

Abstract
Alterations of the dopaminergic system are associated with the cognitive and functional dysfunctions that characterize complex neuropsychiatric disorders. We modeled a dysfunctional dopaminergic system using mice with targeted ablation of dopamine (DA) D2 autoreceptors in mesencephalic dopaminergic neurons. Loss of D2 autoreceptors abolishes D2-mediated control of DA synthesis and release. Here we show that this mutation leads to a profound alteration of the genomic landscape of neurons receiving dopaminergic afferents at distal sites, specifically in the prefrontal cortex. Indeed, we observed a remarkable down-regulation of gene expression in this area of ∼ 2,000 genes, which involves a widespread increase in the histone repressive mark H3K9me2/3. This reprogramming process is coupled to psychotic-like behaviors in the mutant mice. Importantly, chronic treatment with a DA agonist can revert the genomic phenotype. Thus, cortical neurons undergo a profound epigenetic reprogramming in response to dysfunctional D2 autoreceptor signaling leading to altered DA levels, a process that may underlie a number of neuropsychiatric disorders.
Keywords: Dopamine, D2 autoreceptors, mouse model, epigenetics, neuropsychiatric disorders
Introduction
Epigenetic reprogramming is at the heart of cellular differentiation 1. Critical control by chromatin remodeling occurs also in fully differentiated neurons upon pharmacological and behavioral challenges 2. However, how defective neuronal circuits impinge on the specificity and extent of epigenetic programs has not been fully elucidated. Dysfunctions of dopamine (DA) transmission have been causally linked to cognitive and functional impairments proper of a number of neuropsychiatric disorders, such as schizophrenia. Indeed, the DA hypothesis of schizophrenia is one of the most credited hypotheses for the genesis of this disorder, based also on the therapeutic benefits of DA antagonists in patients3-11.
DA release is strictly regulated by convergent signaling from different neurotransmitters12 on dopaminergic neurons as well as by proteins involved in the presynaptic control of this function. The dopamine D2 receptor (D2R) is responsible for inhibiting the synthesis and release of DA from dopaminergic neurons, acting as an autoreceptor 13, 14. Recently we generated site-specific knockout mice, lacking D2R selectively from dopaminergic neurons (hereafter referred as DA-D2RKO) 15. These mice allow addressing in vivo the consequences of D2 autoreceptor deficiency. As expected, these mice are characterized by loss of inhibition of DA synthesis and altered DA release; electrochemical analyses showed diminished DA release after single or multiple pulses 15. Nevertheless, when DA-D2RKO mice are given cocaine, which blocks the DA transporter inhibiting DA reuptake, these mice have a much heightened motor response to the drug indicating increased levels of DA in the striatum. Thus, absence of D2 autoreceptors eliminates the D2R-mediated cell-autonomous control of DA levels both in normal conditions and in response to stimuli15.
In the present study, we used these mice to model a dysfunctional DA system where to analyze the consequences of altered DA levels on areas receiving dopaminergic fibers at the molecular and behavioral levels. With this purpose, we focused on areas involved in neuropsychiatric disorders, the prefrontal cortex and striatum. Our study reveals that dysfunctional control of DA synthesis and release results in an unexpected reprogramming of gene expression in the prefrontal cortex linked to epigenetic modifications of chromatin accompanied by cognitive and behavioral deficits. These studies highlight the critical role of proper DA signaling in the cortex and suggest that in some cases neuropsychiatric disorders might arise from dysfunctional neurotransmission.
Materials and Methods
Detailed descriptions are found in Supplementary Methods (SM)
Animals and treatments
DA-D2RKO 15 and WT littermates were housed under standard conditions (12 h light/dark cycles) with food and water ad libitum. All experiments were performed using 8- to 12-week-old mice (unless otherwise specified) in accordance to the Institutional Animal Care and Use Committee of the University of California Irvine, National Institute of Health, and institutional guidelines. Clozapine (Sigma, USA) was dissolved in 0.1N HCL and then diluted in 0.9% NaCl (adjusted to pH 7), 3mg/kg were administered (i.p) for 21 days. Quinpirole (Sigma, USA) in 0.9% NaCl was injected (i.p) at 0.2 mg/kg for 15 days. Amphetamine (Sigma, USA) in 0.9% NaCl was administered (i.p.) at 1 and 3 mg/kg.
Brains were quickly extracted, placed on ice and tissue punches for RNA, MS and western analyses were obtained from 1mm slices (PFC: Bregma 3.5-2.5 mm), (striatum: Bregma 1.5-0.5mm) using a coronal brain matrix (Harvard apparatus for adult mice ∼30g). Nissl staining of brain sections are described in SM.
RNA preparation and micro-array experiments
Total RNA was extracted using mini RNeasy kit (Qiagen). cDNAs were obtained from 100 ng of total RNA per sample (GeneChip cDNA synthesis Kit; Affymetrix) and hybridized to Mogene (Affymetrix “MoGene 1.0 ST”) by DNA Microarray Core Facility (UCI). Four (PFC) and three (vStr) microarrays/genotype were used. Normalization and analysis were performed using Partek Genomics Suite software, Genecodis software and DAVID (See SM).
qRT-PCR
Total RNA was extracted with TRIzol and cDNAs prepared using M-MLV kit (Invitrogen). 25-50 ng of cDNA/sample were used and PCR performed with iQSYBR Green Supermix (Bio-rad) using the following settings: 95°C for 10min, 40 cycles at 95°C for 15 sec and 60°C for 45sec. See SM for primer sequences.
Western-blot analyses
Frozen tissue punches of the relevant regions were homogenized and boiled in 1% SDS. For clozapine and quinpirole treatments tissues were obtained 24h after the last injection. Extracts (50 μg) were separated on SDS-PAGE and transferred onto membranes. Antibody used were: anti-H3K9me2 (1:5000), anti-H3K9me3 (1:1000) from Active Motif; anti-H3K4me3 (1:1000), anti-H3 (1:1000) from Abcam; anti-GAPDH (1:20,000) Millipore; anti-Akt1 (1:1000) Cell Signaling; anti-NR4A2 (1:1000) Antibody Verify. Secondary anti-rabbit and/or anti-mouse antibodies (1:5000) were from Millipore. Western blots were revealed using ECL Plus (GE Healthcare or Westdura, Pierce). Quantifications were performed using the NIH ImageJ (version 1.42q) software.
Mass spectrometry analyses
PFCs and striatal punches from WT and DA-D2RKO mice were used for MS analyses of histone modifications (n=4/5/genotype/area) performed as previously described 16 (see SM).
Immunohistochemical analyses
Immunostainings were performed as described in SM on 10 μm coronal brain sections using rabbit anti-H3K9me3 (1:500, Active Motif), mouse anti-GAD67 (1:500, Abcam), and mouse anti-NeuN (1:500, Millipore) antibodies followed by anti-rabbit CY3 (1:500, Jackson laboratories) or anti-mouse Alexa-488 (1:800, Jackson Laboratories) or anti-mouse Alexa-546 (1:500, Jackson Laboratories) antibodies. Nuclei were counterstained with Draq5 (1:500, Enzo Life Science). Images were taken at the confocal (DMRE; SP5 Leica) and quantified using LAS AF (Leica) in 3 regions of interest (ROI) of 145x145 μm/image (in Fig. 2f n=16-18 images/mouse 3mice/genotype; in Fig. 3f n=14 images/mouse 3mice/genotype). Data represent ratios of pixels fluorescence/Draq5 per ROIs.
Figure 2. Epigenetic modifications in the PFC of DA-D2RKO mice.
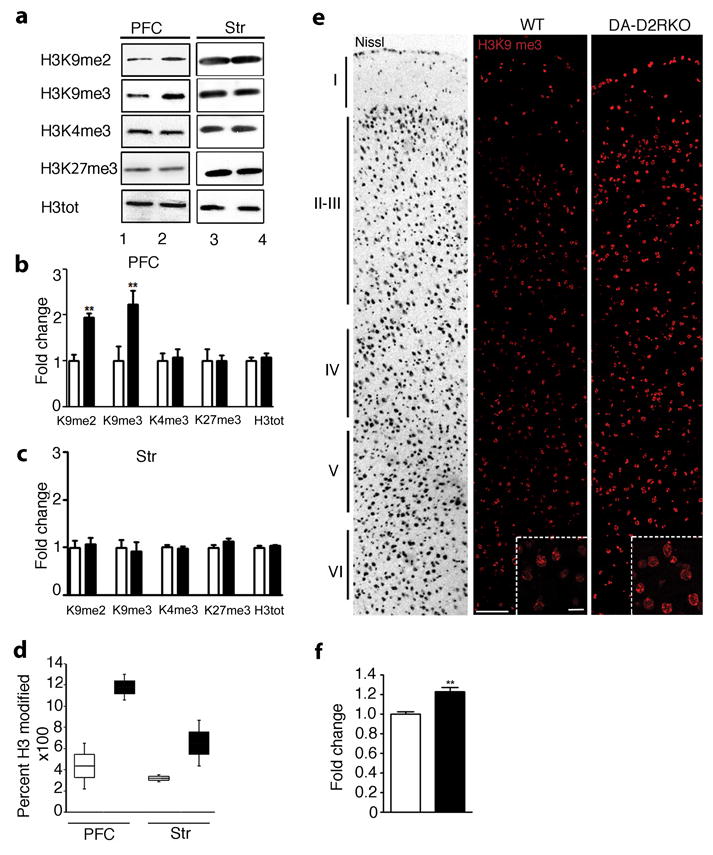
(a) Representative western blot analyses of histone H3K9, K4 and K27 modifications (as indicated) in PFC and striatal extracts from WT (lanes 1, 3) and DA-D2RKO (lanes 2, 4) mice. b-d, f) WT: white bars; DA-D2RKO: black bars. Values are mean ± SEM. (b-c) Quantifications of experiments in (a) (WT values arbitrarily set to 1) show a specific increase of H3K9me2 and me3 in the PFC of mutants (Student's t test: **p<0.01) (n=3WT/5DA-D2RKO/area). Loaded quantities were normalized to H3 total (H3tot) and GAPDH; (d) Box plot representation of Tandem MS/MS analysis of H3K9me2/3 in PFC and striatum extracts of both genotypes (n=4WT/5DA-D2RKO/area). (e) Representative images of Nissl staining of WT PFC (left panel; layers are indicated) and of H3K9me3 immunofluorescence of the same region in WT and DA-D2RKO, as indicated. Inset: magnifications of layer V. Scale bars: 100μm; inset: 20 μm. (f) Quantifications of experiments in (e) (WT intensity set as 1) showed significantly increased H3K9me3 staining in mutant PFCs (Student's t test: **p<0.01) (n=3WT/3DA-D2RKO).
Figure 3. Silencing of Nr4A2 and Akt1 genes in the PFC of DA-D2RKO mice is associated with H3K9me3.
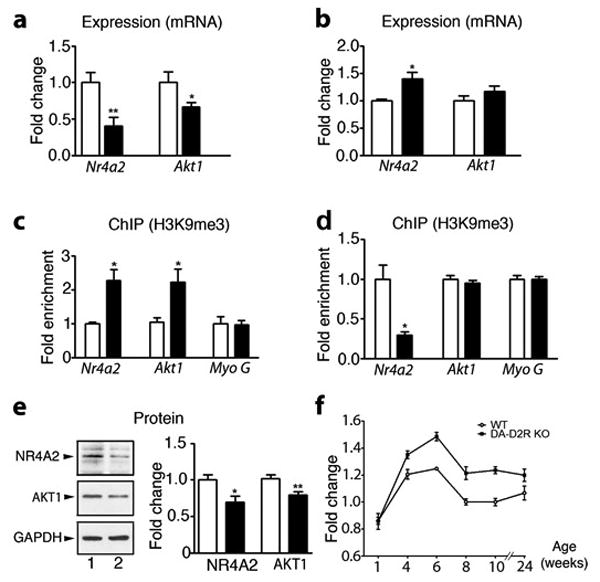
(a-f) WT: white bars; DA-D2RKO: black bars. Values are means ± SEM. (a) qRT-PCR of Nr4a2 and Akt1 mRNA expression in the PFC and (b) striatum (n= 4WT/6DA-D2RKO/area) (WT value set as 1). (Student's t test:*p < 0.05, **p<0.01). (c) Quantitative H3K9me3 ChIP on Nr4a2 (n=4/genotype ), Akt1 (n=3/genotype) and Myoglobin G promoters in PFC and (d) striatum extracts. (Student's t test; *p < 0.05) (e) Western-blot analyses (left) and quantifications (right) of NR4A2 (n=4WT/5DA-D2RKO), AKT 1 (n=6WT/7DA-D2RKO) and GAPDH protein levels in WT (lane 1) and DA-D2RKO mice (lane 2) expressed as fold changes from WT. (Student's t test: *p < 0.05, **p < 0.01). (f) Quantification of H3K9me3 immunostaining of sections from WT and DA-D2RKO PFCs during post-natal life, as indicated. A significant genotype x age interaction is noted (F (5,30)=2.48 p=0.0337) (n=3 mice/genotype).
Chromatin Immunoprecipitation (ChIP)
ChIP experiments were performed as previously described 17. PFC and Str punches from 6 mice/genotype were pooled and DNA sheered by sonication (5 times, 30 sec (40% power) every 30 sec). 5μl of anti-H3K9me3 or anti-IgG antibodies/sample were used. Recovered DNA was subjected to qRT-PCR (98°C 3 min, 40 cycles at 98°C for 30 sec and 60°C for 60 sec). See SM for primer sequences.
Behavioral Experiments
Acoustic Startle reflex (ASR) and Prepulse Inhibition (PPI)
ASR and PPI were performed as previously described 18, 19 on independent mouse groups (See SM).
Motor activity
Mice were habituated for 2 hours to a novel home cage, then administered either saline or amphetamine at 1 or 3 mg/kg (ip) and activity recorded for 1h. Mice injected with either saline or clozapine (3mg/kg) were analyzed in the open field (a white square box of 30x30 cm; 70 lux) and their motor activity analyzed for 15 min (Supplementary Figure 5) 24 h after the last injection. Activity was recorded by a video-tracking system (Viewpoint).
Radial Maze
The 8-arm radial maze test was performed to investigate both spatial and working memory exactly as described 20. Time to complete the task, working memory errors (reentry into a previously visited arm) and reference memory errors (entry into an arm without bait) were recorded and scored for 15 days after 6 days of acquisition.
Statistical analyses
Statistical analyses for micro-arrays were performed as described in Supplementary Methods section. For western-blot, immunohistochemical, qRT-PCR, and ChIP experiments, statistical comparisons were performed using Prism 3.0 (GraphPad) or SPSS 19 softwares and consisted of t-tests, one-way, two-way or two-way with repeated measures analyses of variance as indicated followed by appropriate post hoc analyses. For behavioral experiments: ASR, PPI and radial maze values were corrected by the Mauchly's test if sphericity was violated.
Results
Distinct gene expression profiles in the PFC and striatum of DA-D2RKO mice
To elucidate the impact of a defective DA signaling on gene expression, we performed unbiased micro-array profiling on two areas targeted by the DA mesolimbic pathway, the prefrontal cortex (PFC) and ventral striatum (vStr). Strikingly, analysis of the PFC of DA-D2RKO revealed a robust, widespread down-regulation of gene expression (Figure 1a) (1930 genes (Supplementary Table 1) p<0.05, 8.6 % of the total transcripts of which 1809 genes annotated) as compared to WT (Figure 1b) whereas expression of only 2 genes (<0.009% of transcripts) was up-regulated (Supplementary Table 1). By contrast, analyses of the vStr revealed a total of 132 genes differentially expressed between DA-D2RKO and WT mice (Figure 1b, Supplementary Figure 1a, Supplementary Table 2). Among these, only 20 genes were down-regulated (Supplementary Figure 1b) in DA-D2RKO versus WT vStr, with a negligible overlap between down-regulated genes expressed in both areas (Supplementary Figure 1b). The expression of 112 genes was instead up-regulated in the vStr (p<0.05) (Supplementary Table 2). Gene Ontology (GO) analyses of genes differentially expressed in DA-D2RKO PFC revealed that a preponderant number is involved in the regulation of transcription (240 genes; p= 1.8E-47); importantly, among these genes 15% participate in chromatin remodeling (Figure 1c, Supplementary Table 3). In contrast, GO analyses of the DA-D2RKO vStr identified 15 differentially expressed genes (p=0.042) involved in the regulation of transcription but none in chromatin remodeling (Supplementary Figure 1c; Supplementary Table 4). Pathway comparisons between PFC and vStr of DA-D2RKO mice showed virtual absence of common alterations in specific pathways (Supplementary Figure 2; Supplementary Tables 3, 4). These results demonstrate that ablation of D2 autoreceptors in DAergic neurons generates site-specific modifications of gene expression in the areas targeted by the mesolimbic pathway.
Figure 1. Widespread down-regulation of cortical genes expression in DA-D2RKO mice.
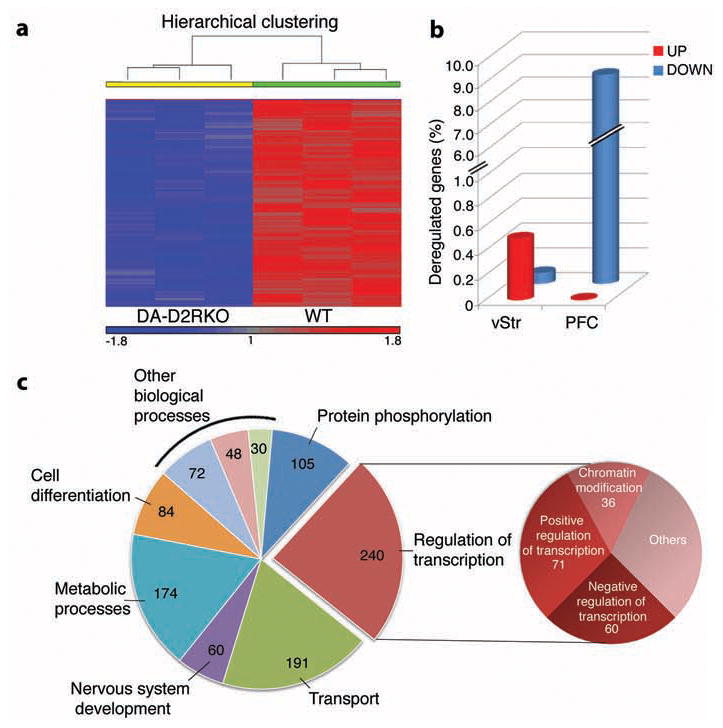
(a) Representative heat map of statistically significant (p<0.05) differentially expressed genes between DA-D2RKO and WT PFCs. (b) Percentage of genes differentially expressed relative to WT in the vStr and PFC of DA-D2RKO. (c) GO analysis of the DA-D2RKO PFC transcriptome. Pie chart (left) of the most significantly enriched biological processes; the pie chart on the right specifies the function of genes contained in the most enriched function: regulation of transcription (p=1.8E-47).
Extensive epigenetic reprogramming in the PFC of DA-D2RKO mice
Transcript down-regulation in the PFC was confirmed for a number of randomly selected genes (Supplementary Table 5). To dissect the molecular mechanisms underlying such widespread effects on gene expression, we analyzed post-translational histone modifications 1, 21-23. H3K9me2/3 24 is a well-characterized repressive mark, with dimethylated (H3K9me2) or trimethylated (H3K9me3) modifications enriched in heterochromatic portions of the genome 25. Western blot analyses of PFC and vStr revealed a significant increase of both H3K9me2 and H3K9me3 specifically in the PFC of DA-D2RKO mice as compared to WT littermates (Figure 2a, b), but not in the striatum (Figure 2a, c); H3K4me3 and H3K27me3 were unmodified in both regions (Figure 2a-c). To unequivocally confirm these results we performed quantitative mass spectrometry (MS) using extracts from the PFC and striatum of DA-D2RKO and WT mice. Interestingly, while the striatum of DA-D2RKO mice showed only a slight increase of H3K9me2/3 as compared to WT samples in MS analyses, we observed a robust increase of H3K9me2/3 specific to the PFC of DA-D2RKO mice as compared to WT PFCs or to the striatum of WT and mutant mice (Figure 2d; Supplementary Figure 3).
Immunofluorescence analyses using H3K9me3 antibodies, supported these findings showing a 20% increase in the overall intensity of H3K9me3 labeling in PFC brain sections of DA-D2R KO mice (Figure 2e, f), with a more sustained increase in deep layers (V and VI) (Figure 2e inset; Supplementary Figure 4a). Double immunofluorescence analyses using the neuronal markers NeuN (Supplementary Figure 4b) or GAD67 to label interneurons (Supplementary Figure 4c) showed that the increased H3K9me3 staining is localized in NeuN+ neurons of DA-D2RKO PFC. Importantly, similar neuronal density was found in the PFC of WT and DA-D2RKO mice (Supplementary Figure 4d).
Silencing histone modifications associated to genes involved in psychiatric disorders
Analyses of functions of genes down-regulated in the PFC (Ingenuity IPA software) revealed the prominent presence of 48 genes previously involved in neurological/psychiatric disorders (p< 10-13) in the DA-D2RKO mice, including 36 genes directly implicated in schizophrenia (Supplementary Table 6). Among these, we selected two genes, Nr4a2 (with highest differential expression from WT; 1.5 fold) and Akt1 (with the lowest differential expression from WT; 1.2 fold) to link the changes in gene expression to chromatin remodeling. These genes were also favored among others because reduced expression of both has been associated with DA-related disorders26,27. Their expression profile (together with that of other randomly selected genes; Supplementary Table 5) confirmed the microarray data, with a decrease of mRNA expression for both genes in the DA-D2RKO PFC as compared to WT (Figure 3a). In contrast to the PFC, comparison of Nr4a2 and Akt1 expression patterns in the striatum of DA-D2RKO and WT mice showed increased Nr4a2 expression (Figure 3b), but no difference for Akt1 expression in mutant mice. Chromatin immunoprecipitation (ChIP) 17 experiments using H3K9me3 antibodies demonstrated a remarkable enrichment of this mark on both Nr4a2 and Akt1 promoters in the PFC of DA-D2RKO mice as compared to WT littermates (Figure 3c). In the striatum H3K9me3 ChIPs showed either reduced (Nr4a2) or similar enrichments (Akt1) of their promoters in DA-D2RKO as compared to WT samples (Figure 3d), mirroring mRNA expression (Figure 3b). A corresponding reduction in the protein levels of NR4A2 and AKT1 was found in PFC protein extracts from DA-D2RKO mice (Figure 3e). Immunofluorescence analyses showed that increased H3K9me3 in the PFC is not present at birth, but it appears postnatally, becomes significant by the 4th week after birth and persists into adulthood (Figure 3f).
Psychotic-like behaviors in DA-D2RKO mice
The PFC-specific widespread epigenetic reprogramming and the newly acquired gene expression profile of the mutant mice prompted us to test DA-D2RKO mice in paradigms associated with psychotic-like behaviors 28. Hyperactivity in a novel environment and heightened response to the motor effect of amphetamines have been used to model psychotic behaviors 28, 29. DA-D2RKO mice show both phenotypes; indeed, they are hyperactive in a novel environment 15, and display an increased motor reactivity to amphetamine administration (Supplementary Figure 5a). Interestingly, hyperactivity in a novel environment can be reversed by administration of clozapine 30, an atypical antipsychotic (Supplementary Figure 5b).
We then evaluated the sensorimotor gating of DA-D2RKO mice by testing the startle reflex and pre-pulse inhibition (PPI) in comparison to WT mice. PPI is normally impaired in animal models of psychosis 29. DA-D2RKO mice exposed to pulses of different intensities showed a significantly amplified startle reflex to high pulse intensities as compared to WT controls (Figure 4a). Importantly, they also showed a significant reduction of PPI as compared to WT mice to the lowest startle-eliciting pulse (Figure 4b).
Figure 4. Behavioral alterations of DA-D2RKO mice.
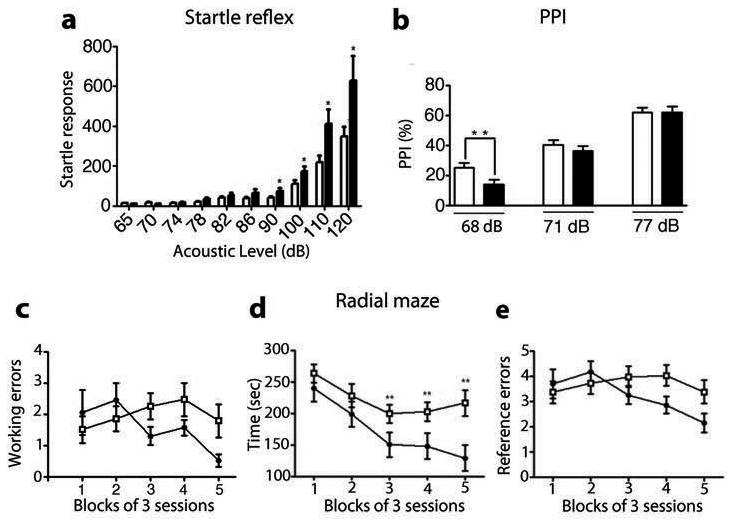
(a-e) WT: white bars; DA-D2RKO: black bars. Values are means ± SEM. (a) Startle reflex in WT and DA-D2RKO mice to pulses of increasing intensity (decibels: dB) indicates significant genotype x pulse intensity interaction (F(1.19, 52.24)= 4.027, p=0.043), genotype effect (F(1,46)= 8.534, p=0.005) and pulse intensity effect (F(1.19, 52.24)= 33.432, p=0.000) Bonferroni's: * p < 0.05. (b) PPI experiments indicates significant genotype x pre-pulse intensity interaction (F(2, 53)= 3.409, p=0.037), and pre-pulse intensity effect (F(2, 53)= 143.555, p=0.000), Bonferroni's:** p < 0.01. (c-e) WT and DA-D2RKO mice were tested in the radial maze for 15 days; results were grouped in 5 blocks of 3 sessions each. Analyses of radial maze results indicate significant differences between WT and DA-D2RKO mice in (c) working memory errors (number of reentries into previous visited arms), genotype x session interaction (F(3.43, 106.33)= 2.828, p=0.035) and (d) in the time to complete the task genotype x session interactions (F(3.07, 95.31) = 3.339, p=0.022), genotype effect (F(1,33)=8.243, p=0.007), and trial effect (F(3.07, 95.31)=20.670, p=0.000); Bonferroni's: *p<0.05, **p<0.01 (e) Analyses of reference memory errors in the radial maze (entrance in an un-baited arm) revealed absence of significant interactions (genotype x trials: F(4, 124)= 2.306, p=0.062; genotype effect: F(1,33)=0.613, p=0.440).
PFC-dependent disorders are characterized by deficits of working memory 31, 32. Thus, DA-D2RKO and WT mice were tested in the radial maze 20. Statistical analyses showed that DA-D2RKO mice had an impaired working memory (Figure 4c, d) but a normal reference memory in this test as compared to WT littermates (Figure 4e). Altogether these studies show that subtle modifications of DA signaling (ablation of D2 autoreceptors) cause PFC-specific cellular and behavioral impairments.
Quinpirole, a D2R agonist reverts epigenetic silencing
Our previous studies using DA-D2RKO mice showed a dual regulation of DA release orchestrated by D2 autoreceptors and by postsynaptic D2R-mediated inhibitory feedbacks on DA neurons 15. We thus analyzed whether chronic postsynaptic D2R stimulation might reverse the cellular phenotype of DA-D2RKO mice. Accordingly, DA-D2RKO mice were daily treated for 2 weeks with the D2R agonist quinpirole. Interestingly, in these conditions we observed a reversal of the PFC H3K9me3 to WT levels (Figure 5a); in agreement with these data, analyses performed at the mRNA level by qRT-PCR showed that quinpirole reestablish the expression levels of selected differentially expressed genes (Supplementary Table 5) in DA-D2RKO PFCs (Supplementary Figure 6) to WT levels. Concomitant to the effect of quinpirole on H3K9me3 levels in DA-D2RKO PFC, we found restored expression of AKT1 and NR4A2 proteins to WT levels (Figure 5c, d). Conversely, chronic clozapine was unable to reverse H3K9me3 (Figure 5b) or to restore protein expression for AKT1 and NR4A2 (Supplementary Figure 7). These results suggest that the PFC phenotype of DA-D2RKO mice is dependent from altered DA-mediated regulation of cortical neurons. In this respect, dysfunctional DA signaling has been associated to human neuropsychiatric disorders 3, 33.
Figure 5. Stimulation of D2Rs reverses the molecular phenotype of DA-D2RKO PFCs.
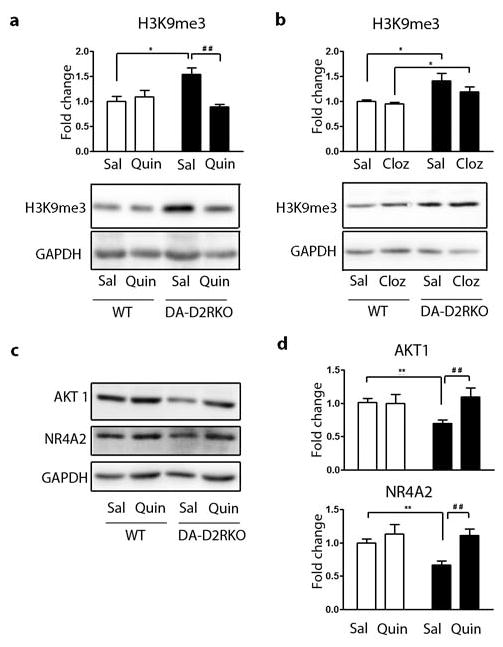
(a,b,d) WT: white bars; DA-D2RKO: black bars. Values are expressed as mean + S.E.M. Quantifications and representative western-blot analyses of H3K9me3 levels in PFC extracts of WT and DA-D2RKO mice chronically treated with saline (Sal) or (a) quinpirole (Quin) or (b) clozapine (Cloz). A significant effect of quinpirole was noted in DA-D2RKO vs WT mice, genotype x treatment interaction (F(1,9)=11.31; p=0.0083) and treatment effect (F(1,9)=6.75; p=0.0289) Bonferroni's: *p<0.05, ## p<0.01. Conversely clozapine revealed no effect of the treatment (F(1,14)=0.61, p=0.4464) but a significant genotype difference (F(1,14)=9.68; p=0.0067). Student's t test: *p<0.05. (c) Representative western-blot analyses of AKT1 and NR4A2 levels in the PFC of WT and DA-D2RKO mice chronically treated with quinpirole and (d) quantifications of experiments (shown in c). Data show that the observed decrease of AKT1 and NR4A2 protein levels in the PFC of DA-D2RKO mice can be rescued upon chronic quinpirole administration to WT levels for AKT1 genotype x treatment interaction (F(1,19)=5.78; p=0.0266) treatment effect (F(1,19)=5.09; p=0.0360) (Bonferroni's: **p<0.01, ## p<0.01); and for NR4A2, genotype (F(1,19)=4.42; p=0.0490) and treatment effect (F(1,19)=11.33; p=0.0032) (Student's t test: **p<0.01, ## p<0.01).
Discussion
Widespread epigenetic changes have been proposed to underlie alterations in gene expression associated to addiction and complex neuropsychiatric disorders, including schizophrenia 2, 22, 23. In this article, we present novel findings showing that altered control of DA synthesis and release due to ablation of D2 autoreceptors from dopaminergic neurons during development leads to major changes in the PFC, an area targeted by DA neurons of the mesolimbic pathway. Interestingly, we report a massive down-regulation of gene expression affecting ∼ 2,000 genes in DA-D2RKO PFC, this effect is paralleled by increased levels of the repressive histone mark H3K9me2/3. GO analyses revealed that a large number of down-regulated PFC genes are involved in transcriptional regulation and chromatin remodeling, including the demethylases KDM4b and KDM4c that specifically target H3K9me3 25, 34 (Supplementary Table 5), as well as genes involved in protein phosphorylation, transport and metabolic processes (Supplementary Table 3). Future studies will address whether reduced expression of these proteins is the leading event of the phenotype observed in DA-D2RKO mice. We show that H3K9me repressive chromatin mark in the PFC of DA-D2RKO mice directly targets at least two genes linked to schizophrenia, Akt1 27 and NR4A2 26. Thus, we establish a direct link between DAergic signaling, chromatin remodeling and the expression of genes implicated in psychiatric disorders. Importantly, we show that exposing mice to chronic treatment with quinpirole reverses histone methylation to normal levels, as reflected by western blot analyses of this modification on the PFC of DA-D2RKO treated mice as well as on the expression of Akt1 and NR4A2. Our findings might contribute to understanding the limited efficacy of antipsychotics in the treatment of schizophrenia 3, 4, 9.
Indeed, it is tempting to speculate that the use of D2R antagonists, while appropriate to treat symptoms associated with heightened DA signaling in the striatum are inefficient at improving PFC-linked functions, likely linked to lowered DA stimulation.
The strikingly different expression profile in the vStr of DA-D2RKO mice, where virtually neither transcriptional silencing nor increase in H3K9me3 are present, stresses the specificity of the widespread reprogramming occurring in PFC neurons and underscores the differences between striatal and cortical circuits. Thus, loss of D2 autoreceptors from DAergic neurons generates profound epigenetic modifications in brain areas targeted by DAergic fibers at distal sites. It is tempting to speculate that a similar reprogramming might operate in human psychiatric disorders.
Supplementary Material
Acknowledgments
Thank to Drs. F. Torri and C. De Mei for assistance and interest in the initial phase of this study; Dr. P. Sassone-Corsi for critical discussions and reading of the manuscript. Supported by NIH grant DA024689 and INSERM-44790 to E.B.
Footnotes
Conflict of interest: The author declare no conflict of interest
References
- 1.Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet. 2003;33 Suppl:245–254. doi: 10.1038/ng1089. [DOI] [PubMed] [Google Scholar]
- 2.Borrelli E, Nestler EJ, Allis CD, Sassone-Corsi P. Decoding the epigenetic language of neuronal plasticity. Neuron. 2008;60(6):961–974. doi: 10.1016/j.neuron.2008.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Howes OD, Kapur S. The dopamine hypothesis of schizophrenia: version III--the final common pathway. Schizophrenia bulletin. 2009;35(3):549–562. doi: 10.1093/schbul/sbp006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kuepper R, Skinbjerg M, Abi-Dargham A. The dopamine dysfunction in schizophrenia revisited: new insights into topography and course. Handb Exp Pharmacol. 2012;(212):1–26. doi: 10.1007/978-3-642-25761-2_1. [DOI] [PubMed] [Google Scholar]
- 5.Lyon GJ, Abi-Dargham A, Moore H, Lieberman JA, Javitch JA, Sulzer D. Presynaptic regulation of dopamine transmission in schizophrenia. Schizophrenia bulletin. 2011;37(1):108–117. doi: 10.1093/schbul/sbp010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miyake N, Thompson J, Skinbjerg M, Abi-Dargham A. Presynaptic dopamine in schizophrenia. CNS neuroscience & therapeutics. 2011;17(2):104–109. doi: 10.1111/j.1755-5949.2010.00230.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heinz A, Romero B, Gallinat J, Juckel G, Weinberger DR. Molecular brain imaging and the neurobiology and genetics of schizophrenia. Pharmacopsychiatry. 2003;36 Suppl 3:S152–157. doi: 10.1055/s-2003-45123. [DOI] [PubMed] [Google Scholar]
- 8.Howes O, Bose S, Turkheimer F, Valli I, Egerton A, Stahl D, et al. Progressive increase in striatal dopamine synthesis capacity as patients develop psychosis: a PET study. Molecular psychiatry. 2011;16(9):885–886. doi: 10.1038/mp.2011.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Howes OD, Egerton A, Allan V, McGuire P, Stokes P, Kapur S. Mechanisms underlying psychosis and antipsychotic treatment response in schizophrenia: insights from PET and SPECT imaging. Current pharmaceutical design. 2009;15(22):2550–2559. doi: 10.2174/138161209788957528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bertolino A, Knable MB, Saunders RC, Callicott JH, Kolachana B, Mattay VS, et al. The relationship between dorsolateral prefrontal N-acetylaspartate measures and striatal dopamine activity in schizophrenia. Biological psychiatry. 1999;45(6):660–667. doi: 10.1016/s0006-3223(98)00380-1. [DOI] [PubMed] [Google Scholar]
- 11.Meyer-Lindenberg A, Miletich RS, Kohn PD, Esposito G, Carson RE, Quarantelli M, et al. Reduced prefrontal activity predicts exaggerated striatal dopaminergic function in schizophrenia. Nature neuroscience. 2002;5(3):267–271. doi: 10.1038/nn804. [DOI] [PubMed] [Google Scholar]
- 12.Rice ME, Patel JC, Cragg SJ. Dopamine release in the basal ganglia. Neuroscience. 2011;198:112–137. doi: 10.1016/j.neuroscience.2011.08.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rouge-Pont F, Usiello A, Benoit-Marand M, Gonon F, Piazza PV, Borrelli E. Changes in extracellular dopamine induced by morphine and cocaine: crucial control by D2 receptors. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2002;22(8):3293–3301. doi: 10.1523/JNEUROSCI.22-08-03293.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Benoit-Marand M, Borrelli E, Gonon F. Inhibition of dopamine release via presynaptic D2 receptors: time course and functional characteristics in vivo. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2001;21(23):9134–9141. doi: 10.1523/JNEUROSCI.21-23-09134.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Anzalone A, Lizardi-Ortiz JE, Ramos M, De Mei C, Hopf FW, Iaccarino C, et al. Dual control of dopamine synthesis and release by presynaptic and postsynaptic dopamine D2 receptors. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2012;32(26):9023–9034. doi: 10.1523/JNEUROSCI.0918-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jasencakova Z, Scharf AN, Ask K, Corpet A, Imhof A, Almouzni G, et al. Replication stress interferes with histone recycling and predeposition marking of new histones. Molecular cell. 2010;37(5):736–743. doi: 10.1016/j.molcel.2010.01.033. [DOI] [PubMed] [Google Scholar]
- 17.Kumar A, Choi KH, Renthal W, Tsankova NM, Theobald DE, Truong HT, et al. Chromatin remodeling is a key mechanism underlying cocaine-induced plasticity in striatum. Neuron. 2005;48(2):303–314. doi: 10.1016/j.neuron.2005.09.023. [DOI] [PubMed] [Google Scholar]
- 18.Paylor R, Crawley JN. Inbred strain differences in prepulse inhibition of the mouse startle response. Psychopharmacology. 1997;132(2):169–180. doi: 10.1007/s002130050333. [DOI] [PubMed] [Google Scholar]
- 19.Errico F, Rossi S, Napolitano F, Catuogno V, Topo E, Fisone G, et al. D-aspartate prevents corticostriatal long-term depression and attenuates schizophrenia-like symptoms induced by amphetamine and MK-801. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2008;28(41):10404–10414. doi: 10.1523/JNEUROSCI.1618-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wood MA, Kaplan MP, Brensinger CM, Guo W, Abel T. Ubiquitin C-terminal hydrolase L3 (Uchl3) is involved in working memory. Hippocampus. 2005;15(5):610–621. doi: 10.1002/hipo.20082. [DOI] [PubMed] [Google Scholar]
- 21.Maze I, Noh KM, Allis CD. Histone regulation in the CNS: basic principles of epigenetic plasticity. Neuropsychopharmacology. 2013;38(1):3–22. doi: 10.1038/npp.2012.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Akbarian S, Huang HS. Epigenetic regulation in human brain-focus on histone lysine methylation. Biological psychiatry. 2009;65(3):198–203. doi: 10.1016/j.biopsych.2008.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Russo SJ, Nestler EJ. The brain reward circuitry in mood disorders. Nat Rev Neurosci. 2013;14(9):609–625. doi: 10.1038/nrn3381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, et al. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129(4):823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 25.Fodor BD, Kubicek S, Yonezawa M, O'Sullivan RJ, Sengupta R, Perez-Burgos L, et al. Jmjd2b antagonizes H3K9 trimethylation at pericentric heterochromatin in mammalian cells. Genes Dev. 2006;20(12):1557–1562. doi: 10.1101/gad.388206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rojas P, Joodmardi E, Hong Y, Perlmann T, Ogren SO. Adult mice with reduced Nurr1 expression: an animal model for schizophrenia. Molecular psychiatry. 2007;12(8):756–766. doi: 10.1038/sj.mp.4001993. [DOI] [PubMed] [Google Scholar]
- 27.Emamian ES, Hall D, Birnbaum MJ, Karayiorgou M, Gogos JA. Convergent evidence for impaired AKT1-GSK3beta signaling in schizophrenia. Nat Genet. 2004;36(2):131–137. doi: 10.1038/ng1296. [DOI] [PubMed] [Google Scholar]
- 28.Arguello PA, Gogos JA. Modeling madness in mice: one piece at a time. Neuron. 2006;52(1):179–196. doi: 10.1016/j.neuron.2006.09.023. [DOI] [PubMed] [Google Scholar]
- 29.Jones CA, Watson DJ, Fone KC. Animal models of schizophrenia. Br J Pharmacol. 2011;164(4):1162–1194. doi: 10.1111/j.1476-5381.2011.01386.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Seeman P. All roads to schizophrenia lead to dopamine supersensitivity and elevated dopamine D2(high) receptors. CNS neuroscience & therapeutics. 2011;17(2):118–132. doi: 10.1111/j.1755-5949.2010.00162.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yoon T, Okada J, Jung MW, Kim JJ. Prefrontal cortex and hippocampus subserve different components of working memory in rats. Learn Mem. 2008;15(3):97–105. doi: 10.1101/lm.850808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Papaleo F, Yang F, Garcia S, Chen J, Lu B, Crawley JN, et al. Dysbindin-1 modulates prefrontal cortical activity and schizophrenia-like behaviors via dopamine/D2 pathways. Molecular psychiatry. 2012;17(1):85–98. doi: 10.1038/mp.2010.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Goto Y, Grace AA. The dopamine system and the pathophysiology of schizophrenia: a basic science perspective. International review of neurobiology. 2007;78:41–68. doi: 10.1016/S0074-7742(06)78002-3. [DOI] [PubMed] [Google Scholar]
- 34.Whetstine JR, Nottke A, Lan F, Huarte M, Smolikov S, Chen Z, et al. Reversal of histone lysine trimethylation by the JMJD2 family of histone demethylases. Cell. 2006;125(3):467–481. doi: 10.1016/j.cell.2006.03.028. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.