V, Tonelli M, Thellung S, et al. Efficacy of novel acridine derivatives in the inhibition of hPrP90-231 prion protein fragment toxicity. Neurotox Res 2011;19:556–574 [DOI] [PubMed] [Google Scholar]
- 33.Yung L, Huang Y, Lessard P, et al. Pharmacokinetics of quinacrine in the treatment of prion disease. BMC Infect Dis 2004;4:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vogtherr M, Grimme S, Elshorst B, et al. Antimalarial drug quinacrine binds to C-terminal helix of cellular prion protein. J Med Chem 2003;46:3563–3564 [DOI] [PubMed] [Google Scholar]
- 35.Li J, Browning S, Mahal SP, Oelschlegel AM, Weissmann C. Darwinian evolution of prions in cell culture. Science 2010;327:869–872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wallace D. Antimalarial agents and lupus. Rheum Dis Clin North Am 1994;20:243–263 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.