

Abstract
The discovery of activating mutations in BRAF at high frequency in cutaneous melanoma opened the door to new treatment options, which have resulted in significantly better patient outcomes. Treatments such as the FDA-approved RAF inhibitor vemurafenib, and the more recently approved dabrafenib and trametinib combination therapy are designed to target the ERK1/2 pathway. Initial success in targeting this pathway is evidenced by the high percentage of melanoma patients who undergo tumor remission. However, the beneficial effects of these targeted therapies are usually short-lived due to the development of resistance, which leads to disease progression. As a result, studies have focused on the acquired forms of resistance that develop following continued exposure to therapy. Conversely, far fewer studies have investigated the adaptive forms of resistance, which activate rapidly, promote cell survival and may underlie the development of acquired resistance by providing melanoma cells the time to develop additional mutations. We provide a detailed review of the known mechanisms of adaptive resistance in melanoma and relate them to similar responses to targeted therapies in other tumor types.
Keywords: Adaptive, BRAF V600E, dabrafenib, vemurafenib
Introduction
The development of selective RAF inhibitors, such as vemurafenib and dabrafenib, has revolutionized the treatment of melanoma patients harboring v-raf murine sarcoma viral oncogene homolog B1 (BRAF) V600E mutations. Despite this major breakthrough, the clinical response of patients to these targeted inhibitors is extremely heterogeneous. In the phase II trial for vemurafenib (ClinicalTrials.gov number, NCT00949702), 6% of patients displayed a complete response, 47% of patients were partial responders, and 14% progressed with no tumor shrinkage (Sosman et al., 2012). A variable response was also observed in the combination therapy phase II trial of the RAF inhibitor dabrafenib plus the mitogen-activated protein kinase kinase (MEK) inhibitor trametinib (ClinicalTrials.gov number, NCT01072175), although the waterfall plot is shifted towards more partial and complete responses (Flaherty et al., 2012). Given the need to predict effective responders, it becomes critical to understand the mechanisms that underlie this heterogeneous response to RAF and MEK inhibitors in order to identify biomarkers that can predict objective responses versus primary resistance in BRAF V600E melanoma patients. We believe that adaptive responses to RAF inhibitors in BRAF V600E melanoma are a major determinant of the susceptibility of a tumor to targeted therapy.
Adaptive responses versus acquired resistance
It is important to differentiate between the different modes of resistance to targeted inhibitors, particularly, between adaptive responses and acquired resistance. We describe adaptive responses as mechanisms that are switched on to compensate for actions of BRAF and MEK inhibition in BRAF V600E melanoma cells. These responses are rapid, occurring within hours of drug treatment, and reversible in that drug removal resets the compensatory signal to its basal state. We postulate that when adaptive responses are exhibited by the majority of cells within the tumor, the degree of tumor shrinkage will be limited. At an individual cell level, adaptive responses promote survival signals that afford the cell time until a bona fide acquired mechanism takes over that allows permanent survival and growth in the presence of inhibitor. Acquired resistance at the level of the tumor refers to lesions that dramatically shrink with RAF inhibitors but subsequently regrow, often at a rapid rate. Outgrowth of cells may be due to the acquisition of a secondary mutation and/or selection of a single cell or small population of cells that harbor a pre-existing genetic alteration that negates the effect of RAF inhibitors. Alterations underlying acquired resistance are stable changes that allow irreversible resistance and often even a growth advantage that is drug dependent (Das Thakur et al., 2013; Hartsough et al., 2014).
In this review, we focus on mechanisms of adaptive response to RAF and MEK inhibitors. We divide these mechanisms into three broad modes: re-setting of extracellular signal-regulated kinase (ERK1/2) pathway activation, up-regulation of receptor tyrosine kinases (RTK) leading to compensatory PI-3K-AKT activation, and changes in metabolic pathways (see Figure 1). For mechanisms of acquired resistance to BRAF inhibitors, we point readers to several recent reviews, which have comprehensively covered this subject (Hartsough et al., 2013; Salama and Flaherty, 2013).
Figure 1. Overview of the adaptive mechanisms to RAF inhibitors in mutant BRAF melanoma.
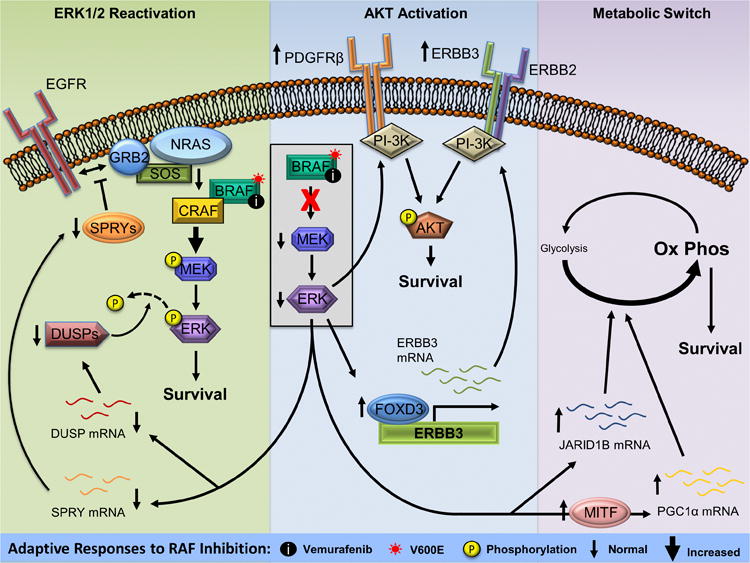
(Left) ERK1/2 pathway inhibition by vemurafenib leads to downregulation of DUSP and SPRY proteins. Loss of SPRY results in more efficient NRAS activation leading to a reactivation of the ERK1/2 pathway. This is enhanced by reduced ERK1/2 dephosphorylation resulting from lower levels of DUSP proteins. (Middle) Vemurafenib treatment increases PDGFRβ and ERBB3 leading to activation of the AKT pathway and promoting resistance to ERK1/2 pathway inhibition. (Right) Increased levels of JARID1B and PGC1α following ERK1/2 pathway inhibition leads to a metabolic switch from glycolysis to oxidative phosphorylation promoting resistance to RAF inhibition. Abbreviations used are: EGFR, epidermal growth factor receptor; GRB2, growth factor receptor-bound protein 2; SOS, son of sevenless; NRAS, neuroblastoma RAS viral oncogene homolog; BRAF, v-Raf murine sarcoma viral oncogene homolog B1; CRAF, v-Raf-1 murine leukemia viral oncogene homolog 1; MEK, mitogen-activated protein kinase kinase; ERK, extracellular signal-regulated kinase; DUSP, dual-specificity phosphatase; SPRY, sprouty; PDGFRβ, platelet-derived growth factor receptor, beta polypeptide; ERBB3, v-erb-b2 avian erythroblastic leukemia viral oncogene homolog 3; ERBB2, v-erb-b2 avian erythroblastic leukemia viral oncogene homolog 2; PI-3K, phosphatidylinositol 3-kinase; AKT, v-akt murine thymoma viral oncogene; FOXD3, forkhead box transcription factor D3; JARID1B, lysine-specific demethylase 5B; MITF, microphthalmia-associated transcription factor; PGC1α, peroxisome proliferator-activated receptor gamma coactivator 1 alpha.
Re-setting of ERK1/2 pathway activation
Mutant BRAF is a potent activator of MEK-ERK1/2 signaling (Davies et al., 2002) and RAF inhibitors efficiently reduce signaling through this pathway. Although often depicted in a simplified linear RAS-RAF-MEK-ERK1/2 model, signaling through this pathway is modulated at multiple levels. Scaffold molecules including kinase suppressor of RAS (KSR) (Morrison, 2001), MEK partner 1 (MP1), and IQ-motif GTPase-activating protein (IQGAP), have been described to control activation at distinct steps in the pathway and/or at subcellular locales (Kolch, 2005). Furthermore, the pathway is finely tuned through the action of negative feedback regulators such as Sprouty (SPRY) and Spred proteins which act at the level of RTK-RAS-RAF signaling (Kim and Bar-Sagi, 2004), and dual-specificity phosphatases (DUSPs) that dephosphorylate the activation loop of ERK1/2 (Owens and Keyse, 2007). In summary, these feedback regulators are responsible for dampening ERK1/2 pathway output.
Reactivation of the ERK1/2 pathway through stable events such as expression of neuroblastoma RAS viral oncogene homolog (NRAS) Q61 mutants or BRAF V600E splice variants is a common occurrence in acquired resistance to RAF inhibitors in melanoma (Nazarian et al., 2010; Poulikakos et al., 2011). Work from the Rosen laboratory indicates that re-setting of ERK1/2 pathway activity also contributes to the initial adaptive response to RAF inhibitors. In microarray studies, Sproutys, SPRY2 and SPRY4, and DUSPs 4 and 6 were identified to be significantly down-regulated by BRAF or MEK inhibition in cultured BRAF V600E melanoma cells (Pratilas et al., 2009). Active, GTP-loaded RAS is often very low when BRAF V600E is expressed but the loss of SPRY2 expression following vemurafenib treatment correlates with an increase in RAS activation (Lito et al., 2012). In turn, RAS activation enhances signaling through the ERK1/2 pathway in a manner dependent on BRAF/v-Raf-1 murine leukemia viral oncogene homolog 1 (CRAF) heterodimers (Lito et al., 2012; Heidorn et al., 2010). It is worth noting that ERK1/2 reactivation occurred in only a subset of the lines tested and was modest in that it did not rebound close to levels associated with full activity (Corcoran et al., 2012; Lito et al., 2012). Conversely, similar studies in BRAF V600E colorectal cancer and thyroid carcinoma cell lines showed robust ERK1/2 reactivation in comparison to BRAF V600E melanomas (Corcoran et al., 2012; Montero-Conde et al., 2013), suggesting that the level of ERK1/2 pathway reactivation differs between lineages and contributes in a significant manner to the poor response of mutant BRAF colorectal cancer and thyroid carcinoma patients to vemurafenib (Montero-Conde et al., 2013). Nonetheless, it is likely that even low level ERK1/2 re-activation and pathway output will contribute to the initial response of melanoma to RAF inhibitors. Targeting MEK downstream of BRAF/CRAF heterodimers using PD0325901 subsequent to RAF inhibitor treatment reduced ERK1/2 rebound and simultaneous treatment of tumor xenografts with both RAF and MEK inhibitors led to more pronounced tumor growth inhibition than either treatment alone (Lito et al., 2012). These data are consistent with the phase I/II results from the recently FDA-approved combination therapy of dabrafenib and trametinib in which progression-free survival and response rates were improved to 9.4 months and 76% respectively with the combination therapy compared to 5.8 months and 54% with dabrafenib monotherapy (Flaherty et al., 2012).
Collectively, these findings emphasize that multiple levels of fine-tuning are associated with the ERK1/2 pathway. Adaptive resetting of ERK1/2 flux occurs in some mutant BRAF melanoma lines following RAF inhibition due to the reduction of negative feedback regulators. Combinatorial targeting of RAF and MEK lessens ERK1/2 reactivation in cell-based studies and increases both the rate and duration of responses in patients.
Adaptive up-regulation of RTKs leading to activation of alternative signaling pathways
Early studies on resistance to vemurafenib in BRAF V600E melanomas indicated the involvement of the RTKs platelet-derived growth factor receptor (PDGFR) and insulin-like growth factor receptor (IGF-1R) (Nazarian et al., 2010; Villanueva et al., 2010). While these studies focused on mechanisms of acquired resistance, there is increasing evidence for up-regulation of RTKs as a rapid adaptive response to RAF inhibitors in BRAF V600E melanomas.
Upregulation of v-erb-b2 avian erythroblastic leukemia viral oncogene homolog 3 (ERBB3)/human epidermal growth factor receptor 3 (HER3) signaling is one example of an adaptive response to BRAF V600E inhibition. ERBB3 is a member of the epidermal growth factor receptor (EGFR)/ERBB family of RTKs (Wilson et al., 2009) but is not well studied compared to other family members. Initial studies showed that the forkhead box transcription factor D3 (FOXD3) is up-regulated following inhibition of the BRAF V600E-MEK-ERK1/2 pathway and modulates the survival of mutant BRAF melanoma cells in response to RAF inhibitors (Basile et al., 2012). Expression microarrays and chromatin immunoprecipitation coupled to DNA sequencing (ChIP-seq) analyses on FOXD3 target genes identified ERBB3 as being upregulated in cells overexpressing FOXD3 or treated with RAF inhibitors (Abel et al., 2013). ERBB3 expression was upregulated rapidly (within 4-6 hours) following exposure of cultured BRAF V600E/D melanoma cells to vemurafenib and was dependent on FOXD3 (Abel et al., 2013). ERBB3 upregulation was associated with enhanced sensitivity to its ligand, neuregulin-1 beta (NRG1), as determined by increased ERBB3 phosphorylation on carboxyl-terminal tyrosine residues upon treatment with exogenous NRG1. Effects were reversible, as removal of vemurafenib reset cells to a state displaying reduced response to NRG1. It is likely that NRG1 is produced mainly by cells in the tumor microenvironment since basal ERBB3 phosphorylation is low in cell monocultures in vitro, but is readily detectable in intradermal xenografts of mutant BRAF melanoma cells in vivo (Abel et al., 2013) and when cells are stimulated by conditioned medium from dermal fibroblasts (unpublished data). Studies with patient samples from RAF inhibitor trials supported the clinical relevance of these findings since ERBB3 phosphorylation was significantly higher in on-treatment samples compared to matched pre-treatment samples (Abel et al., 2013).
Cytoplasmic tyrosine phosphorylation of ERBB3 creates high affinity binding sites for the Src-homology 2 (SH2) domain of the catalytic subunit of phosphatidylinositol 3-kinase (PI-3K). In turn, PI-3K signals through the phosphoinositide-dependent kinase 1-v-akt murine thymoma viral oncogene (AKT) pathway, which is known to protect against the apoptotic actions of RAF inhibitors (Shao and Aplin, 2010; Paraiso et al., 2011; Gopal et al., 2010). Notably, ERBB3 differs from the other members of the family in that it exhibits very low kinase activity (Wilson et al., 2009; Shi et al., 2010). Thus, ERBB3 partners with its family member, v-erb-b2 avian erythroblastic leukemia viral oncogene homolog 2 (ERBB2), to elicit downstream signaling. NRG1-activated ERBB3-ERBB2 complexes led to enhanced growth, proliferation, and viability of RAF- or MEK-inhibited mutant BRAF melanoma cells in vitro. Conversely, vemurafenib combined with ERBB3 depletion or the ERBB2/EGFR inhibitor, lapatanib, to reduce the beneficial effects of NRG1 treatment in multiple BRAF V600E cell lines in vitro and enhanced effects of PLX4720 (the tool compound for vemurafenib) to significantly reduce tumor growth in vivo (Abel et al., 2013). The generation and utility of humanized neutralizing anti-ERBB3 antibodies is currently garnering much interest from the pharmaceutical industry (Gala and Chandarlapaty, 2014). Work currently in submission from our group shows that targeting ERBB3 with neutralizing antibodies in combination with PLX4720 can reduce tumor growth and promote durable responses in vivo (Kugel et al., manuscript under review). Thus, targeting ERBB3 may increase the efficacy of RAF inhibitors.
Although originally implicated in acquired resistance, platelet-derived growth factor receptor (PDGFRβ) is also upregulated as an adaptive response to RAF inhibition. Treatment of mutant BRAF melanoma cells with either vemurafenib, MEK inhibitor (AZD6244) or ERK1/2 inhibitor (FR180204) resulted in increased AKT phosphorylation (Shi et al., 2013). Use of the PDGFRβ inhibitor, sunitinib, indicated that AKT activation was attributed, in part, to the upregulation of PDGFRβ, although sunitinib did not fully inhibit the adaptive increase in AKT activation resulting from vemurafenib treatment. The combination of sunitinib and the EGFR inhibitor, gefitinib, resulted in maximal AKT inhibition, although EGFR expression is typically low in BRAF V600E melanoma (Corcoran et al., 2012; Prahallad et al., 2012). However, the Bernards group described a resistance mechanism to RAF inhibition whereby a subset of cells with elevated expression of EGFR displayed slow growth properties in the absence of inhibitor but were selected for in the presence of vemurafenib. Immunohistochemistry on patient samples confirmed elevated levels of EGFR in a percentage of patients who relapsed while on RAF and/or MEK inhibitor therapy (Sun et al., 2014). The slow growth phenotype of EGFR high-expressing cells suggests that patients displaying elevated EGFR following relapse to ERK1/2 pathway inhibitors would benefit from a “drug holiday” (Sharma et al., 2010). This work describes a mechanism in which the tumor as a whole may “adapt” to ERK1/2 inhibition. However, since the subset of cells express high levels of EGFR prior to RAF inhibition, at the cellular level we view this mechanism as a selection of a population with pre-existing resistance. Overall, these findings suggest that multiple RTKs may contribute to the response to RAF inhibitors in BRAF V600E melanomas and promote initial tumor survival.
The diversity of receptors that play a role in adaptive responses to RAF inhibition highlights the need to identify biomarkers that may serve as predictors for combinatorial targeting of RTKs and the ERK1/2 pathway. As the RTKs presented here predominantly activate the pro-survival PI-3K-AKT pathway, targeting this pathway may be a viable option to overcome RTK activity. However, while the AKT inhibitor MK2206 in combination with vemurafenib (Shi et al., 2013) and previous work showing PI-3K inhibitor in combination with a MEK inhibitor (Smalley et al., 2006) decreased colony growth in vitro compared to treatment with either alone, the utilization of PI-3K and AKT inhibitors in patients has been limited by toxicities (Curry et al., 2013). Furthermore, experience from other tumor types indicates that the efficiency of PI-3K and AKT inhibitors may be hampered by upregulation of RTKs as a compensatory effect (see below).
Adaptive RTK upregulation in response to targeted therapies in non-melanoma tumor types
BRAF mutations are also found in various tumor types besides melanoma. Although approximately 10% of colorectal cancers harbor BRAF V600E perturbations (Davies et al., 2002), vemurafenib is clinically ineffective in these patients compared to effects observed in melanoma (Kopetz et al., 2010). In vitro studies demonstrate that the resistance of BRAF V600E colorectal cancer cell lines is associated with an adaptive mechanism that utilizes intrinsically high levels of phosphorylated EGFR to engage signaling machinery to activate RAS and, consequently, reactivate the ERK1/2 pathway (Corcoran et al., 2012; Prahallad et al., 2012). A secondary mechanism may also be present that leads to an adaptive increase in IGF1R signaling to AKT (Corcoran et al., 2012). Regulation downstream of the receptor is similar to the notion of feedback mechanisms described by Rosen and colleagues in melanoma. In contrast to colorectal cancer cells, mutant BRAF melanoma cells often have low EGFR expression by utilization of other RTKs for adaptive responses in this tumor type. Additionally, BRAF V600E mutations are found in approximately 30% of papillary thyroid carcinomas and, when treated with vemurafenib, these cancers display increased expression and phosphorylation of ERBB3 (Montero-Conde et al., 2013).
Interestingly, adaptive response mechanisms have also been identified following treatment of non-mutant BRAF tumor types with targeted agents other than RAF inhibitors. In ERBB2-amplified breast cancer lines treated with ERBB family tyrosine kinase inhibitors, PI-3K inhibitors and/or AKT inhibitors, there is an induction of multiple phosphorylated RTKs with increased ERBB3 expression/phosphorylation being most notable (Chandarlapaty et al., 2011; Chakrabarty et al., 2012; Sergina et al., 2007; Serra et al., 2011). Importantly, upregulation of ERBB3 is detected in samples from patients treated with the AKT inhibitor GDC-0068 (ClinicalTrials.gov: NCT01090960), demonstrating that compensatory mechanisms occur in a clinical setting (Yan et al., 2013). These adaptive responses may be regulated by the tumor microenvironment. Muranen et al. showed that ovarian and breast cells adherent to the basement membrane extracellular matrix (ECM) displayed resistance to a dual specificity PI-3K/mTOR inhibitor in a 3D spheroid model (Muranen et al., 2012). This apparent ECM-mediated protection was associated with an adaptive signaling program involving multiple RTKs (EGFR, ERBB2 and IGF-1R), alternative signaling pathways (phosphorylation of STAT3 and STAT6) and pro-survival proteins (e.g. Bcl-2). Targeting several of these nodes, specifically EGFR, IGF-1R and Bcl-2, prevented resistance to PI-3K/mTOR inhibitors. Thus, these studies show that adaptive responses modulated by the tumor microenvironment will likely limit the efficacy of PI-3K and AKT inhibitors.
Oxidative metabolic responses
Oxidative phosphorylation, the primary metabolic process in normal human cells, provides a high yield of ATP compared to the alternative metabolic process of glycolysis (Papa et al., 2012). It is now widely accepted that the majority of cancers undergo a metabolic switch, known as the Warburg effect, in which glycolysis is favored over oxidative phosphorylation (Warburg, 1956). The reason for this switch is not completely understood; however, it is not due to loss of oxidative phosphorylation function (Moreno-Sánchez et al., 2007). Recently, an adaptive response to RAF inhibition was identified involving a metabolic reprogramming of mutant BRAF melanoma cells to revert back to oxidative phosphorylation and promote the survival of cells in the presence of drug (Haq et al., 2013). Gene set enrichment analysis (GSEA) on published patient data identified oxidative phosphorylation and ATP synthesis signatures that were increased in patients treated with vemurafenib. Notably, ERK1/2 pathway inhibition increased mRNA levels of the transcriptional coactivator peroxisome proliferator-activated receptor gamma coactivator 1 alpha (PGC1α). This finding was unique to melanomas and was directly controlled by up-regulation of microphthalmia-associated transcription factor (MITF). Overexpression of MITF in cells resulted in a reduction in lactate production, suggesting a decrease in glycolysis. Conversely, knockdown of MITF in patient-derived cell lines led to a significant enrichment of oxidative phosphorylation genes, which are regulated by PGC1α. These data suggest that inhibition of the ERK1/2 pathway in melanoma cells relieves suppression of MITF and PGC1α, which in turn, controls expression of oxidative phosphorylation genes. Notably, high PGC1α levels correlated with poor prognosis in patients (Haq et al., 2013). Reduced sensitivity to pathway inhibition by increased oxidative phosphorylation is mediated by an increase in both mitochondrial number and function. Furthermore, mitochondrial uncouplers and oxidative phosphorylation inhibitors combined with RAF inhibitor to decrease cell viability.
In a separate mechanism, activation of the oxidative phosphorylation pathway was also found in slow-cycling mutant BRAF cells that survived vemurafenib treatment. Levels of the H3K4-demethylase, lysine-specific demethylase 5B (JARID1B), were rapidly increased (within 72 hours) in response to the DNA damaging therapeutic cisplatin or by vemurafenib treatment of mutant BRAF cells in vitro and in xenografts. Increased JARID1B expression was also detected in samples from patients who relapsed while on vemurafenib compared to their matched pre-treatment controls. Knockdown of JARID1B combined with vemurafenib to reduce tumor growth in mice harboring BRAF V600E melanoma cell xenografts. Quantitative proteome profiling identified an increase in proteins associated with oxidative phosphorylation in cells over-expressing JARID1B. Similar to the findings of Haq et al., the oxidative phosphorylation inhibitor, oligomycin, combined with vemurafenib treatment to reduce colony growth in vitro and tumor xenograft growth of mutant BRAF cells. Interestingly, this effect was not dependent on ERK1/2 pathway targeting agents, as the emergence of cells with high JARID1B levels occurred in response to various chemotherapeutics (Roesch et al., 2013). Increased histone demethylase function, specifically that of JARID1A, has been previously implicated in a subpopulation of the EGFR mutant non-small cell lung cancer cells that are resistant to the EGFR inhibitor gefitinib; however, in that study, resistance was not attributed to an increase in oxidative phosphorylation but rather IGF1R-dependent signaling (Sharma et al., 2010).
Collectively, these studies describe a metabolic re-programming of mutant BRAF cells in response to targeted inhibitors as well as standard therapeutics. These findings suggest the potential benefit of targeting oxidative phosphorylation in combination with RAF inhibitors; however, such drugs have toxicity concerns in the clinic (Dykens and Will, 2007) and further refinement is likely to be required before combinatorial treatment is practical.
Conclusions
While the number of effective treatment options for metastatic melanoma has increased dramatically in the last three years, these treatments have revealed multiple resistance mechanisms such as resetting of the ERK1/2 pathway, RTK-mediated AKT activation, and deregulation of metabolic processes, which invariably lead to tumor regrowth. The heterogeneity of these mechanisms highlight the potential issue that within a single patient and possibly within a single tumor, multiple adaptive responses will present a complicated and difficult barrier. The identification and targeting of adaptive response mechanisms presented here may provide at least a partial solution to this problem. Combinatorial targeting of the ERK1/2 pathway and adaptive responses may increase the efficacy of current therapeutics by reducing the number of cells that survive initial drug treatment. This in turn will lower the percentage of surviving cells capable of acquiring additional mutations and is expected to decrease the likelihood of acquired resistance and disease progression. This indicates a strong need for the identification of biomarkers indicative of adaptive mechanisms in order to develop combinatorial treatments to enhance current targeted therapy treatments in mutant BRAF metastatic melanoma patients.
Acknowledgments
We thank Edward Hartsough and Sheera Rosenbaum for feedback on the manuscript. The authors are supported by grants from NIH (R01-CA160495) and the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation to AEA, and a pre-doctoral award from the Joanna M. Nicolay Melanoma Foundation to CHK.
Financial Support: AEA is supported by grants from NIH (R01-CA160495) and the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation to AEA. CHK was awarded a pre-doctoral award from the Joanna M. Nicolay Melanoma Foundation.
Footnotes
Disclosure of Potential Conflicts of Interest: The authors declare they have no competing financial relationships.
References
- Abel EV, Basile KJ, Kugel CH, III, Witkiewicz AK, Le K, Amaravadi RK, Karakousis GC, Xu X, Xu W, Schuchter LM, et al. Melanoma adapts to RAF/MEK inhibitors through FOXD3-mediated upregulation of ERBB3. J Clin Invest. 2013;123:2155–2168. doi: 10.1172/JCI65780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basile KJ, Abel EV, Aplin AE. Adaptive upregulation of FOXD3 and resistance to PLX4032/4720-induced cell death in mutant B-RAF melanoma cells. Oncogene. 2012;31:2471–2479. doi: 10.1038/onc.2011.424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarty A, Sanchez V, Kuba MG, Rinehart C, Arteaga CL. Feedback upregulation of HER3 (ErbB3) expression and activity attenuates antitumor effect of PI3K inhibitors. Proc Natl Acad Sci USA. 2012;109:2718–2723. doi: 10.1073/pnas.1018001108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandarlapaty S, Sawai A, Scaltriti M, Rodrik-Outmezguine V, Grbovic-Huezo O, Serra V, Majumder PK, Baselga J, Rosen N. AKT inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer Cell. 2011;19:58–71. doi: 10.1016/j.ccr.2010.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corcoran RB, Ebi H, Turke AB, Coffee EM, Nishino M, Cogdill AP, Brown RD, Della Pelle P, Dias-Santagata D, Hung KE, et al. EGFR-mediated reactivation of MAPK signaling contributes to insensitivity of BRAF-mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov. 2012;2:227–235. doi: 10.1158/2159-8290.CD-11-0341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curry JL, Torres-Cabala CA, Kim KB, Tetzlaff MT, Duvic M, Tsai KY, Hong DS, Prieto VG. Dermatologic toxicities to targeted cancer therapy: shared clinical and histologic adverse skin reactions. Int J Dermatol. 2013;53:376–384. doi: 10.1111/ijd.12205. [DOI] [PubMed] [Google Scholar]
- Das Thakur M, Salangsang F, Landman AS, Sellers WR, Pryer NK, Levesque MP, Dummer R, McMahon M, Stuart DD. Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature. 2013;494:251–255. doi: 10.1038/nature11814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- Dykens JA, Will Y. The significance of mitochondrial toxicity testing in drug development. Drug Discov Today. 2007;12:777–785. doi: 10.1016/j.drudis.2007.07.013. [DOI] [PubMed] [Google Scholar]
- Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J, Hamid O, Schuchter L, Cebon J, Ibrahim N, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med. 2012;367:1694–1703. doi: 10.1056/NEJMoa1210093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gala K, Chandarlapaty S. Molecular pathways: HER3 targeted therapy. Clin Cancer Res. 2014;20:1410–1416. doi: 10.1158/1078-0432.CCR-13-1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopal YNV, Deng W, Woodman SE, Komurov K, Ram P, Smith PD, Davies MA. Basal and treatment-induced activation of AKT mediates resistance to cell death by AZD6244 (ARRY-142886) in BRAF-mutant human cutaneous melanoma cells. Cancer Res. 2010;70:8736–8747. doi: 10.1158/0008-5472.CAN-10-0902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haq R, Shoag J, Andreu-Perez P, Yokoyama S, Edelman H, Rowe Glenn C, Frederick Dennie T, Hurley Aeron D, Nellore A, Kung Andrew L, et al. Oncogenic BRAF regulates oxidative metabolism via PGC1α and MITF. Cancer Cell. 2013;23:302–315. doi: 10.1016/j.ccr.2013.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartsough E, Shao Y, Aplin AE. Resistance to RAF inhibitors revisited. J Invest Dermatol. 2013;134:319–325. doi: 10.1038/jid.2013.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartsough EJ, Basile K, Aplin AE. Beneficial effects of RAF inhibitor in mutant BRAF splice variant-expressing melanoma. Mol Cancer Res. 2014;0:1–27. doi: 10.1158/1541-7786.MCR-13-0581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidorn SJ, Milagre C, Whittaker S, Nourry A, Niculescu-Duvas I, Dhomen N, Hussain J, Reis-Filho JS, Springer CJ, Pritchard C, et al. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell. 2010;140:209–221. doi: 10.1016/j.cell.2009.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HJ, Bar-Sagi D. Modulation of signalling by Sprouty: a developing story. Nat Rev Mol Cell Biol. 2004;5:441–450. doi: 10.1038/nrm1400. [DOI] [PubMed] [Google Scholar]
- Kolch W. Coordinating ERK/MAPK signalling through scaffolds and inhibitors. Nat Rev Mol Cell Biol. 2005;6:827–837. doi: 10.1038/nrm1743. [DOI] [PubMed] [Google Scholar]
- Kopetz S, Desai J, Chan E, Hecht JR, O'Dwyer PJ, Lee RJ, Nolop KB, Saltz L. PLX4032 in metastatic colorectal cancer patients with mutant BRAF tumors. J Clin Oncol. 2010;28(suppl28):15s. abstract# 3534. [Google Scholar]
- Lito P, Pratilas CA, Joseph EW, Tadi M, Halilovic E, Zubrowski M, Huang A, Wong WL, Callahan MK, Merghoub T, et al. Relief of profound feedback inhibition of mitogenic signaling by RAF inhibitors attenuates their activity in BRAFV600E melanomas. Cancer Cell. 2012;22:668–682. doi: 10.1016/j.ccr.2012.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montero-Conde C, Ruiz-Llorente S, Dominguez JM, Knauf JA, Viale A, Sherman EJ, Ryder M, Ghossein RA, Rosen N, Fagin JA. Relief of feedback inhibition of HER3 transcription by RAF and MEK inhibitors attenuates their antitumor effects in BRAF-mutant thyroid carcinomas. Cancer Discov. 2013;3:520–533. doi: 10.1158/2159-8290.CD-12-0531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Sánchez R, Rodríguez-Enríquez S, Marín-Hernández A, Saavedra E. Energy metabolism in tumor cells. FEBS J. 2007;274:1393–1418. doi: 10.1111/j.1742-4658.2007.05686.x. [DOI] [PubMed] [Google Scholar]
- Morrison DK. KSR: a MAPK scaffold of the Ras pathway? J Cell Sci. 2001;114:1609–1612. doi: 10.1242/jcs.114.9.1609. [DOI] [PubMed] [Google Scholar]
- Muranen T, Selfors Laura M, Worster Devin T, Iwanicki Marcin P, Song L, Morales Fabiana C, Gao S, Mills Gordon B, Brugge Joan S. Inhibition of PI3K/mTOR leads to adaptive resistance in matrix-attached cancer cells. Cancer Cell. 2012;21:227–239. doi: 10.1016/j.ccr.2011.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazarian R, Shi H, Wang Q, Kong X, Koya RC, Lee H, Chen Z, Lee MK, Attar N, Sazegar H, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010;468:973–977. doi: 10.1038/nature09626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens DM, Keyse SM. Differential regulation of MAP kinase signalling by dual-specificity protein phosphatases. Oncogene. 2007;26:3203–3213. doi: 10.1038/sj.onc.1210412. [DOI] [PubMed] [Google Scholar]
- Papa S, Martino P, Capitanio G, Gaballo A, Rasmo D, Signorile A, Petruzzella V. The oxidative phosphorylation system in mammalian mitochondria. In: Scatena R, et al., editors. Advances in Mitochondrial Medicine. Netherlands: Springer Netherlands; 2012. pp. 3–37. [DOI] [PubMed] [Google Scholar]
- Paraiso KHT, Xiang Y, Rebecca VW, Abel EV, Chen YA, Munko AC, Wood E, Fedorenko IV, Sondak VK, Anderson ARA, et al. PTEN loss confers BRAF inhibitor resistance to melanoma cells through the suppression of BIM expression. Cancer Res. 2011;71:2750–2760. doi: 10.1158/0008-5472.CAN-10-2954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulikakos PI, Persaud Y, Janakiraman M, Kong X, Ng C, Moriceau G, Shi H, Atefi M, Titz B, Gabay MT, et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E) Nature. 2011;480:387–390. doi: 10.1038/nature10662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prahallad A, Sun C, Huang S, Di Nicolantonio F, Salazar R, Zecchin D, Beijersbergen RL, Bardelli A, Bernards R. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature. 2012;483:100–103. doi: 10.1038/nature10868. [DOI] [PubMed] [Google Scholar]
- Pratilas CA, Taylor BS, Ye Q, Viale A, Sander C, Solit DB, Rosen N. V600EBRAF is associated with disabled feedback inhibition of RAF-MEK signaling and elevated transcriptional output of the pathway. Proc Natl Acad Sci USA. 2009;106:4519–4524. doi: 10.1073/pnas.0900780106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roesch A, Vultur A, Bogeski I, Wang H, Zimmermann Katharina M, Speicher D, Korbel C, Laschke Matthias W, Gimotty Phyllis A, Philipp Stephan E, et al. Overcoming intrinsic multidrug resistance in melanoma by blocking the mitochondrial respiratory chain of slow-cycling JARID1B high cells. Cancer Cell. 2013;23:811–825. doi: 10.1016/j.ccr.2013.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salama AKS, Flaherty KT. BRAF in melanoma: current strategies and future directions. Clin Cancer Res. 2013;19:4326–4334. doi: 10.1158/1078-0432.CCR-13-0779. [DOI] [PubMed] [Google Scholar]
- Sergina NV, Rausch M, Wang D, Blair J, Hann B, Shokat KM, Moasser MM. Escape from HER-family tyrosine kinase inhibitor therapy by the kinase-inactive HER3. Nature. 2007;445:437–441. doi: 10.1038/nature05474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serra V, Scaltriti M, Prudkin L, Eichhorn PJA, Ibrahim YH, Chandarlapaty S, Markman B, Rodriguez O, Guzman M, Rodriguez S, et al. PI3K inhibition results in enhanced HER signaling and acquired ERK dependency in HER2-overexpressing breast cancer. Oncogene. 2011;30:2547–2557. doi: 10.1038/onc.2010.626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao Y, Aplin AE. Akt3-mediated resistance to apoptosis in B-RAF–targeted melanoma cells. Cancer Res. 2010;70:6670–6681. doi: 10.1158/0008-5472.CAN-09-4471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma SV, Lee DY, Li B, Quinlan MP, Takahashi F, Maheswaran S, McDermott U, Azizian N, Zou L, Fischbach MA, et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell. 2010;141:69–80. doi: 10.1016/j.cell.2010.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi F, Telesco SE, Liu Y, Radhakrishnan R, Lemmon MA. ErbB3/HER3 intracellular domain is competent to bind ATP and catalyze autophosphorylation. Proc Natl Acad Sci USA. 2010;107:7692–7697. doi: 10.1073/pnas.1002753107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H, Hong A, Kong X, Koya RC, Song C, Moriceau G, Hugo W, Yu CC, Ng C, Chodon T, et al. A novel AKT1 mutant amplifies an adaptive melanoma response to BRAF inhibition. Cancer Discov. 2013;4:69–79. doi: 10.1158/2159-8290.CD-13-0279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smalley KSM, Haass NK, Brafford PA, Lioni M, Flaherty KT, Herlyn M. Multiple signaling pathways must be targeted to overcome drug resistance in cell lines derived from melanoma metastases. Mol Cancer Ther. 2006;5:1136–1144. doi: 10.1158/1535-7163.MCT-06-0084. [DOI] [PubMed] [Google Scholar]
- Sosman JA, Kim KB, Schuchter L, Gonzalez R, Pavlick AC, Weber JS, McArthur GA, Hutson TE, Moschos SJ, Flaherty KT, et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N Engl J Med. 2012;366:707–714. doi: 10.1056/NEJMoa1112302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun C, Wang L, Huang S, Heynen GJJE, Prahallad A, Robert C, Haanen J, Blank C, Wesseling J, Willems SM, et al. Reversible and adaptive resistance to BRAF(V600E) inhibition in melanoma. Nature. 2014;508:118–122. doi: 10.1038/nature13121. [DOI] [PubMed] [Google Scholar]
- Villanueva J, Vultur A, Lee JT, Somasundaram R, Fukunaga-Kalabis M, Cipolla AK, Wubbenhorst B, Xu X, Gimotty PA, Kee D, et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell. 2010;18:683–695. doi: 10.1016/j.ccr.2010.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg O. On the origin of cancer cells. Science. 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- Wilson KJ, Gilmore JL, Foley J, Lemmon MA, Riese Ii DJ. Functional selectivity of EGF family peptide growth factors: Implications for cancer. Pharmacol Ther. 2009;122:1–8. doi: 10.1016/j.pharmthera.2008.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Y, Serra V, Prudkin L, Scaltriti M, Murli S, Rodriguez O, Guzman M, Sampath D, Nannini M, Xiao Y, et al. Evaluation and clinical analyses of downstream targets of the Akt inhibitor GDC-0068. Clin Cancer Res. 2013;19:6976–6986. doi: 10.1158/1078-0432.CCR-13-0978. [DOI] [PubMed] [Google Scholar]