

Abstract
The serologic hallmark of primary biliary cirrhosis (PBC), the antimitochondrial response to the E2 component of the pyruvate dehydrogenase complex (PDC-E2), has unique features, including continuous high titers of IgM and IgG reactivity throughout all stages of disease, capable not only of target enzyme inhibition, but also cross-reactive with chemical xenobiotics that share molecular homology with the inner lipoyl domain of PDC-E2; such chemicals have been proposed as potential etiological agents. We have used flow cytometry and ELISPOT to examine B cell subsets in 59 subjects, including 28 with PBC, 13 with PSC and 18 healthy controls. Strikingly, in PBC, although there were no significant differences in B cell phenotype subpopulations, 10% of the total IgG and IgA plasmablast population and 23% of the IgM plasmablast population were uniquely reactive with PDC-E2, detected in the CXCR7+CCR10low plasmablast population. In contrast, plasmablast reactivity to a control antigen, tetanus toxoid, was minimal and similar in all groups. Additionally, we isolated plasmablast-derived polyclonal antibodies and compared reactivity with plasma-derived antibodies and noted a distinct non-circulating tissue source of xenobiotic cross-reacting antibodies. The high levels of autoantigen specific peripheral plasmablasts indicate recent activation of naive or memory B cells and a continuous and robust activation. The presence of CXCR7+CCR10low PDC-E2-specific ASCs suggests a mechanistic basis for the migration of circulating antigen specific plasmablasts to the mucosal epithelial ligands CXCL12 and CCL28. In conclusion, our findings suggest a sustained rigorous B cell response in PBC, likely activated and perpetuated by cognate autoantigen.
Keywords: Plasmablast, autoantigen-specific, PDC-E2, autoreactive B cell, antibody cross-reactivity
Terminally differentiated antibody secreting B cells, or plasma cells, that localize to the bone marrow are the primary source of serum antibodies (1, 2). Upon antigen specific stimulation at the site of exposure, naïve or memory B cells enter secondary lymphoid organs through high endothelial venules and are activated in the presence of specialized CD4 helper T cells. The activated B cells proliferate and differentiate into plasmablasts within the germinal centers of local lymph nodes. At approximately day 6-8 after antigen exposure, antibody-secreting plasmablasts leave the germinal centers to transiently enter the circulation (3). Depending on the expression of trafficking receptors, these plasmablasts thence quickly leave the circulation and migrate to bone marrow or target organs where they further develop into plasma cells (4). The presence of circulating plasmablasts is an essential intermediate stage of B cell development, indicative of recent activation of naïve or memory B cells (3, 5).
Antibody responses to environmental xenobiotics that bear structural similarity to host autoantigens have been proposed as a mechanism for the breach of tolerance to PDC-E2 (6, 7). This is supported by our previous finding that sera from PBC patients react not only with the primary autoantigen PDC-E2, but also with environmental chemicals that are structurally related to lipoic acid (6, 8). It is at present unclear whether the serum antibody against PDC-E2 in PBC results from de novo activated autoantigen-specific B cells, or from B cells previously primed by xenobiotics when self-tolerance was originally compromised. The goal of the current study was to explore the relationship between B cell immunity and PBC pathogenesis by examining plasmablasts for frequency, isotype class, and patterns of autoantibody reactivity. We report herein that there is a striking frequency of circulating plasmablasts that are specific for PDC-E2, but not xenobiotics in patients with PBC, consistent with ongoing activation of autoantigen-specific B cells by cognate antigen.
Materials and Methods
Human participants and blood samples
The subjects studied herein included patients with PBC (n=28), with primary sclerosing cholangitis (PSC) (n=13) and healthy individuals (n=18). The diagnosis of all patients was based on established criteria for PBC and PSC (9-11). As expected, the majority (24/28) of PBC cases were women and 78.6% (22/28) were on ursodeoxycholic acid therapy as the sole treatment at the time of enrollment. The clinical characteristics of study population are listed in Table 1. All participants provided written informed consent and the study protocol was approved by the Institutional Review Board at the University of California, Davis prior to initiation of study. A heparinized venous blood sample was collected and used for isolating plasma, PBMC and B cells. PBMC were isolated from blood utilizing Histopaque-1.077. B cells were isolated by negative selection with RosetteSep™ Human B cell Enrichment cocktail (Stemcell Technologies, Vancouver, BC, Canada); the purity was 70-80%.
Table 1. Clinical characteristics of study population.
| PBC (N=28) |
PSC (N=13) |
Healthy control (N=18) |
|
|---|---|---|---|
| Mean age, years | 59.1 | 43.4 | 48.2 |
| (Range) | (36-82) | (23-67) | (33-68) |
| Male | 4 | 6 | 7 |
| Female | 24 | 7 | 11 |
| Disease stage* | |||
| Stage I-II | 21 | n/a | n/a |
| Stage III-IV | 7 | n/a | n/a |
| AMA Positive | 28 | 0 | 0 |
Generation of plasmablast-derived polyclonal antibodies (PPAb)
PPAb were generated (5). In brief, freshly isolated B cells were re-suspended in complete medium (RPMI 1640 supplemented with 10% heat inactivated fetal calf serum, 100 units/ml of penicillin G and 100 μg/ml of streptomycin) at 3 million cells per ml and incubated in a 5% CO2 humidified atmosphere at 37 °C for 7 days. Conditioned media containing secreted PPAb were collected and kept at −80 °C until use.
Enumeration of total and antigen-specific antibody secreting cells (ASCs)
Total and antigen-specific IgA, IgG or IgM ASCs were enumerated using ELISPOT. For detecting total ASCs, 96-well MultiScreen HTS plates (Millipore, Billerica, MA, USA) were coated with 100μl of affinity purified goat anti-human IgA+IgG+IgM (H+L) (KPL) at a concentration of 4 μg/ml in PBS. For antigen-specific ASCs, wells were coated with 100μl of either recombinant GST-PDC-E2 fusion protein (12) or Tetanus toxoid (TT) (Pfenex, San Diego, CA, USA) at a concentration of 10 μg/ml in PBS. Wells coated with recombinant GST (10 μg/ml) or PBS served as negative controls. Plates were incubated overnight at 4 °C and then blocked for 2 h at 37 °C. Freshly isolated B cells were re-suspended at 1×106 cells/ml containing alkaline phosphatase conjugated goat antibodies against human IgA (α), human IgG (γ) and human IgM at 0.2 μg/ml, respectively, and dispensed into the coated/blocked wells at two fold serial dilutions in a volume of 100 μl and incubated for 6 h at 37 °C in 5% CO2. Plates were washed with PBS and developed with blue alkaline phosphatase. The total number of IgG, IgA and IgM ASCs in each well was determined by counting the spots. Spots detected in the negative control (GST or PBS) wells were subtracted from the counts of GST-PDC-E2- or TT-coated wells, respectively.
B cell analysis by flow cytometry
PBMC freshly isolated from whole blood were first incubated with anti-mouse CD16/32 to block the Fc receptor, then stained with fluorochrome-conjugated antibodies including FITC-conjugated IgD, PE-conjugated anti-CD27, APC-conjugated anti-CD3, and Alexa750–conjugated anti-CD19 (Biolegend, San Diego, CA, USA). Stained cells were analyzed using a FACScan flow cytometer. Data was acquired on a total of 10,000 events and analyzed utilizing CELLQUEST software. Known positive and negative controls were used throughout.
ELISA for plasma antibodies and PPAbs
The antibody reactivity against recombinant PDC-E2, TT or the xenobiotics 2-octynoic acid-conjugated bovine serum albumin (2OA-BSA) and 6, 8-bis (acetylthio) octanoic acid-conjugated BSA (SAc-BSA) was determined by ELISA (13, 14). 2OA-BSA and SAc-BSA were synthesized (6, 15). Briefly, each well of 96-well ELISA plates was coated with either 0.5 μg of recombinant PDC-E2 protein, 1.0μg of TT, 0.5 μg of 2OA-BSA or 0.5 μg of SAc-BSA in 100 μl of carbonate buffer (pH 9.6) and incubated overnight at 4°C. Plates were washed with PBS containing 0.05% Tween-20 (PBST) (Fisher Scientific, Pittsburgh, PA, USA), then blocked with 200 μl per well of 1% BSA in PBS, for 1 h at room temperature and the fluid discarded. One hundred μl of diluted plasma (1:2000) or PPAb supernatant (1:10) to be tested was added to each well in triplicate. The plates were incubated at room temperature for 1 h, washed with PBST for at least three times and incubated for 1 h with a mixture of horseradish peroxidase (HRP)-conjugated anti-human IgG, IgM, and IgA (Invitrogen, Carlsbad, CA, USA), or each individual antibody, at predetermined optimized dilution, washed, and developed with BD OptEIA Substrate. Optical density (OD) was read at 450 nm and the mean +/- SD calculated.
Characteristics of PDC-E2-specific ASCs
Freshly isolated B cells were treated with Human Fc Receptor Binding Inhibitor to block the Fc receptor prior to staining. B cells were then stained with FITC-conjugated CD27, PE-conjugated anti-CCR10, PE-Cy7-conjugated anti-CD20, Percp-Cy5.5-conjugated anti-CD38, APC-conjugated anti-CXCR7, Alexa700–conjugated anti-CD3 (Biolegend, San Diego, CA, USA) and Alexa750–conjugated anti-CD19. Cell sorting was performed using FACS-Aria. Appropriate known positive and negative controls were used throughout. Sorted CD3−CD19+CD20−CD27hiCD38hiCXCR7+CCR10low cells were suspended in complete medium containing alkaline phosphatase conjugated goat antibodies against human IgA, human IgG and human IgM (all from KPL, Gaithersburg, Maryland, USA) at 0.2 μg/ml, respectively, and then seeded in 96-well MultiScreen HTS plates (Millipore, Billerica, MA, USA) coated with 100μl of affinity purified goat anti-human IgA+IgG+IgM (H+L) (KPL) at a concentration of 4 μg/ml in PBS. For antigen-specific ASCs, wells were coated with 100μl of recombinant GST-PDC-E2 fusion protein. Plates were incubated for 16 h at 37 °C under 5% CO2 prior to development with blue alkaline phosphatase substrate. The number of total IgG/IgA/IgM ASCs and PDC-E2 specific ASCs in each well was thence determined as noted earlier.
Statistical Analysis
Data were analyzed using the one-way ANOVA followed by Tukey's test or with paired t-test as appropriate. A p-value of 0.05 or less was considered statistically significant.
Results
Characterization of the B cell population
In the first phase of this work, and as positive controls for all the data thereafter, anti-mitochondrial antibodies (AMA), measured as autoantigen PDC-E2-specific IgA, IgG and IgM antibodies, were quantitated in plasma (Figure 1). Flow cytometry was thence utilized to examine the frequencies of total B cells as well as specific B cell subsets in PBMC from PBC and controls. There were no significant differences detected in the frequencies of total B cells, naïve B cells, class-unswitched memory B cells, and class-switched memory B cells between PBC and healthy controls (Table 2). These data are important as they indicate that there are no significant disturbances in the major circulating B cell subpopulations in PBC. In comparison, the frequency of total B cells was significantly higher in PBMC from PSC compared with healthy controls. However, there were no significant differences in the frequencies of the four B cell subsets between PSC and healthy control groups (Table 2).
Figure 1.
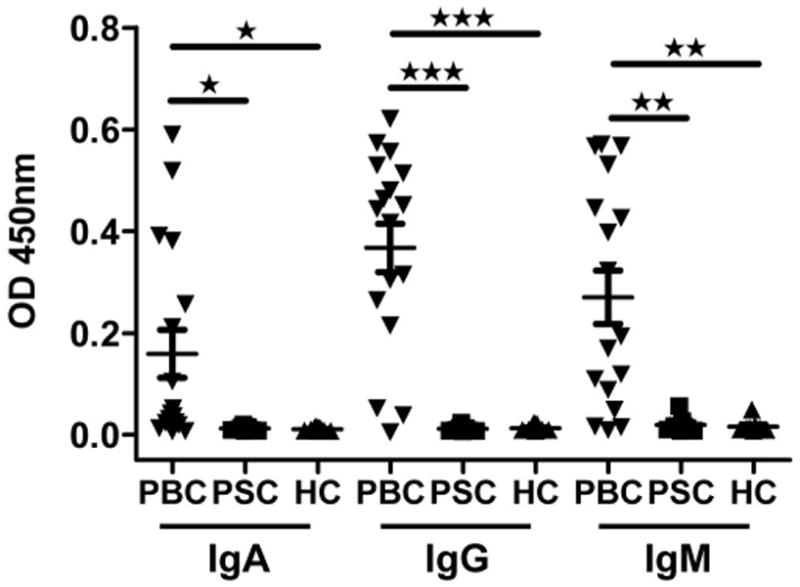
Plasma antibody reactivity to the autoantigen PDC-E2. Plasma samples from PBC patients (n=17), PSC patients (n=7) and healthy controls (n=8) were tested by ELISA for IgA, IgG and IgM isotype specific antibody activity against recombinant PDC-E2. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Table 2. Frequencies of B cells and B cell subsets.
| PBC (n=12) |
PSC (n=6) |
Healthy controls (n=7) |
|
|---|---|---|---|
| B cells in PBMC, % | 7.5 ± 0.7 | 13.8 ± 1.2* | 7.4 ± 0.6 |
| B cell subsets in B cells | |||
| Naïve B cells, % | 59.2 ± 5.9 | 69.5 ± 11.3 | 65.2 ± 8.3 |
| Class-unswitched memory B cells, % | 18.0 ± 4.4 | 8.4 ± 2.8 | 9.1 ± 2.3 |
| Class-switched memory B cells, % | 13.2 ± 1.9 | 13.0 ± 6.0 | 21.6 ± 6.90 |
PBMC samples were analyzed by flow cytometry for total B cells (CD3−CD19+), naïve B cells (CD3−CD19+CD27−IgD+), class-unswitched memory B cells (CD3−CD19+CD27+IgD+) and class-switched memory B cells (CD3−CD19+CD27+IgD−).
significantly higher than healthy controls and PBC.
Ongoing in vivo activation of PDC-E2-specific B cells in PBC
As noted in Figure 2, there was a significantly higher frequency of PDC-E2 specific IgM, IgG and IgA antibody secreting cells (ASCs) in the B cells from the PBC patients with average values as high as 23% for IgM ASCs and approximately 10% for either IgG or IgA ASCs (p< 0.01 compared to controls) (Figure 2). There was no correlation between the values noted and disease stage or serum AMA levels (data not shown). GST-specific ASCs were not detected in any of the PBC B cell samples indicating that the ASCs detected were specific for PDC-E2. As a further control when the total number of ASCs was considered, there were no significant differences in the number of IgM, IgG or IgA secreting cells between PBC, PSC, or the healthy controls (Figure 2).
Figure 2.
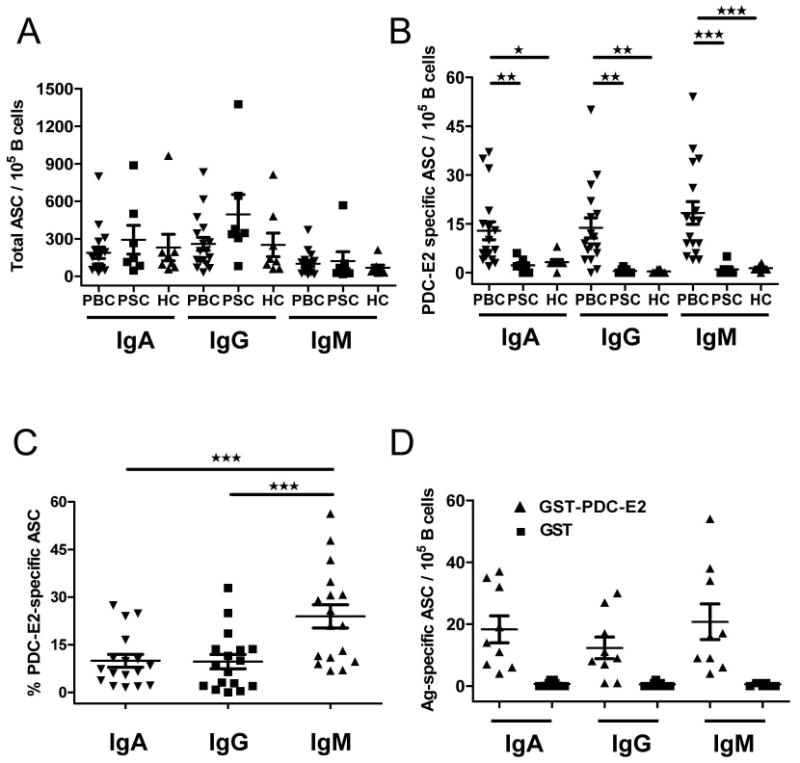
Frequencies of circulating total and PDC-E2-specific ASCs. Isolated B cells were assayed with ELISPOT to enumerate total and antigen-specific IgA, IgG and IgM ASCs. A. Frequency of total IgA, IgG and IgM ASCs in PBC patients (n=17), PSC patients (n=7) and healthy controls (n=8). B. Frequency of PDC-E2-specific IgA, IgG and IgM ASCs. C. Percentage of PDC-E2-specific ASCs in total ASCs of PBC patients. D. Frequency of ASCs from PBC patients (n=9) detected with ELISPOT plates coated with recombinant GST-PDC-E2 fusion protein or recombinant GST protein. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
To confirm that this significant elevation of PDC-E2 specific ASCs were in vivo activated B cells at the time the blood samples were collected rather than memory B cells activated ex vivo during the blood sample processing and analysis, we analyzed B cells from a subset of PBC or PSC patients with ELISPOT plates coated with TT. All patients had ELISA antibodies for TT by ELISA (Figure 3A), indicating past exposure to TT and priming of TT-specific memory B cells. In agreement with a previous report (16), TT-specific ASCs were only detected at a minimum level in B cells. In contrast, PDC-E2-specific ASCs were detectable, at strikingly high levels, in the same B cell preparations (Figure 3B). These results support the thesis that PDC-E2-specific ASCs represent newly activated plasmablasts in vivo, rather than due to ex vivo re-activation of the pool of PDC-E2-specific memory B cells.
Figure 3.
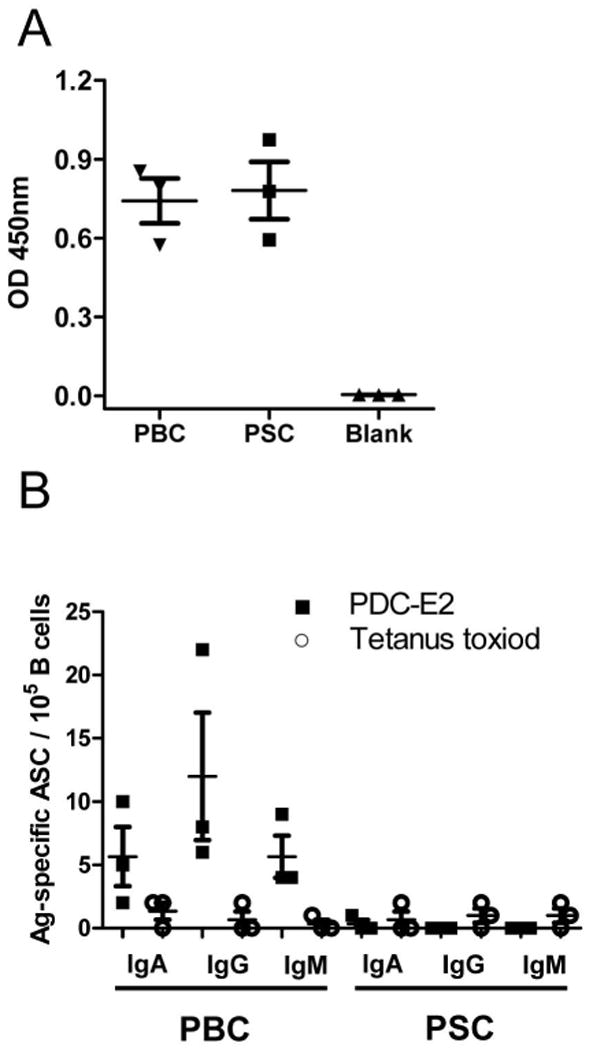
TT-specific plasma antibodies and circulating ASCs. Plasma and B cells from PBC patients (n=3) and PSC patients (n=3) were tested by ELISA or ELISPOT, respectively. A. TT-specific plasma antibodies. The background reading was obtained from blank wells without addition of plasma samples. B. Detection of TT-specific ASCs and PDC-E2-specific ASCs by ELISPOT.
PDC-E2-specific ASCs express tissue-specific homing receptors CXCR7 and CCR10
By flow cytometry, the majority of plasmablasts (defined as CD19+CD20−CD27hiCD38hi) expressed both CXCR7 and CCR10 (Figure 4A). The MFI of CCR10 on plasmablasts was significantly lower than that on CD19+CD20+ B cells (MFI: 1852 ± 315 vs. 33123 ± 3654 n=5; p< 0.0001). Next we enumerated total and PDC-E2-specific IgA/IgG/IgM ASCs in the sorted CD3−CD19+CD20−CD27hiCD38hiCXCR7+CCR10low population by ELISPOT. PDC-E2-specific ASCs were detected in the sorted total ASC population at frequencies consistent with our observation in bulk B cells (Figure 4B), indicating that PDC-E2-specific ASCs express the trafficking receptor phenotype CXCR7+CCR10low.
Figure 4.
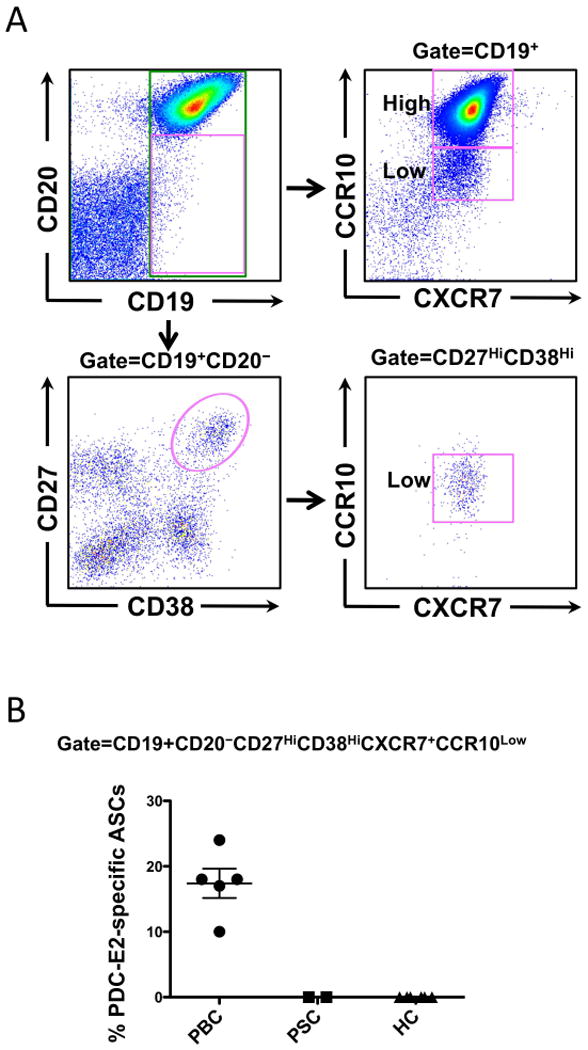
PDC-E2-specific ASCs express homing receptors CXCR7 and CCR10. Enriched B cells from PBC (n=5), and controls (n=8), including PSC (n=2) and healthy (n=6) were used to sort the CD3−CD19+CD20−CD27hiCD38hiCXCR7+CCR10low plasmablast population. A. Flow cytometric gating strategy analysis and sorting of plasmablasts. B. Total and PDC-E2-specific IgA/IgG/IgM ASCs in the sorted CXCR7+CCR10Low plasmablast population were enumerated by ELISPOT.
Antigen specificity of antibodies produced by the in vivo activated B cells in PBC
To characterize the antibodies produced by the in vivo activated B cells in PBC, we took advantage of our ability to define the heterogeneous AMA populations, which include the presence of AMA directed to PDC-E2, and AMA directed to two representative xenobiotics, 2-octynoic acid (2OA) and 6, 8-bis (acetylthio) octanoic acid (SAc), both putative etiological agents of PBC. We compared antigen specificity of plasmablasts-derived antibodies (PPAb) to antibodies in plasma from the same patients (Figure 5). Plasma antibodies from patients with PBC, but not controls, reacted to PDC-E2, 2-OA and Sac (Figure 5A). However, the PPAb from PBC reacted with PDC-E2 but did not reveal detectable reactivity against the two xenobiotics (Figure 5B). When the binding reactivity to the two xenobiotics, as measured by the OD450nm value, was normalized to that of PDC-E2 and then compared between the plasma and PPAb samples of the PBC patients, this cross-reactivity was significantly higher in plasma than in PPAb (Figure 5C). Taken together, these results indicate that in contrast to the plasma antibodies that react with both PDC-E2 and xenobiotics, the antibodies secreted from the newly in vivo activated B cells of PBC patients are specific for the autoantigen PDC-E2 but do not recognize the xenobiotics 2OA and SAc.
Figure 5.
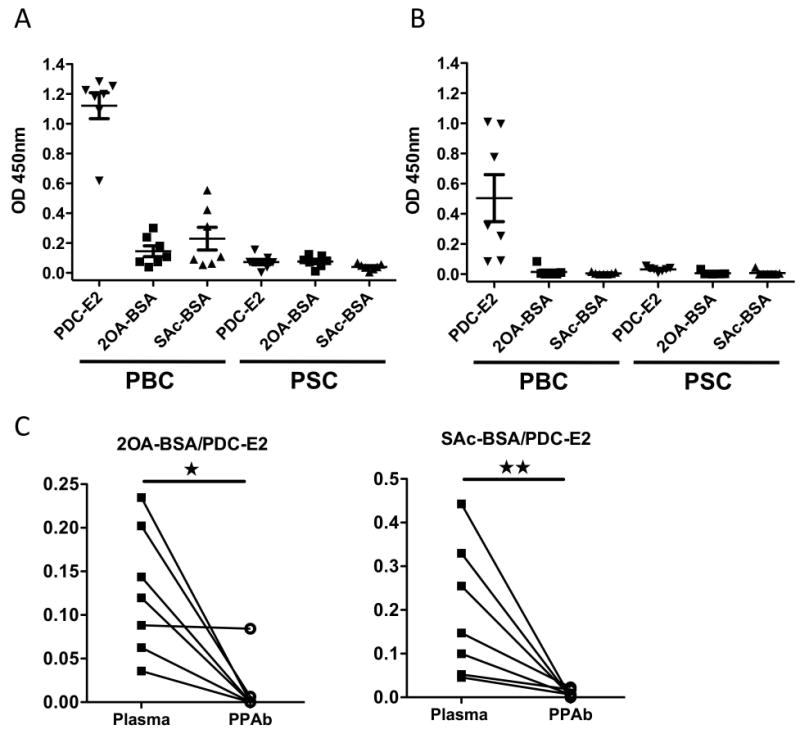
PDC-E2- and xenobiotic-specific antibody reactivity in plasma and PPAb. Plasma and PPAb samples from PBC (n=7) and PSC (n=7) patients were analyzed by ELISA for antibodies binding to recombinant PDC-E2 or the xenobiotics 2OA-BSA and SAc-BSA. All plasma samples were diluted 1:2000. All PPAb samples were diluted 1:10. Binding reactivity was detected with a mixture of conjugated anti-human IgA, IgG and IgM secondary antibodies. A. Plasma antibody binding to recombinant PDC-E2 and xenobiotics. B. PPAb binding to recombinant PDC-E2 and xenobiotics. C. Comparison of xenobiotic cross-reactivity, defined as the xenobiotic-specific binding (value of OD450nm) normalized to PDC-E2-specific binding of each sample, between plasma and PPAb of the PBC patients. *, p < 0.05; **, p < 0.01.
Discussion
We analyzed B cells and B cell subsets at various stages of differentiation in the peripheral blood of patients with PBC. In particular we have focused on autoantigen-specific plasmablasts which represent recently activated autoreactive B cells and determined their frequency and antibody reactivity. Importantly, our data reveal high levels of PDC-E2 specific plasmablasts, detected in the CXCR7+CCR10low population, and constitute 10% of circulating IgA and IgG plasmablasts as well as 23% of circulating IgM plasmablasts. Previous studies have indicated that following vaccination, large numbers of plasmablasts enter the circulation at days 6-8, but then quickly disappear from peripheral blood (3). Hence the presence of antigen-specific plasmablasts in the periphery is an indicator of recent activation of naïve or memory B cells by their cognate antigens. Such high levels of PDC-E2-specific IgG, IgA and in particular IgM plasmablasts in our PBC cohort, that includes patients at all disease stages, suggest a substantial and sustained activation of autoreactive B cells, in particular naïve B cells.
We also demonstrated that in contrast to the plasma samples from PBC patients that are reactive to the autoantigen PDC-E2 and two representative xenobiotics 2OA and SAc, the antibodies derived from the circulating plasmablasts are specific for the autoantigen but do not cross-react with the xenobiotics. These findings may be interpreted as consistent with the view that there must exist two different sources of autoantibodies in plasma, those that continue to be synthesized by plasma cells derived from circulating plasmablasts and are of recent origin and others that are a result of B cells that are non-circulating and presumably are of an earlier lineage. There is no reason to expect that the development of an antibody response to xenobiotics would differ from that of autoantigens. These findings suggest that the plasma contains antibodies produced by long-lived antibody secreting cells that are the result of an earlier time of exposure and stimulation. Whether this would be as far back as the original occasion of tolerance breakdown is unknown, but these findings support the idea that xenobiotics act as a “hit and run” initiator of tolerance breakdown and then are no longer present (5, 17, 18). Tissue resident plasma cells continue to secrete specificities that react with xenobiotics but strictly PDC-E2-specific antibodies are generated by an ongoing process, presumably related to continuing tissue damage and release of autoantigen (19).
If xenobiotics are indeed the source of the initial breakdown of self-tolerance in PBC, the question needs to be asked whether targeting this subpopulation of B cells with anti-xenobiotic specificities would be valuable. This, first of all requires that we identify the tissue source where such B cells reside. Furthermore, this would likely only be the case if such antibodies exacerbated the disease state or prevented a state of normal tolerance being re-established. Along these lines, it is important to note the potential role of the human equivalent of non-recirculating murine B-1a cells (20) that have been shown to be activated and regulated by CD1d restricted natural killer T cells (21). It is thus possible that urogenital exposure to xenobiotics, or cross reactive E. Coli infection, via their cross-reactivity to the inner lipoyl domain would also contribute to NK T cell activation, including interaction with other lymphoid subpopulations (14).
In SLE, there is a significant correlation between the frequency of CD27highCD20−CD19dim plasmablasts and disease activity index or IgG anti-double-stranded DNA antibody levels (22). In contrast, there was no association found between autoantibodies and plasmablasts in other systemic rheumatic autoimmune diseases (23). In our study herein, we note that data do not reflect a correlation between the frequency of circulating plasmablasts and AMA titer. Thus, our findings suggest that the serologic markers of PBC are not directly associated with changes in frequency or phenotype of B cell subpopulations. However, we have previously reported that in PBC, liver CD20 B cells, which are a precursor of plasma cells, were found in scattered locations or occasionally forming follicle-like aggregations; in contrast, there was a unique and distinct coronal arrangement of CD38 plasma cells around the intrahepatic ducts. Patients with PBC who manifest this unique coronal arrangement were those with significantly higher titers of anti-mitochondrial antibodies. These data point to a role for autoantigen-specific B cells in the specific destruction of intrahepatic bile ducts in PBC (19).
CCR10 is expressed by circulating IgA ASCs and tissue IgA+ plasma cells in the salivary glands, small and large intestines (24). In addition, CXCR7, whose ligand CXCL12 is highly induced in lymph nodes, lung, liver and bone marrow (25), is elevated in inflammation (25). Moreover, CXCL12 and CCL28 are strongly expressed by proliferating bile ducts (26-29), suggesting that circulating ASCs can be attracted specifically toward proliferating bile ducts under chronic inflammatory conditions. Our data on PDC-E2-specific plasmablasts expressing CXCR7 and CCR10 suggests a role of chemokine receptors in the migration of autoantigen-specific plasmablasts.
Despite the poorly understood pathogenic mechanisms governing initiation of autoimmunity and development of disease in PBC (30), clinical case reports, genome-wide association studies, epigenetic and epidemiological studies, as well as animal model studies have, in concert, identified a complex interplay between environmental agents (such as xenobiotics and bacteria) and genetic susceptibility (31-33). Two chemical xenobiotics, 2-OA and SAc that react with PBC sera, are among the candidates identified in our laboratory as potential environmental risk factors for PBC (6, 34). In the current study, we demonstrated that while plasma from AMA-positive PBC patients displayed strong cross-reactivity against PDC-E2 as well as SAc-, and 2-OA-conjugates, PPAb from these patients recognize PDC-E2 but not the two xenobiotics. In contrast to antigen-specific circulating plasmablasts that represent recently activated B cells, the plasma antibodies are primarily produced by long-lived bone marrow plasma cells and likely reflect antigen exposure that occurred decades ago (5, 17, 18).
We have previously proposed a hypothetical etiology of PBC based on molecular mimicry (6). Initial exposure to xenobiotic-modified PDC-E2 leads to a primary antibody response against the xenobiotic antigens, which cross react with the self-antigen based on the antigenic similarity between the xenobiotics and the lipoyl domain of PDC-E2, leading to the breakdown of self-tolerance to the mitochondrial PDC-E2 and damage of small BECs (35). Subsequently, through the process of affinity maturation in which mutated B-cell clones with high affinity for PDC-E2 in apoptotic BECs (36) are repeatedly selected for further expansion (37), their reactivity to the original priming xenobiotics, which are antigenically similar but distinct to PDC-E2, would be eventually lost. This mechanism is in agreement with our previous findings that sera from PBC contain a heterogeneous population of antibodies with different levels of cross-reactivity between PDC-E2 autoantigen and xenobiotics (6), and is now further supported by our current study which demonstrate that newly activated autoreactive B cells in patients with established PBC do not recognize xenobiotics. Epidemiological studies indicate that female PBC patients have a higher incidence of recurrent urinary tract infections (38), which is frequently caused by E. coli. E. coli PDC-E2 shares homologous amino acid sequences with human PDC-E2. We recently reported that E. coli infection induces autoimmune cholangitis and AMA in the non-obese diabetic (NOD) mouse model (14). Therefore exposure to E. coli is another potential factor that potentially contributes to ongoing activation of PDC-E2-specific plasmablasts, along with constant exposure to cognate antigen.
Acknowledgments
Financial Support: Funded by National Institutes of Health grants DK39588 and DK067003.
Abbreviations
- AMA
antimitochondrial antibody
- PDC-E2
E2 subunit of pyruvate dehydrogenase
- PBC
primary biliary cirrhosis
- ELISPOT
Enzyme-Linked Immunospot Assay
- PSC
primary sclerosing cholangitis
- ASC
antibody secreting cell
- GST
glutathione S-transferase
- TT
tetanus toxoid
- 2OA
2-octynoic acid
- BSA
Bovine Serum Albumin
- SAc
6, 8-bis (acetylthio) octanoic acid
- MFI
mean fluorescent intensity
Contributor Information
Jun Zhang, Email: lxtzhang@ucdavis.edu.
Weici Zhang, Email: ddzhang@ucdavis.edu.
Patrick S.C. Leung, Email: psleung@ucdavis.edu.
Christopher L. Bowlus, Email: clbowlus@ucdavis.edu.
Sandeep Dhaliwal, Email: sandeep.dhaliwal@ucdmc.ucdavis.edu.
Ross L. Coppel, Email: Ross.Coppel@monash.edu.
Aftab A. Ansari, Email: pathaaa@emory.edu.
Guo-Xiang Yang, Email: gxyang@ucdavis.edu.
Jinjun Wang, Email: yjwang@ucdavis.edu.
Thomas P. Kenny, Email: tpkenny@ucdavis.edu.
Xiao-Song He, Email: xiaosong@stanford.edu.
Ian R. Mackay, Email: ian.mackay@med.monash.edu.
M. Eric Gershwin, Email: megershwin@ucdavis.edu.
References
- 1.Gardam S, Brink R. Non-Canonical NF-kappaB Signaling Initiated by BAFF Influences B Cell Biology at Multiple Junctures. Front Immunol. 2014;4:509. doi: 10.3389/fimmu.2013.00509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bergmann B, Grimsholm O, Thorarinsdottir K, Ren W, Jirholt P, Gjertsson I, Martensson IL. Memory B cells in mouse models. Scand J Immunol. 2013;78:149–156. doi: 10.1111/sji.12073. [DOI] [PubMed] [Google Scholar]
- 3.Wrammert J, Smith K, Miller J, Langley WA, Kokko K, Larsen C, Zheng NY, Mays I, Garman L, Helms C, et al. Rapid cloning of high-affinity human monoclonal antibodies against influenza virus. Nature. 2008;453:667–671. doi: 10.1038/nature06890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kunkel EJ, Butcher EC. Chemokines and the tissue-specific migration of lymphocytes. Immunity. 2002;16:1–4. doi: 10.1016/s1074-7613(01)00261-8. [DOI] [PubMed] [Google Scholar]
- 5.He XS, Sasaki S, Narvaez CF, Zhang C, Liu H, Woo JC, Kemble GW, Dekker CL, Davis MM, Greenberg HB. Plasmablast-derived polyclonal antibody response after influenza vaccination. J Immunol Methods. 2011;365:67–75. doi: 10.1016/j.jim.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen RC, Naiyanetr P, Shu SA, Wang J, Yang GX, Kenny TP, Guggenheim KC, Butler JD, Bowlus C, Tao MH, et al. Antimitochondrial antibody heterogeneity and the xenobiotic etiology of primary biliary cirrhosis. Hepatology. 2013;57:1498–1508. doi: 10.1002/hep.26157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang J, Budamagunta MS, Voss JC, Kurth MJ, Lam KS, Lu L, Kenny TP, Bowlus C, Kikuchi K, Coppel RL, et al. Antimitochondrial antibody recognition and structural integrity of the inner lipoyl domain of the E2 subunit of pyruvate dehydrogenase complex. J Immunol. 2013;191:2126–2133. doi: 10.4049/jimmunol.1301092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leung PS, Lam K, Kurth MJ, Coppel RL, Gershwin ME. Xenobiotics and autoimmunity: does acetaminophen cause primary biliary cirrhosis? Trends Mol Med. 2012;18:577–582. doi: 10.1016/j.molmed.2012.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lindor KD, Gershwin ME, Poupon R, Kaplan M, Bergasa NV, Heathcote EJ American Association for Study of Liver, D. Primary biliary cirrhosis. Hepatology. 2009;50:291–308. doi: 10.1002/hep.22906. [DOI] [PubMed] [Google Scholar]
- 10.Chapman R, Fevery J, Kalloo A, Nagorney DM, Boberg KM, Shneider B, Gores GJ American Association for Study of Liver, D. Diagnosis and management of primary sclerosing cholangitis. Hepatology. 2010;51:660–678. doi: 10.1002/hep.23294. [DOI] [PubMed] [Google Scholar]
- 11.Wang Q, Selmi C, Zhou X, Qiu D, Li Z, Miao Q, Chen X, Wang J, Krawitt EL, Gershwin ME, et al. Epigenetic considerations and the clinical reevaluation of the overlap syndrome between primary biliary cirrhosis and autoimmune hepatitis. J Autoimmun. 2013;41:140–145. doi: 10.1016/j.jaut.2012.10.004. [DOI] [PubMed] [Google Scholar]
- 12.Tartakovsky F, Worman HJ. Detection of Gp210 autoantibodies in primary biliary cirrhosis using a recombinant protein containing the predominant autoepitope. Hepatology. 1995;21:495–500. [PubMed] [Google Scholar]
- 13.Leung PS, Iwayama T, Coppel RL, Gershwin ME. Site-directed mutagenesis of lysine within the immunodominant autoepitope of PDC-E2. Hepatology. 1990;12:1321–1328. doi: 10.1002/hep.1840120612. [DOI] [PubMed] [Google Scholar]
- 14.Wang J, Yang GX, Zhang W, Lu L, Tsuneyama K, Kronenberg M, Vela JL, Lopez-Hoyos M, He XS, Ridgway WM, et al. Escherichia coli infection induces autoimmune cholangitis and antimitochondrial antibodies in NOD.B6 (Idd10/Idd18) mice: Clin Exp Immunol - used Patrick's E. Coli 9-9-13.enl. Clin Exp Immunol. 2013 doi: 10.1111/cei.12224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wakabayashi K, Lian ZX, Leung PS, Moritoki Y, Tsuneyama K, Kurth MJ, Lam KS, Yoshida K, Yang GX, Hibi T, et al. Loss of tolerance in C57BL/6 mice to the autoantigen E2 subunit of pyruvate dehydrogenase by a xenobiotic with ensuing biliary ductular disease. Hepatology. 2008;48:531–540. doi: 10.1002/hep.22390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee FE, Halliley JL, Walsh EE, Moscatiello AP, Kmush BL, Falsey AR, Randall TD, Kaminiski DA, Miller RK, Sanz I. Circulating human antibody-secreting cells during vaccinations and respiratory viral infections are characterized by high specificity and lack of bystander effect. J Immunol. 2011;186:5514–5521. doi: 10.4049/jimmunol.1002932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Slocombe T, Brown S, Miles K, Gray M, Barr TA, Gray D. Plasma cell homeostasis: the effects of chronic antigen stimulation and inflammation. J Immunol. 2013;191:3128–3138. doi: 10.4049/jimmunol.1301163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Amanna IJ, Carlson NE, Slifka MK. Duration of humoral immunity to common viral and vaccine antigens. N Engl J Med. 2007;357:1903–1915. doi: 10.1056/NEJMoa066092. [DOI] [PubMed] [Google Scholar]
- 19.Takahashi T, Miura T, Nakamura J, Yamada S, Miura T, Yanagi M, Matsuda Y, Usuda H, Emura I, Tsuneyama K, et al. Plasma cells and the chronic nonsuppurative destructive cholangitis of primary biliary cirrhosis. Hepatology. 2012;55:846–855. doi: 10.1002/hep.24757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Duan B, Morel L. Role of B-1a cells in autoimmunity. Autoimmun Rev. 2006;5:403–408. doi: 10.1016/j.autrev.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 21.Tazawa H, Irei T, Tanaka Y, Igarashi Y, Tashiro H, Ohdan H. Blockade of invariant TCR-CD1d interaction specifically inhibits antibody production against blood group A carbohydrates. Blood. 2013;122:2582–2590. doi: 10.1182/blood-2012-02-407452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jacobi AM, Mei H, Hoyer BF, Mumtaz IM, Thiele K, Radbruch A, Burmester GR, Hiepe F, Dorner T. HLA-DRhigh/CD27high plasmablasts indicate active disease in patients with systemic lupus erythematosus. Ann Rheum Dis. 2010;69:305–308. doi: 10.1136/ard.2008.096495. [DOI] [PubMed] [Google Scholar]
- 23.Ten Boekel E, Siegert CE, Vrielink GJ, Van Dam VC, Ceelen A, De Kieviet W. Analyses of CD27++ plasma cells in peripheral blood from patients with bacterial infections and patients with serum antinuclear antibodies. J Clin Immunol. 2007;27:467–476. doi: 10.1007/s10875-007-9099-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kunkel EJ, Kim CH, Lazarus NH, Vierra MA, Soler D, Bowman EP, Butcher EC. CCR10 expression is a common feature of circulating and mucosal epithelial tissue IgA Ab-secreting cells. J Clin Invest. 2003;111:1001–1010. doi: 10.1172/JCI17244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sanchez-Martin L, Sanchez-Mateos P, Cabanas C. CXCR7 impact on CXCL12 biology and disease. Trends Mol Med. 2013;19:12–22. doi: 10.1016/j.molmed.2012.10.004. [DOI] [PubMed] [Google Scholar]
- 26.Wald O, Pappo O, Safadi R, Dagan-Berger M, Beider K, Wald H, Franitza S, Weiss I, Avniel S, Boaz P, et al. Involvement of the CXCL12/CXCR4 pathway in the advanced liver disease that is associated with hepatitis C virus or hepatitis B virus. Eur J Immunol. 2004;34:1164–1174. doi: 10.1002/eji.200324441. [DOI] [PubMed] [Google Scholar]
- 27.Terada R, Yamamoto K, Hakoda T, Shimada N, Okano N, Baba N, Ninomiya Y, Gershwin ME, Shiratori Y. Stromal cell-derived factor-1 from biliary epithelial cells recruits CXCR4-positive cells: implications for inflammatory liver diseases. Lab Invest. 2003;83:665–672. doi: 10.1097/01.lab.0000067498.89585.06. [DOI] [PubMed] [Google Scholar]
- 28.Shimoda S, Selmi C, Gershwin ME. Fractalkine and other chemokines in primary biliary cirrhosis. Int J Hepatol. 2012;2012:102839. doi: 10.1155/2012/102839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eksteen B, Miles A, Curbishley SM, Tselepis C, Grant AJ, Walker LS, Adams DH. Epithelial inflammation is associated with CCL28 production and the recruitment of regulatory T cells expressing CCR10. J Immunol. 2006;177:593–603. doi: 10.4049/jimmunol.177.1.593. [DOI] [PubMed] [Google Scholar]
- 30.Lleo A, Liao J, Invernizzi P, Zhao M, Bernuzzi F, Ma L, Lanzi G, Ansari AA, Coppel RL, Zhang P, et al. Immunoglobulin M levels inversely correlate with CD40 ligand promoter methylation in patients with primary biliary cirrhosis. Hepatology. 2012;55:153–160. doi: 10.1002/hep.24630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Smyk D, Rigopoulou EI, Baum H, Burroughs AK, Vergani D, Bogdanos DP. Autoimmunity and environment: am I at risk? Clin Rev Allergy Immunol. 2012;42:199–212. doi: 10.1007/s12016-011-8259-x. [DOI] [PubMed] [Google Scholar]
- 32.McNally RJ, James PW, Ducker S, James OF. Seasonal variation in the patient diagnosis of primary biliary cirrhosis: further evidence for an environmental component to etiology. Hepatology. 2011;54:2099–2103. doi: 10.1002/hep.24597. [DOI] [PubMed] [Google Scholar]
- 33.Leung PS, Wang J, Naiyanetr P, Kenny TP, Lam KS, Kurth MJ, Gershwin ME. Environment and primary biliary cirrhosis: electrophilic drugs and the induction of AMA. J Autoimmun. 2013;41:79–86. doi: 10.1016/j.jaut.2012.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Amano K, Leung PS, Rieger R, Quan C, Wang X, Marik J, Suen YF, Kurth MJ, Nantz MH, Ansari AA, et al. Chemical xenobiotics and mitochondrial autoantigens in primary biliary cirrhosis: identification of antibodies against a common environmental, cosmetic, and food additive, 2-octynoic acid. J Immunol. 2005;174:5874–5883. doi: 10.4049/jimmunol.174.9.5874. [DOI] [PubMed] [Google Scholar]
- 35.Jin Q, Moritoki Y, Lleo A, Tsuneyama K, Invernizzi P, Moritoki H, Kikuchi K, Lian ZX, Hirschfield GM, Ansari AA, et al. Comparative analysis of portal cell infiltrates in antimitochondrial autoantibody-positive versus antimitochondrial autoantibody-negative primary biliary cirrhosis. Hepatology. 2012;55:1495–1506. doi: 10.1002/hep.25511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lleo A, Selmi C, Invernizzi P, Podda M, Coppel RL, Mackay IR, Gores GJ, Ansari AA, Van de Water J, Gershwin ME. Apotopes and the biliary specificity of primary biliary cirrhosis. Hepatology. 2009;49:871–879. doi: 10.1002/hep.22736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Scholz JL, Cancro MP. Resolve, revise, and relax: the 3 Rs of B cell repertoire adjustment. Immunol Lett. 2012;143:2–8. doi: 10.1016/j.imlet.2012.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gershwin ME, Selmi C, Worman HJ, Gold EB, Watnik M, Utts J, Lindor KD, Kaplan MM, Vierling JM, Group UPE. Risk factors and comorbidities in primary biliary cirrhosis: a controlled interview-based study of 1032 patients. Hepatology. 2005;42:1194–1202. doi: 10.1002/hep.20907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stravitz RT, Lefkowitch JH, Fontana RJ, Gershwin ME, Leung PS, Sterling RK, Manns MP, Norman GL, Lee WM. Autoimmune acute liver failure: proposed clinical and histological criteria. Hepatology. 2011;53:517–526. doi: 10.1002/hep.24080. [DOI] [PMC free article] [PubMed] [Google Scholar]