

Abstract
SIRT1 is a NAD+-dependent deacetylase that plays important roles in many cellular processes. SIRT1 activity is uniquely controlled by a C-terminal regulatory segment (CTR). Here we present crystal structures of the catalytic domain of human SIRT1 in complex with the CTR in an open apo form and a closed conformation in complex with a cofactor and a pseudo-substrate peptide. The catalytic domain adopts the canonical sirtuin fold. The CTR forms a β hairpin structure that complements the β sheet of the NAD+-binding domain, covering an essentially invariant, hydrophobic surface. The apo form adopts a distinct open conformation, in which the smaller subdomain of SIRT1 undergoes a rotation with respect to the larger NAD+-binding subdomain. A biochemical analysis identifies key residues in the active site, an inhibitory role for the CTR, and distinct structural features of the CTR that mediate binding and inhibition of the SIRT1 catalytic domain.
Keywords: X-ray crystallography, conformational plasticity, enzyme peptide substrate interaction, mutational analysis, enzyme regulation
Introduction
Sirtuins are a large family of protein-modifying enzymes highly conserved throughout bacteria, archaea, and eukaryotes. The founding member of the sirtuin family, silent information regulator 2 (Sir2), was first identified through a genetic screen in Saccharomyces cerevisiae as necessary for silencing of the mating-type information locus, HM.1 Three additional sirtuin genes were subsequently discovered in S. cerevisiae,2,3 the products of which collectively act along with Sir2 to silence transcription at telomeres and rDNA clusters as well as HM loci.4–7 Later work showed Sir2 and its homologs to function primarily as nicotinamide adenine dinucleotide (NAD+)-dependent deacetylases,8–10 with specific family members reported to possess mono-ADP ribosyl transferase,11–16 demalonylase, or desuccinylase activity.17
In the sirtuin deacetylation reaction, the substrate acetyl group is transferred onto the ribose moiety of NAD+, generating nicotinamide (NAM) and 2′-O-acetyl-adenosine disphosphoribose (2-O′-AADPR).18 While the details of this reaction are still a matter of debate, a proposed mechanism that proceeds through an adenosine diphosphoribose (ADPR)-peptidyl-imidate intermediate is consistent with available biochemical data.19 Crystal structures of a number of sirtuin family members have provided insight into substrate and cofactor binding as well as catalysis.11,20–40
The mammalian genome encodes seven different sirtuins (SIRT1-7), with varying subcellular localizations and chemical activities.17,41 The direct human homolog of S. cerevisiae Sir2 is SIRT1. SIRT1 deacetylates a wide range of substrates, including p53, NF-κB, FOXO transcription factors, and PGC-1α, with roles in cellular processes ranging from energy metabolism to cell survival.42 As such, SIRT1 is implicated in a wide range of human diseases and is a prominent therapeutic target.
Despite progress over the last decade, relatively little is known about the regulatory mechanism of SIRT1. Like all sirtuins, SIRT1 is strongly inhibited by NAM through a base-exchange mechanism that reforms cleaved NAD+.43 Active Regulator of SIRT1 (AROS) and Deleted in Breast Cancer 1 (DBC1) have been identified as endogenous proteins that promote or inhibit SIRT1 activity, respectively.44–46 Additionally, various regions in the long and mostly unstructured N- and C-termini that flank the SIRT1 catalytic domain have been shown to affect SIRT1 deacetylation activity.47,48
To shed light on the regulation of human SIRT1 activity, we have determined the crystal structure of SIRT1 in complex with its C-terminal regulatory segment (CTR) in its apo form and in a quaternary complex with the NAD+ hydrolysis product ADPR and a substrate-mimicking peptide at 2.65 Å and 1.85 Å resolution, respectively. The structures reveal that the CTR binds at the lower edge of the larger NAD+-binding domain, complementing the central parallel β sheet of its Rossmann fold. The substrate-bound closed state completely encapsulates the cofactor and forms a binding site with a hydrophobic tunnel for the substrate residue that leads to a shielded active site in the interior of the enzyme. The overall conformation and mode of substrate binding confirms previous predictions of how human SIRT1 interacts with peptide substrates. In the absence of bound cofactor and substrate, the smaller domain of the SIRT1 catalytic domain undergoes a striking ~25° rotation that is accompanied by an ~15 Å shift of the residues of the domain, generating a wide open interdomain groove, while the larger domain and CTR interface remain mostly unchanged. A mutational analysis identifies key residues for enzymatic activity of SIRT1 and supports the previously proposed imidate reaction mechanism. Further biochemical experiments establish an inhibitory role for the CTR and define corresponding binding and inhibitory regions. Our results provide a promising avenue for the development of novel SIRT1 activators that take advantage of the distinct features of the catalytic domain-CTR interface.
Results
Reconstitution of active SIRT1 and structure determination
Our attempts to express various fragments of the catalytic domain of Homo sapiens SIRT1 in bacteria yielded protein prone to aggregation. Based on previous findings that a C-terminal region is required for SIRT1 activity,47,48 we generated a series of expression constructs for various C-terminal fragments that were tested for their ability to interact with the catalytic domain. We identified residues 234 to 510 and 641 to 665 of the catalytic domain (CAT) and the C-terminal regulatory segment (CTR), respectively, which formed a heterodimeric complex as determined by size exclusion chromatography (Fig. 1a, b). Coexpression of the two SIRT1 fragments greatly improved the solubility, stability, and behavior of the catalytic domain in solution (Tables S1 and S2). An analysis by size exclusion chromatography coupled to multiangle light scattering (SEC-MALS) revealed that the heterodimer is monomeric in solution with a measured molecular mass of 34.8 kDa (theoretical 35.1 kDa) (Fig. 1c). We refer to this complex as the SIRT1CAT•CTR heterodimer in the following text.
Fig. 1.

Biochemical analysis of the SIRT1CAT•CTR complex. (a) Domain structure. Blue, NAD+-binding domain; yellow, helical module; green, Zn2+-binding module; red, pseudo-substrate peptide (T, tail); purple, C-terminal regulatory segment (CTR); grey, predicted unstructured regions (U). The bars above the domain structure mark the crystallized fragments. (b) Size-exclusion chromatography interaction analysis of SIRT1CAT with SUMO-CTR. The analyzed proteins and complexes are indicated in each gel filtration profile. For analysis of complex formation, the SUMO-CTR was mixed at approximately 2-fold molar excess of SIRT1CAT and injected onto a Superdex 200 10/300 GL gel filtration column. Gray bars and colored lines designate the analyzed fractions. Molecular mass standards and the positions of the proteins are indicated. The asterisk indicates a degraded SUMO-CTR fragment. (c) Multiangle light scattering (MALS) analysis of the SIRT1CAT•CTR heterodimer. The differential refractive index is plotted against the elution volume from a Superdex 200 10/300 GL gel filtration column and overlaid with the determined molecular mass for the peak.
Crystals of the apo form of SIRT1CAT•CTR appeared in the tetragonal space group P43212, contained four copies of the heterodimer in the asymmetric unit, and diffracted to 2.65 Å resolution. We also obtained crystals of the SIRT1CAT•CTR heterodimer in the presence of NAD+ in the trigonal space group P3221 that diffracted to 1.85 Å and contained one copy of the complex in the asymmetric unit. Both structures were solved by single-wavelength anomalous dispersion (SAD) using anomalous X-ray diffraction data obtained by taking advantage of the endogenous bound Zn2+ ions. The final models essentially contain all residues of both protein fragments in the substrate- and cofactor-bound states and possess excellent stereochemical parameters. The models were refined to Rwork and Rfree values of 23.0 % and 26.5 % (apo form) and 16.8 % and 18.9 % (quaternary complex), respectively. For details of the data collection and refinement statistics, see Table 1.
Table 1.
Crystallographic analysis
| Data collection | ||
| Protein | SIRT1•CTR•ADPR•Substrate | SIRT1•CTR apo |
| PDB code | 4KXQ | 4IF6 |
| Synchrotron | SSRL | SSRL |
| Beamline | BL12-2 | BL12-2 |
| Space group | P3221 | P43212 |
| Cell parameters | ||
| a, b, c (Å) | 92.7, 92.7, 97.7 | 115.8, 115.8, 350.5 |
| α, β, γ (°) | 90.0, 90.0, 120.0 | 90.0, 90.0, 90.0 |
| Zn peak | Zn peak | |
| Wavelength (Å) | 1.28210 | 1.2676 |
| Resolution (Å) | 50.0 – 1.85 | 50.0 – 2.65 |
| Rsym (%)b | 4.3 (62.6) | 8.8 (94.3) |
| <I>/<σI>b | 34.3 (2.4) | 18.3 (1.8) |
| Completeness (%)b | 100.0 (99.9) | 97.1 (92.8) |
| No. observations | 406,347 | 541,806 |
| No. unique reflections | 80,328 (8,010) | 68,583 (6,409) |
| Redundancy | 5.1 (4.7) | 7.9 (7.8) |
| Refinement | ||
| Resolution (Å) | 50.0 – 1.85 | 50.0 – 2.65 |
| No. reflections total | 80,282 | 67,954 |
| No. reflections test set | 3,824 (4.76%) | 1982 (2.92%) |
| Rwork/Rfree (%) | 16.8/18.9 | 23.0/26.5 |
| No. atoms | 2,842 | 9,424 |
| Protein | 2,484 | 9,374 |
| Ligand | 47 | 4 |
| Water | 311 | 46 |
| B-factors | ||
| Protein | 39.4 | 30.3 |
| Ligand | 33.8 | 60.6 |
| Water | 45.1 | 8.2 |
| R.m.s. deviations | ||
| Bond lengths (Å) | 0.009 | 0.002 |
| Bond angles (°) | 1.2 | 0.630 |
| Ramachandran plotc | ||
| Favored (%) | 97.7 | 96.1 |
| Additionally allowed (%) | 2.3 | 3.9 |
| Outliers (%) | 0.0 | 0.0 |
| MolProbity score | 1.11 | 2.04 |
SSRL, Stanford Synchrotron Radiation Lightsource
Highest resolution shell is shown in parentheses
As determined by MolProbity60
Structural overview
The SIRT1 catalytic domain possesses the canonical sirtuin fold as first described in the archaeal Sir2•Af1•NAD+ complex structure,25 composed of a larger NAD+-binding subdomain with a Rossmann fold and a smaller subdomain composed of a helical module and a Zn2+-binding module, which are both insertions in the NAD+-binding domain.
The 25-residue CTR can be divided into a larger N-terminal region, followed by a shorter C-terminal extension, which each make distinct interactions with the catalytic domain. The N-terminal region forms a β hairpin that complements the central parallel β sheet of the Rossmann fold domain with two additional strands. This interface between the catalytic domain and the CTR is large, hydrophobic, and highly conserved in evolution. The C-terminal CTR extension forms additional contacts with the helical module that are only observed in one of the two crystallized states.
Despite the fact that we obtained crystals of SIRT1CAT•CTR in the presence of NAD+, the heterodimer is bound to adenosine diphosphate ribose (ADPR), likely a product of non-enzymatic NAD+ hydrolysis or disorder of the nicotinamide moiety. Surprisingly, the heterodimer engages the extended tail region of the catalytic domain of an adjacent molecule in the crystal lattice, yielding a quaternary substrate-mimicking complex. When compared to the substrate- and cofactor-bound state of the heterodimer, the apo form adopts a wide-open conformation, in which the smaller subdomain undergoes a striking ~25° rotation, leading to the disruption of all interactions between the large and small subdomains and the formation of an ~15 Å wide interdomain cleft. As such, we refer to these two states as “open” and “closed” throughout the text. This conformational change leads to the exposure of the hydrophobic interior and the cofactor-binding site of the enzyme in the open state, and to the disruption of the substrate-binding groove.
The structure of SIRT1 catalytic domain
The SIRT1 catalytic core is composed of 277 residues and is divided into two subdomains, a larger NAD+-binding domain, which adopts a Rossmann fold, and a smaller domain, which is generated by two insertions in the NAD+-binding domain, a helical module (residues 269 to 324) and a Zn2+-binding module (residues 362 to 419) (Figs. 2 and S1). The NAD+-binding domain is composed of a central six-stranded parallel β sheet, comprising strands β1-3 and β7-9, and eight α helices, αA, αB, αG, αH, and αJ-M that pack against the β sheet core of the domain (Figs. 2 and S1). The helical module is composed of four α helices, αC-F, and the Zn2+-binding module is composed of three β strands, β4-6, and a single α helix, αI. The Zn2+ ion is tetrahedrally coordinated by four invariant cysteine residues, Cys371, Cys374, Cys395, and Cys398. An extensive hydrophobic interface holds the helical and Zn2+-binding modules together, generating the tightly associated small subdomain of the catalytic core. In total, 40 residues, primarily of helices αD, αE, αF and the αI-β6 region, are involved in the interaction between the two modules, burying ~1,800 Å2 of surface area. The relative orientation and the interaction between the large and small domains are dramatically different in the open and closed states and are described in detail below.
Fig. 2.

Structure of H. sapiens SIRT1. (a) Structure of the quaternary SIRT1CAT•CTR•ADPR•Substrate complex in ribbon representation, colored as in Fig. 1a. (b) Structure of the SIRT1CAT•CTR heterodimer, displayed in the same orientation as in panel (b).90°-rotated views are shown on the right. See also Figs. S1 and S3.
Interaction between SIRT1CAT and the CTR
The larger N-terminal region of the CTR (residues 641 to 653) forms a β hairpin that binds to the lower edge of the NAD+-binding domain of SIRT1CAT, complementing the central six-stranded parallel β sheet of the Rossmann fold with two additional β strands, β10 and β11 (Fig. 3). The β hairpin covers an essentially invariant hydrophobic patch, which is generated by residues of helices αA and αL, β strand β9, and the β8-β9 connector, forming an entirely hydrophobic interface that is composed of 17 and 11 residues of SIRT1CAT and CTR, respectively. In total, the two proteins bury ~1,150 Å2 of surface area. The CTR-binding site on the Rossmann fold is located close to the binding site for the adenine ring of NAD+ (termed the A pocket) and the CTR directly interacts with Arg466 of the β8-β9 loop, which forms extensive van der Waals contacts with one face of the adenine ring (Fig. 3). CTR binding does not alter the conformation of the catalytic domain, as evidenced by a root-mean-square deviation (RMSD) of ~0.6 Å (276 Cα atoms) between the closed state and a recently determined structure of the catalytic domain in the absence of the CTR in complex with a small molecule inhibitor (Fig. S2a).39 In the closed state the C-terminal extension of the CTR (residues 654 to 660) undergoes a conformational change and forms additional interactions with the helical module (see below).
Fig. 3.
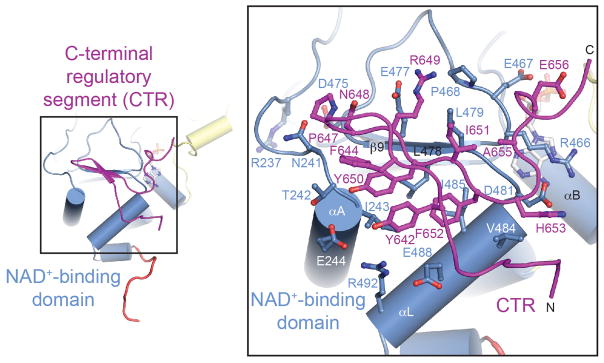
Interface between SIRT1CAT and the C-terminal regulatory segment. Ribbon representation of the interface between the SIRT1CAT and the CTR colored according to Fig. 1a. The inset illustrates the position of the CTR-binding site on the larger NAD+-binding domain and its interacting residues and is expanded on the right. Residues participating in the interface are shown in ball-and-stick representation.
Substrate interaction
To shed light on the conformational changes that SIRT1 undergoes upon cofactor binding, we also crystallized SIRT1CAT•CTR in the presence of NAD+. While we were unable to obtain crystals of SIRT1CAT•CTR in the presence of an acetylated substrate peptide, the structure surprisingly revealed that SIRT1CAT•CTR engages the extended seven-residue tail segment (residues 504 to 510) of an adjacent heterodimer in the crystal (Figs. 2 and S3a). The sequence of the tail segment, PVKLSEI, resembles the sequence and chemical nature of the SIRT1 substrate region in p53, HKKAcLMF, but possesses a hydrophobic leucine residue in place of the substrate acetyl-lysine residue that is inserted into the hydrophobic tunnel which leads to the internal active site of the enzyme (Fig. 4). The tail region binds SIRT1CAT•CTR by forming anti-parallel β sheet-like interactions on one side with the β7-αK connector (residues 444 to 446) of the Rossmann fold domain and on the other side with the Phe-Gly-Glu (FGE) motif (residues 414 to 416) of the Zn2+-binding module (Fig. 4a, b). While substrate binding is primarily achieved by backbone hydrogen bonding, the residues of both regions are evolutionarily highly conserved, suggesting that different substrates interact with SIRT1 in a similar manner. Overall, the trans binding of the pseudo substrate tail region to SIRT1CAT•CTR faithfully mimics the “β staple” interaction that was previously observed in the structure of the non-physiological ternary complex between the thermophilic bacterium Thermotoga maritima TmSir2, ADPR, and a six-residue p53 peptide (Fig. 4).29 Confirmation of these results will require additional structural analysis of a SIRT1 substrate interaction.
Fig. 4.

Structural comparison of H. sapiens SIRT1 to a bacterial Sir2 substrate complex. (a) A ribbon representation of SIRT1CAT•CTR•ADPR•Substrate (left, colored as in Fig. 1a), T. maritima Sir2 in complex with a p53 peptide (middle), and a superposition of the two (right). (b) A stick representation of the pseudo-substrate and p53 substrate peptide from the corresponding structures in panel (a) and their superposition. The PDB code of the TmSir2 structure is 2H59.29 (c) Final 2|fo|-|fc| electron density map around the ADPR and pseudo-substrate peptide rendered at 1.0 σ. See also Fig. S4.
While we crystallized SIRT1CAT•CTR in the presence of NAD+, the electron density unambiguously establishes that only the NAD+ hydrolysis product ADPR, which lacks nicotinamide, is bound (Fig. 4c). The presence of ADPR may be the result of slow non-enzymatic hydrolysis of NAD+, or disorder of the nicotinamide moiety.49,50 The ADPR molecule is well ordered and makes extensive hydrogen bond and van der Waals interactions with the Rossmann fold domain, essentially identical to those seen in other previously determined sirtuin structures.20,21,27,29,30,39,51
In order to determine whether the trans interaction between the tail region of one SIRT1CAT•CTR with the substrate-binding site of another SIRT1CAT•CTR may be physiologically relevant, we tested whether SIRT1CAT•CTR is capable of forming dimers or other higher order oligomers in solution in the presence of NAD+. A SEC-MALS analysis revealed that SIRT1CAT•CTR remains monomeric in the presence of 5 mM NAD+ with no higher order oligomeric states detectable at concentrations as high as 20 mg/ml (Fig. S4). Further studies are required to address whether the tail region is important for oligomerization and regulation of SIRT1 in vivo, especially in the context of the full-length protein and oligomeric substrates.
Conformational changes of SIRT1 upon cofactor and substrate binding
We have determined the structure of human SIRT1 in two different states, the apo form of SIRT1CAT•CTR (open state), and a quaternary complex of SIRT1CAT•CTR with ADPR and a pseudo-substrate peptide (closed state) (Fig. 2). The four copies of the apo form in the asymmetric unit make different crystal packing interactions and align with a maximum RMSD of ~0.5 Å2 over 286 Cα atoms, suggesting that SIRT1 possesses a distinct open conformation that is independent of crystal packing interactions (Fig. S3b). Moreover, the tail region that engages an adjacent SIRT1CAT•CTR in the closed state is disordered in all four copies of the open state, further demonstrating that it is not a structural element of the catalytic core.
A comparison between the open and closed states reveals the conformational changes that SIRT1 undergoes upon substrate and cofactor binding. The larger NAD+-binding domain, together with the N-terminal region of the CTR, does not undergo any major structural changes, as illustrated by an RMSD of ~0.5 Å2 over 170 Cα atoms (Fig. 5). In the closed state, the smaller domain forms an extensive interface with the larger domain, resulting in the complete closure of the interdomain groove and the formation of a hydrophobic tunnel that engages the substrate residue (Fig. 5d). In the absence of cofactor and substrate, SIRT1 adopts an open state with a large interdomain groove, exposing the invariant hydrophobic interior and the NAD+-binding site (Fig. 5e and 5f). Overall, the smaller domain rotates by ~25° with a maximum displacement of the domain atoms of ~15 Å. Altogether, the two modules that generate the small subdomain rotate essentially as a rigid body with only minor changes to the backbone and side-chain conformations as illustrated by an RMSD of ~1.3 Å2 over 114 Cα atoms. Notable exceptions are the αB-αC interdomain connector and helix αC, which undergo an additional conformational change to form a lid over the cofactor, leading to its encapsulation, and the FGE motif of the β6-αJ connector which forms the upper β strand of the substrate-binding β staple. The lower β strand of the β staple that is formed by the β7-αK connector of the Rossmann fold only undergoes minor conformational changes. Whereas the N-terminal region of the CTR is static, the C-terminal extension adopts a distinct conformation in the closed state that allows for the formation of a salt bridge between Glu656 and Arg276 of the helical module.
Fig. 5.

Structural comparison of open and closed SIRT1 states. (a) Ribbon representations of the closed SIRT1CAT•CTR•ADPR•Substrate state, (b) the open SIRT1CAT•CTR conformation, and (c) a superposition of the two. (d, e) Surface representations of the corresponding structures in panels (a) and (b). A 90°-rotated view is shown on the right. (f) A model of pseudo-substrate peptide and ADPR binding to the open SIRT1CAT•CTR state. A 90°-rotated view is shown on the right. See also Fig. S2.
Taken together, SIRT1 undergoes dramatic conformational changes upon substrate and cofactor binding. The open state observed here for SIRT1 is substantially more open than previously determined apo states of other sirtuins, suggesting that each member of the sirtuin family adopts a unique open state, while the closed states are essentially identical.
Mutational analysis
To determine the importance of various residues for the deacetylase activity of SIRT1, we generated mutants based on the structures presented here, a multi-species sequence alignment (Fig. S1), and previous biochemical and structural work.18 We mutated seven residues in the SIRT1 active site that are evolutionarily invariant in bacteria, archaea, and eukaryotes, and which have been shown in other sirtuins to either capture the nicotinamide released from the NAD+ cofactor (Ser265, Asn346, Ile347, and Asp348), bind and orient the NAD+ cofactor (Gln345), mediate the interaction with the substrate peptide (Phe414), or are catalytically required to abstract a proton from the activated NAD+ (His363) (Fig. 6a). To clarify the role of the CTR in SIRT1 regulation, we generated an additional five mutants in the SIRT1CAT-CTR interface. These included two residues of the catalytic domain which contact both the CTR and the adenine ring of ADPR (Arg466 and Asp481), two residues that form a salt bridge between the catalytic domain and the CTR in the closed conformation (Arg276 and Glu656), as well as a truncation of the twelve C-terminal residues of the CTR (CTR3ΔC) (Fig. 6b). Of the twelve mutants, ten were indistinguishable from the wild-type SIRT1CAT•CTR heterodimer in their behavior on a gel filtration column. Two mutants, N346A and D348N, were insoluble following recombinant expression and could therefore not be tested. The ten SIRT1CAT•CTR mutants (S265A, R276A, Q345A, I347A, H363A, F414A, R466A, D481A, E656A, and CTR3ΔC) along with the crystallized wild-type SIRT1CAT•CTR heterodimer and SIRT1CAT in the absence of the CTR, were tested in two different in vitro deacetylase activity assays that employ modified, acetylated p53-based peptides (Fig. S5). The results of both assays were consistent and are summarized in Fig. 6c.
Fig. 6.

Mutational analysis. (a) Ribbon representation of the SIRT1CAT•CTR•ADPR•Substrate complex, with a stick representation of all mutated active side residues. The Zn2+-binding and helical modules were removed for clarity. (b) Ribbon representation of the SIRT1CAT•CTR•ADPR•Substrate complex, with a stick representation of all mutated SIRT1CAT-CTR interface residues. (c) Deacetylase activity of SIRT1 proteins and mutants towards modified p53-based peptides. Activity was determined in a luminescence (red) or fluorescence (blue) based assay and normalized against wild-type SIRT1CAT activity. Each data point represents the mean of at least three independent measurements. Error bars represent standard deviations. See also Fig. S5.
All five active site mutants display either weak (S265A) or no deacetylase activity (Q345A, I347A, H363A, and F414A), demonstrating their involvement in the catalytic reaction. These results are in line with previous studies on other sirtuins and support the previously proposed ADPR-peptidyl-imidate mechanism for the deacetylation reaction.19 The two mutants R466A and D481A, in which the wild-type residues are sandwiched between the ADPR and the C-terminal extension of the CTR, yield proteins with substantially reduced catalytic activity, highlighting their role in co-factor binding.
SIRT1CAT alone, being prone to aggregation, showed variability in activity between experiments, and, hence, any results obtained in the absence of CTR need to be interpreted with caution. However, despite its fragility, SIRT1CAT in isolation consistently displayed higher activity compared to the SIRT1CAT•CTR heterodimer, indicating an inhibiting effect of the CTR. This inhibitory effect is mediated by the C-terminal 10 residues of the CTR (CTR3ΔC), as evidenced by the loss of SIRT1CAT inhibition in the SIRT1CAT•CTR3ΔC heterodimer. Similar results were obtained for the R276A and E656A mutants, indicating that the salt bridge between these residues is the primary determinant of inhibition by the CTR. The R276A mutant did not significantly increase the activity of SIRT1CAT in the absence of the CTR, suggesting that the primary means through which this residue regulates activity is through its interaction with the CTR. Moreover, both Arg276 and Glu656 are invariant in mammalian SIRT1 enzymes, indicating that this regulatory feature is evolutionarily conserved.
Biochemical analysis of the SIRT1CAT-CTR interaction
As a result of the observed inhibitory effect of the CTR on SIRT1 activity, we sought to determine whether the CTR is a persistent structural feature of the catalytic domain, as previously observed for the tail region of the catalytic domain of S. cerevisiae Sir2 (Fig. S2b).40 Due to the fact that SIRT1CAT is prone to aggregation, we were unable to determine the dissociation constant between SIRT1CAT and the CTR by isothermal titration calorimetry. However, we tested whether the CTR can be displaced from the SIRT1CAT•CTR complex. To test this hypothesis we incubated the SIRT1CAT•CTR heterodimer with SUMO-tagged CTR and monitored exchange by size-exclusion chromatography. Indeed, a SUMO-CTR can displace the CTR from a preformed SIRT1CAT•CTR complex (Fig. 7a). We additionally incubated SUMO-CTR with a GST-CTR•SIRT1CAT complex and performed pull-down experiments. Similarly, increasing amounts of SUMO-CTR resulted in decreasing amounts of pulled-down SIRT1CAT (Fig. 7b). To further probe the SIRT1CAT•CTR interaction, we designed mutations at the binding interface and assayed their ability to form a complex in a GST pull-down assay (Fig. 7c). Mutation of three hydrophobic CTR residues (Phe644, Tyr650, Ile651) to alanine or aspartate abolished CTR binding to SIRT1CAT. Mutation of Tyr642 to alanine resulted in reduced binding that was completely lost upon mutation to aspartate. Based on these results we conclude that the CTR binding may represent a regulated interaction to inhibit SIRT1 activity.
Fig. 7.

Exchange of the C-terminal regulatory segment. (a) Size-exclusion chromatography interaction analysis of the SIRT1CAT•CTR heterodimer with SUMO-CTR. The analyzed proteins and complexes are indicated in each gel filtration profile. For analysis of complex formation, SUMO-CTR was mixed at approximately 2-fold molar excess and injected onto a Superdex 200 10/300 GL gel filtration column. Gray bars and colored lines designate the analyzed fractions. Molecular mass standards and the positions of the proteins are indicated. Untagged CTR is too small to be visualized on the SDS-PAGE. (b) GST pull-downs of a preformed GST-CTR•SIRT1CAT complex with increasing amounts of SUMO-CTR. The loaded (top) and pulled-down (bottom) samples are visualized by SDS-PAGE, followed by Coomassie brilliant blue staining. As reference, the first two lanes of each gel contain the loaded samples of GST-CTR•SIRT1CAT and SUMO-CTR. The relative molar amounts of GST-CTR•SIRT1CAT and SUMO-CTR, molecular mass standards and the positions of the proteins are indicated. (c) CTR mutational analysis. His-tagged SIRT1CAT and GST-tagged CTR variants were coexpressed in E. coli and the resulting soluble lysate fraction was analyzed in GST pull-down assays. Samples were visualized by SDS-PAGE, followed by Coomassie brilliant blue staining. Molecular mass standards and the positions of the proteins are indicated. The asterisk indicates a degraded GST-CTR fragment
Thermostability
During our in vitro deacetylation assays, we noticed a rapid decay of enzymatic activity and a variability between different purifications for SIRT1CAT, which was not observed for SIRT1CAT•CTR. In addition, we obtained substantially lower yields for SIRT1CAT in isolation relative to the coexpressed SIRT1CAT•CTR complex (Table S2). These findings prompted us to explore whether the CTR stabilizes the catalytic domain. We found that the SIRT1CAT•CTR heterodimer was substantially more resistant to temperature denaturation than SIRT1CAT in isolation or in the presence of a non-interacting CTR mutant. Specifically, whereas SIRT1CAT already begins to precipitate at 30 °C, the SIRT1CAT•CTR heterodimer remains completely soluble up to 39 °C (Fig. 8a). In line with these results, the isolated SIRT1 catalytic domain displays an almost complete loss of activity when incubated for two hours at 37 °C, whereas activity is maintained for the SIRT1CAT•CTR heterodimer (Fig. 8b). These data support a stabilizing role for the CTR on the SIRT1 catalytic domain and provide an explanation for previous difficulties in characterizing the biochemical and structural properties of SIRT1.
Fig. 8.

Thermostability assay. (a) SIRT1CAT was incubated alone and in the presence of SUMO-CTR or a non-interacting SUMO-CTRY650D, I651D mutant at the indicated temperatures for one hour and pelleted by centrifugation. Pellet and supernatant fractions were visualized by SDS-PAGE, followed by Coomassie brilliant blue staining. Molecular mass standards and the positions of the proteins are indicated. (b) Deacetylase activity of SIRT1CAT and the SIRT1CAT•CTR heterodimer under normal conditions (red) and following incubation at 37 °C for approximately 2 hours prior to the assay (blue). Activity was determined in a fluorescence-based assay and normalized against wild-type SIRT1CAT activity. Each data point represents the mean of at least three independent measurements. Error bars represent standard deviations. The loss of deacetylase activity is attributed to the aggregation of the catalytic domain.
Discussion
We have determined the crystal structure of the catalytic domain of human SIRT1 in complex with a C-terminal regulatory segment (CTR) in two different conformations, the apo form of the heterodimer and a quaternary complex with ADPR and a pseudo-substrate, the tail of another SIRT1CAT that mimics a p53 substrate sequence. The comparison of the two structures reveals that the interdomain angle and association between the two domains of the catalytic core are dramatically different upon substrate and cofactor binding. Further conformational changes occur in the C-terminal extension of the CTR that inhibit the activity of the catalytic domain.
The observation that sirtuins engage unique regions flanking the canonical catalytic domain has been made previously in the structure of S. cerevisiae Hst2.26 In that case, a homotrimer has been observed in the crystal and in solution, which is mediated by an N-terminal region that interacts with an adjacent catalytic domain in a substrate-mimicking fashion. In human SIRT1, a C-terminal seven-residue tail segment that is disordered in the apo state engages an adjacent molecule in the NAD+-bound closed state. The overall mode of interaction faithfully mimics the interaction between a p53 substrate peptide, the main difference being that a hydrophobic leucine residue, instead of an acetylated lysine residue, is inserted into the hydrophobic tunnel that leads to the internal active site of the enzyme. The substrate-mimicking interaction observed here might be useful for the development of competitive SIRT1 inhibitors.
Our biochemical analysis expands upon recent findings on the regulatory nature of the CTR.47,48 Namely, we establish an inhibitory role for the CTR primarily mediated by the formation of a salt bridge between the C-terminal extension and the helical module that is only observed in our closed SIRT1CAT•CTR•ADPR•Substrate structure. We propose that this interaction tunes the activity of the catalytic domain by influencing the conformational changes that are required for substrate and cofactor turnover (Fig. 9). This is in contrast to recent work stating that the CTR is required for SIRT1 activity.47 However, we believe this conclusion may be a result of the propensity of the SIRT1CAT to aggregate in solution in the absence of the CTR. Despite the overall hydrophobic nature and large surface of the SIRT1CAT-CTR interface, we demonstrate that the SIRT1CAT-CTR interaction is not merely a structural augmentation of the catalytic domain, but rather possesses the necessary characteristics of a regulated interaction. Such a regulatory mechanism could involve the phosphorylation of the CTR, which contains four predicted phosphorylation sites (Tyr650, Tyr658, Ser659, and Ser661).52
Fig. 9.
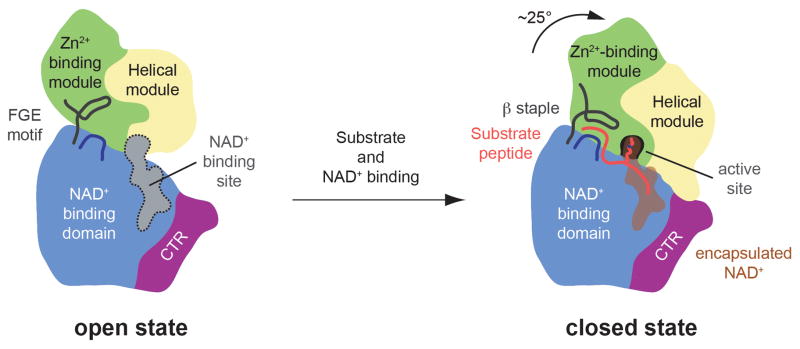
Model for the conformational changes and regulation of SIRT1. A cartoon representation of the apo SIRT1CAT•CTR heterodimer (left) and the SIRT1CAT•CTR•ADPR•Substrate complex (right), colored as in Fig. 1a. Substrate and co-factor binding leads to the closure of the SIRT1 catalytic domain. The substrate peptide is primarily bound by backbone interactions and the formation of a three-stranded anti-parallel β staple. The hydrophobic acetylated lysine substrate residue reaches into the secluded internal active site through the hydrophobic tunnel where it is oriented in close proximity to the activated NAD+. In the closed state, the CTR forms a salt-bridge with the helical module, thereby reducing the efficiency of catalysis.
Our structural and functional analysis presented here allow us to propose a model for the regulation of SIRT1 and provide the framework for the development of potent and selective SIRT1 activity modulators that promise to be a new therapeutic avenue for the treatment of a variety of human diseases.
Methods
Protein expression and purification
DNA fragments of H. sapiens SIRT1 were amplified by PCR and cloned into the vector pGEX-6P-1 (GE Healthcare), a modified pET28a vector (Novagen) that contained a PreScission protease site after the N-terminal hexahistidine tag (pET28a-PreS),53 and a modified pET28b vector that contained an N-terminal hexahistidine-SUMO tag (pET28b-SUMO).54 SIRT1 mutants were generated by QuikChange mutagenesis (Stratagene) and confirmed by DNA sequencing. Details of all constructs are listed in Table S1.
All proteins were expressed in E. coli BL21-CodonPlus(DE3)-RIL cells (Stratagene) grown in LB media supplemented with appropriate antibiotics. Protein expression was induced at an OD600 of approximately 0.6 with 500 μM IPTG for 16–18 hours at 18 °C. Cells were harvested by centrifugation.
The SIRT1CAT•CTR complex and all SIRT1CAT•CTR mutants were made by coexpressing SIRT1CAT in pET28a-PreS and CTR in pGEX-6P-1 (Table S2). Cells were resuspended in a buffer containing 20 mM Tris, pH 8.0, 100 mM NaCl, 5 mM DTT, 2 μM bovine lung aprotinin (Sigma), and complete EDTA-free protease inhibitor cocktail (Roche), and flash frozen in liquid nitrogen. Thawed cells were lysed with a cell disrupter (Avestin) and the lysate was centrifuged for 1 hour at 40,000 x g. The cleared lysate was applied to a glutathione sepharose column equilibrated in 20 mM Tris, pH 8.0, 100 mM NaCl, and 5 mM DTT (GE Healthcare) and eluted via a glutathione gradient. Pooled fractions were cleaved with PreScission protease (GE Healthcare) for 12 hours and further purified over an additional glutathione sepharose column. The protein was concentrated and injected onto a HiLoad 16/60 Superdex 75 column (GE Healthcare) equilibrated in 20 mM Tris, pH 8.0, 100 mM NaCl, and 5 mM DTT. Pooled fractions were concentrated to 12.5 mg/ml and used in subsequent crystallization trials or for in vitro activity assays. The GST-tagged SIRT1CAT•CTR used in pull-down experiments was purified in the same manner, omitting the protease cleavage step.
SIRT1CAT in isolation, and all SIRT1CAT mutants, were made by expressing SIRT1CAT in pET28a-PreS. Cells were resuspended in a buffer containing 20 mM Tris, pH 8.0, 500 mM NaCl, 5 mM β-mercaptoethanol, 2 μM bovine lung aprotinin (Sigma), and complete EDTA-free protease inhibitor cocktail (Roche), and flash frozen in liquid nitrogen. Thawed cells were lysed with a cell disrupter and the lysate was centrifuged for 1 hour at 40,000 x g. The cleared lysate was loaded onto a Ni-NTA column (Qiagen) and eluted via an imidazole gradient. Protein-containing fractions were pooled, dialyzed against a buffer containing 20 mM Tris, pH 8.0, 100 mM NaCl, and 5 mM DTT, and cleaved with PreScission protease for 12 hours. Cleaved protein was bound to a HiTrap Q HP (GE Healthcare) column and eluted via a NaCl gradient, concentrated, and injected onto a HiLoad 16/60 Superdex 75 column equilibrated in 20 mM Tris, pH 8.0, 100 mM NaCl, and 5 mM DTT. SUMO-CTR, and all SUMO-CTR mutants, followed the same purification as SIRT1CAT in isolation.
Structure determination and refinement
Crystals of SIRT1CAT•CTR were grown at 21 °C in hanging drops containing 1 μl of the protein and 1 μl of a reservoir solution consisting of 100 mM Tris, pH 7.4, and 11 % PEG 20,000. Crystals grew in the tetragonal space group P43212 and reached a maximum size of approximately 700 μm × 100 μm × 100 μm within a week. Crystals of SIRT1CAT•CTR•ADPR were grown in similar conditions in the presence of a two-fold molar excess of NAD+ (Sigma), grew in the trigonal space group P3221, and reached a maximum size of approximately 300 μm × 100 μm × 100 μm within two weeks. For cryo protection, crystals were stabilized in 100 mM Tris, pH 7.4, 13 % PEG 20,000, and 20 % ethylene glycol (added in 1 % increments). X-ray diffraction data were collected at 100 K at beamline 12-2 at the Stanford Synchrotron Radiation Lightsource (SSRL), and at beamline 8.2.2 at the Advanced Light Source (ALS) at Lawrence Berkeley National Laboratory (LBNL). X-ray intensities were processed using the HKL2000 denzo/scalepack package.55
Both structures were solved by single wavelength anomalous dispersion (SAD) using X-ray diffraction data collected utilizing the anomalous scattering of the endogenous Zn2+ ions. Heavy metal sites were identified with SHELXD.56 Phases were calculated in SHARP,57 followed by density modification in DM58 with solvent flattening, histogram matching, and NCS averaging. This procedure yielded excellent electron density maps that allowed for the placement of the coordinates of the S. cerevisiae Sir2 (PDB code 2HJH). Iterative rounds of model building and refinement in Coot59 and PHENIX60 yielded final models in which all residues of both protein fragments were resolved in the substrate- and cofactor-bound states with the exception of the C-terminal tails of the catalytic domain (residues 503 to 510; apo state) and CTR (residues 661 to 665; both states). The final Rwork and Rfree values of the apo and the substrate- and cofactor-bound states of SIRT1 were 23.0 % and 26.5 % and 15.6 % and 18.7 %, respectively. Both models possess excellent stereochemical parameters with no residues in the generously allowed or disallowed regions of the Ramachandran plot as assessed with PROCHECK and MolProbity.61,62 For details of the data collection and refinement statistics, see Table 1.
Analytical size-exclusion chromatography
Protein interaction experiments were carried out on a Superdex 200 10/300 GL gel filtration column (GE Healthcare) equilibrated in a buffer containing 20 mM Tris, pH 8.0, 100 mM NaCl, and 5 mM DTT. SIRT1CAT and the SIRT1CAT•CTR heterodimer were mixed with an approximately two-fold molar excess of SUMO-tagged CTR, and incubated for 30 minutes on ice. Complex formation was monitored by injection of the preincubated proteins and the individual components in isolation, and confirmed by SDS-PAGE of the protein-containing fractions, followed by Coomassie brilliant blue staining.
GST pull-down assays
12.5 μg of a GST-CTR•SIRT1CAT complex was mixed with varying amounts of SUMO-CTR, added to approximately 20 μl of Glutathione Sepharose beads (GE Healthcare) equilibrated in a buffer containing 20 mM Tris, pH 8.0, 100 mM NaCl, and 5 mM DTT, and incubated on ice for 1 hour. The beads were isolated via centrifugation at 750 x g, and washed five times by resuspension in 200 μL of buffer, followed by subsequent centrifugation. All samples were analyzed by SDS-PAGE, followed by Coomassie brilliant blue staining. For CTR mutational analysis, lysate from E. coli transformed with the indicated constructs was added to 250 μl of beads.
Deacetylase activity assays
Deacetylase activity of SIRT1 proteins was determined using the SIRT-Glo (Promega) and Fluor de Lys SIRT1 (Enzo) assays, according to the manufacturers’ protocols. SIRT-Glo reactions were carried out with 1 μM of pure protein and were incubated at room temperature for 45 minutes. Fluor de Lys reactions contained 0.2 μM pure protein, 75 μM substrate, and 3 mM NAD+, were incubated at 37 °C for 45 minutes, and developed at room temperature for 1 hour. Luminescence (SIRT-Glo) and fluorescence (Fluor de Lys) were measured using a Flexstation-3 microplate reader (Molecular Devices) and autofluorescence was subtracted.
Thermostability Assay
15 μg of SIRT1CAT in isolation or mixed with an approximately two fold molar excess of SUMO-CTR or SUMO-CTRY650D, I651D was incubated at the indicated temperatures for 1 hour. Samples were pelleted by centrifugation at 30,000 x g and analyzed by SDS-PAGE followed by Coomassie brilliant blue staining.
Multiangle light scattering
Purified proteins were characterized by multiangle light scattering following size-exclusion chromatography.63 Proteins were injected onto a Superdex 200 10/300 GL gel filtration chromatography column equilibrated in a buffer containing 20 mM Tris, pH 8.0, 100 mM NaCl, and 5 mM DTT. The chromatography system was connected in series with an 18-angle light-scattering detector (DAWN HELEOS II; Wyatt Technology), a dynamic light-scattering detector (DynaPro Nanostar; Wyatt Technology), and a refractive index detector (Optilab t-rEX; Wyatt Technology). Data were collected every 1 second at a flow rate of 0.5 mL/min at 25 °C. Data analysis was carried out using the program ASTRA 6, yielding the molar mass and mass distribution (polydispersity) of the sample.
Illustration and figures
The sequence alignment of SIRT1 was generated using ClustalX and colored with Alscript.64,65 Figures were generated using PyMOL (www.pymol.org). Electrostatic potential was calculated with Adaptive Poisson-Boltzmann Solver (APBS) software.66
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Highlights.
Crystal structures of the human SIRT1 catalytic domain in complex with its CTR
Conformational changes upon substrate and co-factor binding elucidated
Mutagenesis identifies key residues for catalysis and CTR-mediated inhibition
CTR binding pocket provides opportunity for the development of novel therapeutics
Acknowledgments
We thank Erik W. Debler, Daniel H. Lin, Alina Patke, Pete Stavropoulos, Tobias Stuwe, and Yunji Wu for critical reading of the manuscript, Leslie N. Collins for technical support, David King for mass spectrometry analysis, Anders Näär for providing material, and Jens Kaiser and the scientific staff of SSRL beamline 12-2, and Corie Ralston and scientific staff of ALS beamline 8.2.2 for their support with X-ray diffraction measurements. We acknowledge the Gordon and Betty Moore Foundation for their support of the Molecular Observatory at the California Institute of Technology. The operations at the SSRL are supported by the Department of Energy and by the National Institutes of Health. AMD is supported by a National Institutes of Health Research Service Award (5 T32 GM07616). AH was supported by the Albert Wyrick V Scholar Award of the V Foundation for Cancer Research, the 54th Mallinckrodt Scholar Award of the Edward Mallinckrodt, Jr. Foundation, and a Kimmel Scholar Award of the Sidney Kimmel Foundation for Cancer Research.
Footnotes
Accession numbers
The atomic coordinates and structure factors of the H. sapiens SIRT1CAT•CTR heterodimer and the quaternary SIRT1CAT•CTR•ADPR•Substrate complex have been deposited in the Protein Data Bank under accession codes 4IF6 and 4KXQ.
References
- 1.Klar AJ, Fogel S, Macleod K. MAR1-a Regulator of the HMa and HMalpha Loci in SACCHAROMYCES CEREVISIAE. Genetics. 1979;93:37–50. doi: 10.1093/genetics/93.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Haber JE, George JP. A mutation that permits the expression of normally silent copies of mating-type information in Saccharomyces cerevisiae. Genetics. 1979;93:13–35. doi: 10.1093/genetics/93.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rine J, Strathern JN, Hicks JB, Herskowitz I. A suppressor of mating-type locus mutations in Saccharomyces cerevisiae: evidence for and identification of cryptic mating-type loci. Genetics. 1979;93:877–901. doi: 10.1093/genetics/93.4.877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aparicio OM, Billington BL, Gottschling DE. Modifiers of position effect are shared between telomeric and silent mating-type loci in S. cerevisiae. Cell. 1991;66:1279–87. doi: 10.1016/0092-8674(91)90049-5. [DOI] [PubMed] [Google Scholar]
- 5.Bryk M, Banerjee M, Murphy M, Knudsen KE, Garfinkel DJ, Curcio MJ. Transcriptional silencing of Ty1 elements in the RDN1 locus of yeast. Genes Dev. 1997;11:255–69. doi: 10.1101/gad.11.2.255. [DOI] [PubMed] [Google Scholar]
- 6.Fritze CE, Verschueren K, Strich R, Easton Esposito R. Direct evidence for SIR2 modulation of chromatin structure in yeast rDNA. EMBO J. 1997;16:6495–509. doi: 10.1093/emboj/16.21.6495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smith JS, Boeke JD. An unusual form of transcriptional silencing in yeast ribosomal DNA. Genes Dev. 1997;11:241–54. doi: 10.1101/gad.11.2.241. [DOI] [PubMed] [Google Scholar]
- 8.Imai S, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature. 2000;403:795–800. doi: 10.1038/35001622. [DOI] [PubMed] [Google Scholar]
- 9.Landry J, Sutton A, Tafrov ST, Heller RC, Stebbins J, Pillus L, Sternglanz R. The silencing protein SIR2 and its homologs are NAD-dependent protein deacetylases. Proc Natl Acad Sci USA. 2000;97:5807–11. doi: 10.1073/pnas.110148297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smith JS, Brachmann CB, Celic I, Kenna MA, Muhammad S, Starai VJ, Avalos JL, Escalante-Semerena JC, Grubmeyer C, Wolberger C, Boeke JD. A phylogenetically conserved NAD+-dependent protein deacetylase activity in the Sir2 protein family. Proc Natl Acad Sci USA. 2000;97:6658–63. doi: 10.1073/pnas.97.12.6658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garcia-Salcedo JA, Gijon P, Nolan DP, Tebabi P, Pays E. A chromosomal SIR2 homologue with both histone NAD-dependent ADP-ribosyltransferase and deacetylase activities is involved in DNA repair in Trypanosoma brucei. EMBO J. 2003;22:5851–62. doi: 10.1093/emboj/cdg553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liszt G, Ford E, Kurtev M, Guarente L. Mouse Sir2 homolog SIRT6 is a nuclear ADP-ribosyltransferase. J Biol Chem. 2005;280:21313–20. doi: 10.1074/jbc.M413296200. [DOI] [PubMed] [Google Scholar]
- 13.Tsang AW, Escalante-Semerena JC. CobB, a new member of the SIR2 family of eucaryotic regulatory proteins, is required to compensate for the lack of nicotinate mononucleotide:5,6-dimethylbenzimidazole phosphoribosyltransferase activity in cobT mutants during cobalamin biosynthesis in Salmonella typhimurium LT2. J Biol Chem. 1998;273:31788–94. doi: 10.1074/jbc.273.48.31788. [DOI] [PubMed] [Google Scholar]
- 14.Kowieski TM, Lee S, Denu JM. Acetylation-dependent ADP-ribosylation by Trypanosoma brucei Sir2. J Biol Chem. 2008;283:5317–26. doi: 10.1074/jbc.M707613200. [DOI] [PubMed] [Google Scholar]
- 15.Merrick CJ, Duraisingh MT. Plasmodium falciparum Sir2: an unusual sirtuin with dual histone deacetylase and ADP-ribosyltransferase activity. Eukaryot Cell. 2007;6:2081–91. doi: 10.1128/EC.00114-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Haigis MC, Mostoslavsky R, Haigis KM, Fahie K, Christodoulou DC, Murphy AJ, Valenzuela DM, Yancopoulos GD, Karow M, Blander G, Wolberger C, Prolla TA, Weindruch R, Alt FW, Guarente L. SIRT4 inhibits glutamate dehydrogenase and opposes the effects of calorie restriction in pancreatic beta cells. Cell. 2006;126:941–54. doi: 10.1016/j.cell.2006.06.057. [DOI] [PubMed] [Google Scholar]
- 17.Du J, Zhou Y, Su X, Yu JJ, Khan S, Jiang H, Kim J, Woo J, Kim JH, Choi BH, He B, Chen W, Zhang S, Cerione RA, Auwerx J, Hao Q, Lin H. Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science. 2011;334:806–9. doi: 10.1126/science.1207861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sauve AA, Wolberger C, Schramm VL, Boeke JD. The biochemistry of sirtuins. Annu Rev Biochem. 2006;75:435–65. doi: 10.1146/annurev.biochem.74.082803.133500. [DOI] [PubMed] [Google Scholar]
- 19.Sauve AA, Celic I, Avalos J, Deng H, Boeke JD, Schramm VL. Chemistry of gene silencing: the mechanism of NAD+-dependent deacetylation reactions. Biochemistry. 2001;40:15456–63. doi: 10.1021/bi011858j. [DOI] [PubMed] [Google Scholar]
- 20.Avalos JL, Bever KM, Wolberger C. Mechanism of sirtuin inhibition by nicotinamide: altering the NAD(+) cosubstrate specificity of a Sir2 enzyme. Mol Cell. 2005;17:855–68. doi: 10.1016/j.molcel.2005.02.022. [DOI] [PubMed] [Google Scholar]
- 21.Avalos JL, Boeke JD, Wolberger C. Structural basis for the mechanism and regulation of Sir2 enzymes. Mol Cell. 2004;13:639–48. doi: 10.1016/s1097-2765(04)00082-6. [DOI] [PubMed] [Google Scholar]
- 22.Avalos JL, Celic I, Muhammad S, Cosgrove MS, Boeke JD, Wolberger C. Structure of a Sir2 enzyme bound to an acetylated p53 peptide. Mol Cell. 2002;10:523–35. doi: 10.1016/s1097-2765(02)00628-7. [DOI] [PubMed] [Google Scholar]
- 23.Finnin MS, Donigian JR, Pavletich NP. Structure of the histone deacetylase SIRT2. Nat Struct Biol. 2001;8:621–5. doi: 10.1038/89668. [DOI] [PubMed] [Google Scholar]
- 24.Jin L, Wei W, Jiang Y, Peng H, Cai J, Mao C, Dai H, Choy W, Bemis JE, Jirousek MR, Milne JC, Westphal CH, Perni RB. Crystal structures of human SIRT3 displaying substrate-induced conformational changes. J Biol Chem. 2009;284:24394–405. doi: 10.1074/jbc.M109.014928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Min J, Landry J, Sternglanz R, Xu RM. Crystal structure of a SIR2 homolog-NAD complex. Cell. 2001;105:269–79. doi: 10.1016/s0092-8674(01)00317-8. [DOI] [PubMed] [Google Scholar]
- 26.Zhao K, Chai X, Clements A, Marmorstein R. Structure and autoregulation of the yeast Hst2 homolog of Sir2. Nat Struct Biol. 2003;10:864–71. doi: 10.1038/nsb978. [DOI] [PubMed] [Google Scholar]
- 27.Zhao K, Chai X, Marmorstein R. Structure of the yeast Hst2 protein deacetylase in ternary complex with 2′-O-acetyl ADP ribose and histone peptide. Structure. 2003;11:1403–11. doi: 10.1016/j.str.2003.09.016. [DOI] [PubMed] [Google Scholar]
- 28.Zhao K, Harshaw R, Chai X, Marmorstein R. Structural basis for nicotinamide cleavage and ADP-ribose transfer by NAD(+)-dependent Sir2 histone/protein deacetylases. Proc Natl Acad Sci USA. 2004;101:8563–8. doi: 10.1073/pnas.0401057101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hoff KG, Avalos JL, Sens K, Wolberger C. Insights into the sirtuin mechanism from ternary complexes containing NAD+ and acetylated peptide. Structure. 2006;14:1231–40. doi: 10.1016/j.str.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 30.Chang JH, Kim HC, Hwang KY, Lee JW, Jackson SP, Bell SD, Cho Y. Structural basis for the NAD-dependent deacetylase mechanism of Sir2. J Biol Chem. 2002;277:34489–98. doi: 10.1074/jbc.M205460200. [DOI] [PubMed] [Google Scholar]
- 31.Szczepankiewicz BG, Dai H, Koppetsch KJ, Qian D, Jiang F, Mao C, Perni RB. Synthesis of carba-NAD and the structures of its ternary complexes with SIRT3 and SIRT5. J Org Chem. 2012;77:7319–29. doi: 10.1021/jo301067e. [DOI] [PubMed] [Google Scholar]
- 32.Zhou Y, Zhang H, He B, Du J, Lin H, Cerione RA, Hao Q. The bicyclic intermediate structure provides insights into the desuccinylation mechanism of human sirtuin 5 (SIRT5) J Biol Chem. 2012;287:28307–14. doi: 10.1074/jbc.M112.384511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schuetz A, Min J, Antoshenko T, Wang CL, Allali-Hassani A, Dong A, Loppnau P, Vedadi M, Bochkarev A, Sternglanz R, Plotnikov AN. Structural basis of inhibition of the human NAD+-dependent deacetylase SIRT5 by suramin. Structure. 2007;15:377–89. doi: 10.1016/j.str.2007.02.002. [DOI] [PubMed] [Google Scholar]
- 34.Cosgrove MS, Bever K, Avalos JL, Muhammad S, Zhang X, Wolberger C. The structural basis of sirtuin substrate affinity. Biochemistry. 2006;45:7511–21. doi: 10.1021/bi0526332. [DOI] [PubMed] [Google Scholar]
- 35.Zhu AY, Zhou Y, Khan S, Deitsch KW, Hao Q, Lin H. Plasmodium falciparum Sir2A preferentially hydrolyzes medium and long chain fatty acyl lysine. ACS Chem Biol. 2012;7:155–9. doi: 10.1021/cb200230x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bheda P, Wang JT, Escalante-Semerena JC, Wolberger C. Structure of Sir2Tm bound to a propionylated peptide. Protein Sci. 2011;20:131–9. doi: 10.1002/pro.544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hawse WF, Hoff KG, Fatkins DG, Daines A, Zubkova OV, Schramm VL, Zheng W, Wolberger C. Structural insights into intermediate steps in the Sir2 deacetylation reaction. Structure. 2008;16:1368–77. doi: 10.1016/j.str.2008.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sanders BD, Zhao K, Slama JT, Marmorstein R. Structural basis for nicotinamide inhibition and base exchange in Sir2 enzymes. Mol Cell. 2007;25:463–72. doi: 10.1016/j.molcel.2006.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhao X, Allison D, Condon B, Zhang F, Gheyi T, Zhang A, Ashok S, Russell M, MacEwan I, Qian Y, Jamison JA, Luz JG. The 2.5 A crystal structure of the SIRT1 catalytic domain bound to nicotinamide adenine dinucleotide (NAD+) and an indole (EX527 analogue) reveals a novel mechanism of histone deacetylase inhibition. J Med Chem. 2013;56:963–9. doi: 10.1021/jm301431y. [DOI] [PubMed] [Google Scholar]
- 40.Hsu HC, Wang CL, Wang M, Yang N, Chen Z, Sternglanz R, Xu RM. Structural basis for allosteric stimulation of Sir2 activity by Sir4 binding. Genes Dev. 2013;27:64–73. doi: 10.1101/gad.208140.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Haigis MC, Sinclair DA. Mammalian sirtuins: biological insights and disease relevance. Annu Rev Pathol. 2010;5:253–95. doi: 10.1146/annurev.pathol.4.110807.092250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guarente L. Sirtuins in aging and disease. Cold Spring Harb Symp Quant Biol. 2007;72:483–8. doi: 10.1101/sqb.2007.72.024. [DOI] [PubMed] [Google Scholar]
- 43.Sauve AA, Schramm VL. Sir2 regulation by nicotinamide results from switching between base exchange and deacetylation chemistry. Biochemistry. 2003;42:9249–56. doi: 10.1021/bi034959l. [DOI] [PubMed] [Google Scholar]
- 44.Kim EJ, Kho JH, Kang MR, Um SJ. Active regulator of SIRT1 cooperates with SIRT1 and facilitates suppression of p53 activity. Mol Cell. 2007;28:277–90. doi: 10.1016/j.molcel.2007.08.030. [DOI] [PubMed] [Google Scholar]
- 45.Kim JE, Chen J, Lou Z. DBC1 is a negative regulator of SIRT1. Nature. 2008;451:583–6. doi: 10.1038/nature06500. [DOI] [PubMed] [Google Scholar]
- 46.Zhao W, Kruse JP, Tang Y, Jung SY, Qin J, Gu W. Negative regulation of the deacetylase SIRT1 by DBC1. Nature. 2008;451:587–90. doi: 10.1038/nature06515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kang H, Suh JY, Jung YS, Jung JW, Kim MK, Chung JH. Peptide switch is essential for Sirt1 deacetylase activity. Mol Cell. 2011;44:203–13. doi: 10.1016/j.molcel.2011.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pan M, Yuan H, Brent M, Ding EC, Marmorstein R. SIRT1 contains N- and C-terminal regions that potentiate deacetylase activity. J Biol Chem. 2012;287:2468–76. doi: 10.1074/jbc.M111.285031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.French JB, Cen Y, Sauve AA. Plasmodium falciparum Sir2 is an NAD+-dependent deacetylase and an acetyllysine-dependent and acetyllysine-independent NAD+ glycohydrolase. Biochemistry. 2008;47:10227–39. doi: 10.1021/bi800767t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sanders BD, Jackson B, Marmorstein R. Structural basis for sirtuin function: what we know and what we don’t. Biochim Biophys Acta. 2010;1804:1604–16. doi: 10.1016/j.bbapap.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gertz M, Fischer F, Nguyen GT, Lakshminarasimhan M, Schutkowski M, Weyand M, Steegborn C. Ex-527 inhibits Sirtuins by exploiting their unique NAD+-dependent deacetylation mechanism. Proc Natl Acad Sci U S A. 2013;110:E2772–81. doi: 10.1073/pnas.1303628110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Blom N, Gammeltoft S, Brunak S. Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J Mol Biol. 1999;294:1351–62. doi: 10.1006/jmbi.1999.3310. [DOI] [PubMed] [Google Scholar]
- 53.Hoelz A, Nairn AC, Kuriyan J. Crystal structure of a tetradecameric assembly of the association domain of Ca2+/calmodulin-dependent kinase II. Mol Cell. 2003;11:1241–51. doi: 10.1016/s1097-2765(03)00171-0. [DOI] [PubMed] [Google Scholar]
- 54.Mossessova E, Lima CD. Ulp1-SUMO crystal structure and genetic analysis reveal conserved interactions and a regulatory element essential for cell growth in yeast. Mol Cell. 2000;5:865–76. doi: 10.1016/s1097-2765(00)80326-3. [DOI] [PubMed] [Google Scholar]
- 55.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 56.Sheldrick GM. A short history of SHELX. Acta Crystallogr A. 2008;64:112–22. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 57.Bricogne G, Vonrhein C, Flensburg C, Schiltz M, Paciorek W. Generation, representation and flow of phase information in structure determination: recent developments in and around SHARP 2.0. Acta Crystallogr D. 2003;59:2023–30. doi: 10.1107/s0907444903017694. [DOI] [PubMed] [Google Scholar]
- 58.CCP4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D. 1994;50:760–3. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 59.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 60.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Laskowski RA, Macarthur MW, Moss DS, Thornton JM. Procheck - a Program to Check the Stereochemical Quality of Protein Structures. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- 62.Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB, Snoeyink J, Richardson JS, Richardson DC. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007;35:W375–W383. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wyatt PJ. Multiangle light scattering: The basic tool for macromolecular characterization. Instrum Sci Technol. 1997;25:1–18. [Google Scholar]
- 64.Jeanmougin F, Thompson JD, Gouy M, Higgins DG, Gibson TJ. Multiple sequence alignment with Clustal X. Trends Biochem Sci. 1998;23:403–5. doi: 10.1016/s0968-0004(98)01285-7. [DOI] [PubMed] [Google Scholar]
- 65.Barton GJ. ALSCRIPT: a tool to format multiple sequence alignments. Protein Eng. 1993;6:37–40. doi: 10.1093/protein/6.1.37. [DOI] [PubMed] [Google Scholar]
- 66.Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci USA. 2001;98:10037–41. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.