

Abstract
The ventral tegmental area (VTA) is required for the rewarding and motivational actions of opioids and activation of dopamine neurons has been implicated in these effects. The canonical model posits that opioid activation of VTA dopamine neurons is indirect, through inhibition of GABAergic inputs. However, VTA dopamine neurons also express postsynaptic μ-opioid peptide (MOP) receptors. We report here that in Sprague Dawley rat, the MOP receptor-selective agonist DAMGO (0.5–3 μm) depolarized or increased the firing rate of 87 of 451 VTA neurons (including 22 of 110 dopamine neurons). This DAMGO excitation occurs in the presence of GABAA receptor blockade and its EC50 value is two orders of magnitude lower than for presynaptic inhibition of GABA release on to VTA neurons. Consistent with a postsynaptic channel opening, excitations were accompanied by a decrease in input resistance. Excitations were blocked by CdCl2 (100 μm, n = 5) and ω-agatoxin-IVA (100 nm, n = 3), nonselective and Cav2.1 Ca2+ channel blockers, respectively. DAMGO also produced a postsynaptic inhibition in 233 of 451 VTA neurons, including 45 of 110 dopamine neurons. The mean reversal potential of the inhibitory current was −78 ± 7 mV and inhibitions were blocked by the K+ channel blocker BaCl2 (100 μm, n = 7). Blockade of either excitation or inhibition unmasked the opposite effect, suggesting that MOP receptors activate concurrent postsynaptic excitatory and inhibitory processes in most VTA neurons. These results provide a novel direct mechanism for MOP receptor control of VTA dopamine neurons.
Keywords: calcium channel, midbrain, opioid, VTA
Introduction
While μ-opioid peptide (MOP) receptors are widely distributed in the brain, their expression in the ventral tegmental area (VTA) is required for MOP receptor-mediated positive reinforcement. Local microinfusion of MOP receptor agonists into the VTA produces conditioned place preference (Bozarth and Wise, 1984) and rodents self-administer MOP receptor agonists directly into the VTA (Bozarth and Wise, 1981; but see Jhou et al., 2012). Importantly, the rewarding effect of systemically administered MOP receptor agonists is blocked by VTA inactivation of neurons, injection of MOP receptor-selective antagonists, and downregulation of MOP receptor expression (Olmstead and Franklin, 1997; Moaddab et al., 2009; Zhang et al., 2009). Because selective activation of VTA dopamine neurons produces positive reinforcement (Tsai et al., 2009; Witten et al., 2011; Steinberg et al., 2013) and dopamine receptor antagonists in the ventral striatum can block reinforcement produced by VTA MOP receptor activation in opioid-dependent rodents (Shippenberg et al., 1993; Nader and van der Kooy, 1997; Laviolette et al., 2002), it is widely accepted that MOP receptor agonists produce positive motivational actions through excitation of VTA dopamine neurons.
Although the synaptic and local circuit mechanisms responsible for MOP receptor reinforcement are unknown, the dominant current view is that the excitatory action of MOP receptors on VTA dopamine neurons is indirect, due to somadendritic hyperpolarization of local GABA interneurons or inhibition of GABA release from terminals. Several lines of indirect evidence support this idea: local GABA neurons synapse on and inhibit VTA dopamine neurons (Johnson and North, 1992a; Omelchenko and Sesack, 2009; van Zessen et al., 2012), a subset of VTA GABA neurons is hyperpolarized by MOP receptor activation (Steffensen et al., 2006; Chieng et al., 2011; Margolis et al., 2012), and GABA terminals in the VTA are inhibited by MOP receptor-selective agonists (Johnson and North, 1992a; Bonci and Williams, 1997; Matsui and Williams, 2011; Hjelmstad et al., 2013). Therefore, the local circuitry required for disinhibition exists in the VTA. However, there is no direct evidence that inhibition of GABA inputs by MOP receptor agonists is required for excitation of dopamine neurons. Arguments supporting the disinhibition model depend in part upon the assumption that the VTA consists of only two types of neurons: GABA interneurons uniformly inhibited by MOP receptor agonists and dopamine projection neurons not directly affected by MOP receptor agonists. In fact, MOP receptor agonists have a variety of synaptic actions on different neuronal types in the VTA. MOP receptor agonists inhibit GABA inputs to both dopamine and nondopamine VTA neurons (Margolis et al., 2008; Xia et al., 2011; Hjelmstad et al., 2013). MOP receptor activation also inhibits glutamate release onto most VTA neurons (Bonci and Malenka, 1999; Margolis et al., 2005). Even more problematic, direct inhibition of VTA dopamine neurons by MOP receptor agonists has been observed in guinea pigs, mice, and rats (Cameron et al., 1997; Margolis et al., 2003; Ford et al., 2006). To directly examine the synaptic mechanism(s) by which MOP receptor agonists excite VTA dopamine neurons, we systematically investigated the effect of the MOP receptor-selective agonist DAMGO across a large sample of neurons throughout the VTA.
Materials and Methods
Animal care and all experimental procedures were in accordance with guidelines from the National Institutes of Health and approved in advance by the Ernest Gallo Clinic and Research Center and University of California, San Francisco institutional animal care and use committees.
Slice preparation and electrophysiology.
Recordings were made in control male Sprague Dawley rats (postnatal day 22–56). A small number of experiments were completed in adult male Sprague Dawley rats (250–350 g). Some data were obtained in neurons also used for previously reported experiments (Margolis et al., 2003, 2006, 2012). Rats were thoroughly anesthetized with isoflurane and then decapitated. Horizontal brain slices (150 μm thick) were prepared using a vibratome (Leica Instruments). Slices were prepared in ice-cold Ringer's solution (in mm: 119 NaCl, 2.5 KCl, 1.3 MgSO4, 1.0 NaH2PO4, 2.5 CaCl2, 26.2 NaHCO3, and 11 glucose saturated with 95% O2–5% CO2) and allowed to recover at 33–35°C for ≥1 h. Slices were visualized under a Zeiss Axioskop or Axioskop FS 2 Plus with differential interference contrast optics and near infrared illumination, using a Zeiss Axiocam MRm and Axiovision 4 (Zeiss) or Microlucida (MBF Biosciences) software. Whole-cell recordings were made at 33°C using 2.5–5 MΩ pipettes containing the following (in mm): 123 K-gluconate, 10 HEPES, 0.2 EGTA, 8 NaCl, 2 MgATP, 0.3 Na3GTP, and 0.1% biocytin, pH 7.2, osmolarity adjusted to 275. Liquid junction potentials were not corrected during recordings. Hyperpolarization-activated cation current (Ih) was measured by voltage-clamping cells and stepping from −60 to −40, −50, −70, −80, −90, −100, −110, and −120 mV. Input (Ri) and series resistances were monitored with hyperpolarizing pulses (0.1 Hz) throughout each experiment.
Recordings were made using an Axopatch 1-D (Molecular Devices), filtered at 2 kHz, and collected at 5 kHz, or filtered at 5 kHz, and collected at 20 kHz using IGOR Pro (Wavemetrics). For all current-clamp experiments, I = 0; for voltage-clamp experiments, V = −60 or −70 mV. Most VTA neurons were selected in an unbiased manner from throughout the VTA by superimposing a grid on the slice, numbering each grid location, and using a random number generator to choose the grid location for recording. The closest healthy cell to the randomly generated grid location was patched.
In all cases, action potentials (APs) were collected by holding the cell in current clamp at I = 0, and only spontaneously occurring APs collected within the first 2 min of gaining whole-cell access were analyzed for waveform data. Neurons were required to be firing spontaneously and stably for ≥2 min with firing rates >0.25 Hz to be included in the firing rate data.
Agonists, antagonists, salts, ATP, and GTP were obtained from Sigma-Aldrich or Tocris Bioscience. ω-Agatoxin-IVA was purchased from Alomone Labs.
Cell-type identification and immunocytochemistry.
All cells were labeled with biocytin during whole-cell recording. Slices were fixed immediately after recording in 4% formaldehyde for 2 h and then stored at 4°C in PBS. Slices were preblocked for 2 h at room temperature in PBS plus 0.2% BSA and 5% normal goat serum, then incubated at 4°C with a rabbit anti-TH polyclonal antibody (1:100). Slices were then washed thoroughly in PBS with 0.2% BSA before being agitated overnight at 4°C with Cy5 anti-rabbit secondary antibody (1:100) and FITC streptavidin (6.5 μl/ml). Sections were rinsed and mounted on slides using Bio-Rad Fluoroguard Antifade Reagent mounting media and visualized under a Zeiss LSM 510 META microscope or with an Axioskop FS2 Plus with an Axiocam MRm running Neurolucida (MBF Biosciences). Primary antibodies were obtained from Millipore Bioscience Research Reagents or Millipore, secondary antibodies were obtained from Jackson ImmunoResearch Laboratories, and all other reagents were obtained from Sigma Chemical.
In neurons where immunocytochemistry was inconclusive, Ih− neurons were classified as nondopaminergic (Margolis et al., 2006). While false negatives are possible using immunocytochemical techniques, we have previously demonstrated that the experimental methods for both recording and cytochemistry used here produce reliable TH results (Margolis et al., 2010). Further, our percentage of TH+ neurons out of the total population of cytochemically identified neurons when Ih− neurons are included in the total (52%, 110 of 211 neurons; Table 1) is remarkably similar to those obtained in the systematic anatomical analysis of sections cytochemically processed for TH and NeuN (Margolis et al., 2006).
Table 1.
Postsynaptic MOP receptor agonist effects do not sort by neurotransmitter content
| Excited/depolarized | Inhibited/hyperpolarized | No change | |
|---|---|---|---|
| TH+ | |||
| n (110) | 22 (20%) | 45 (41%) | 43 (39%) |
| Mean membrane potential change in quiescent neurons | 4.2 ± 1.2 mV (n = 12) |
−3.6 ± 0.6 mV (n = 24) |
|
| TH− | |||
| n (61) | 9 (15%) | 34 (56%) | 18 (30%) |
| Mean membrane potential change in quiescent neurons | 3.6 ± 1.3 mV (n = 5) |
−5.5 ± 0.9 mV (n = 30) |
|
| Ih− | |||
| n (40) | 11 (28%) | 18 (45%) | 11 (28%) |
| Mean membrane potential change in quiescent neurons | 4.6 ± 1.5 mV (n = 11) |
−4.3 ± 1.0 mV (n = 9) |
|
| Adult | |||
| n (8) | 4 | 3 | 1 |
Single-cell qRT-PCR.
Individual neurons were recorded in whole-cell configuration for ≥3 min. At the termination of recording, the cytoplasm of the neuron was aspirated into the recording pipette, the pipette was retracted from the slice, and the pipette contents were ejected into an RNase-free centrifuge tube prechilled to −20°C. Samples were stored at −80°C until they were processed. cDNA was synthesized from single-cell VTA neurons using the MessageBOOSTER cDNA Synthesis from Cell Lysates Kit (MBCL90310, Epicentre) according to the manufacturer's protocol. Real-time PCR was performed using the Power SYBR Green qPCR Master Mix (Applied Biosystems). The PCR template source was 4 μl of 10× diluted first-strand cDNA. Amplification was performed with an ABI Prism 7900HT sequence detection system (Applied Biosystems). After an initial denaturation step at 95°C for 10 min, amplification was performed using 45 cycles of denaturation (95°C for 15 s), annealing (55°C for 30 s), and extension (72°C for 30 s). We amplified GAPDH, a housekeeping gene, as control. The data were analyzed using the sequence detection system software (version 2.2.1, Applied Biosystems). The software generates the baseline-subtracted amplification plot of normalized reporter values (ΔRn) versus cycle number. The amplification threshold was set at 6–7 of ΔRn linear dynamic range (50–60% of maximum ΔRn). The fractional cycle at which the intersection of amplification threshold and the plot occurs is defined as the threshold cycle (Ct-value) for the plot. Samples that gave a Ct-value within 45 cycles were considered to be positive for the mRNA expression. The samples for which Ct-values were not observed within 45 cycles (i.e., undetected) were considered to be negative for the mRNA expression. The OPRM1 primers were confirmed as specific by comparing brain tissue samples from wild-type (WT) mice and OPRM1-knock-out (KO) mice. These samples were quantified using ImageJ band intensity analysis following gel visualization. Using this approach, midbrain and cortex WT samples were measured at 35.3 and 29.1 mean band intensity, respectively, compared with 3.7 and −1.9 mean band intensity in samples from the same brain regions in the KO mice. Further, RNase/DNase-free water yielded 0.0 signal for all primers.
The forward (F) and reverse (R) primers are as follows: GAPDH-F, TCAAGAAGGTGGTGAAGCAG; GAPDH-R, AGGTGGAAGAATGGGAGTTG; OPRM1-F, TCGGTCTGCCTGTAATGTTC; OPRM1-R, CAGATTTTGAGCAGGTTCTCC; TH-F, AGGGGTACAAAACCCTCCTC; TH-R, CGCACAAAATACTCCAGGTG.
Data analysis.
At least 10 APs from each neuron were averaged together, and the resulting trace was measured between when the membrane potential (Vm) crossed threshold (when the slope of the Vm first rise exceeded 5 V/s for data collected at 5 kHz or 10 V/s for data collected at 20 kHz) to when the Vm recrossed the threshold following the AP peak. Ih magnitude was measured as the difference between the initial capacitative response to a voltage step from −60 to −120 mV and the final current during the same 200 ms step. Neurons were considered Ih− if the slope of the I–V curve for hyperpolarizing steps from −60 to −90, −100, −110, and −120 mV was 0. The reported firing rates are averages of the instantaneous firing rate over at least 2 and up to 10 min, usually at the beginning of the experiment.
Results are presented as mean ± SEM. DAMGO effects were statistically evaluated in each neuron by binning data into 30 s data points and comparing the last eight baseline data points to the last eight data points during drug application using Student's unpaired t test. In neurons that were firing spontaneously, firing rate was analyzed. In neurons that were quiescent, membrane potential was analyzed. In experiments where DAMGO was applied multiple times, this analysis was applied to each individual DAMGO application, where baseline data were the 4 min preceding each individual DAMGO application. Therefore, when DAMGO was applied in the presence of blockers, the baseline period consisted of 4 stable minutes of recording during aCSF plus blocker application. Effects of blockers were assessed with Student's paired t tests comparing the response to the first DAMGO application in control aCSF compared with the second DAMGO application in the presence of the blocker. To assess whether intracellular GDP-β-s interfered with DAMGO-induced effects, we performed permutation analysis of the SDs of changes in Vm with DAMGO of quiescent neurons. The one-tailed p value is given because the hypothesis being tested is directional: did the GDP-β-s make the variability of responses smaller? Statistical comparisons between groups of neurons were made using one-way ANOVAs. p < 0.05 was required for significance in all analyses.
Results
Using whole-cell current-clamp recording, we measured responses of VTA neurons to bath application of the MOP receptor agonist DAMGO. Significant numbers of VTA neurons were either excited or inhibited: 87 of 451 (19%) of VTA neurons showed a depolarization or an increase in firing rate in response to a saturating dose of DAMGO (500 nm–3 μm; Fig. 1; Fig. 3). The EC50 value was in the low nanomolar range (Fig. 5I). Surprisingly, a larger percentage (233 of 451, 52%) was inhibited (hyperpolarized or decreased firing rate) by DAMGO (500 nm–3 μm; Figs. 2, 3). The dose–response curve for inhibitions overlapped that of the excitations (EC50 value in the low nanomolar range; Fig. 5I). Furthermore, contrary to previous interpretations, the proportions of excited and inhibited VTA neurons and the magnitudes of these effects were similar in identified dopamine (TH+) and nondopamine (TH−) neurons (Table 1). We also observed inhibitions and excitations in neurons that were Ih− (Table 1), which are uniformly TH− (Margolis et al., 2006), and possibly GABAergic or glutamatergic. Both excitations and inhibitions were reversed by the MOP receptor-selective antagonist d-Phe-Cys-Tyr-d-Trp-Arg-Pen-Thr-NH2 (CTAP, 100–500 nm; Figs. 1D, 2C), indicating that both effects require actions at the MOP receptor. There was no apparent topographic organization within the VTA of neurons exhibiting excitations or inhibitions (Fig. 4). While most observations were made in young rats (postnatal days 22–56), we confirmed these observations in VTA tissue taken from adult rats (250–350 g; Table 1).
Figure 1.
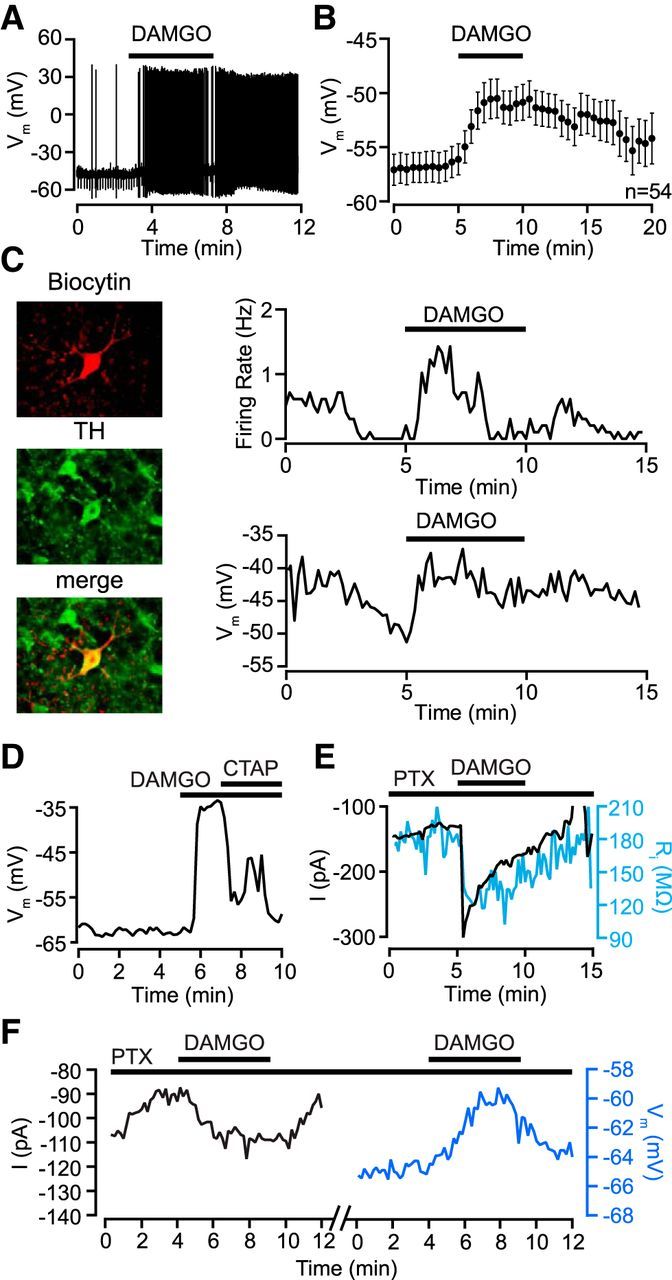
MOP receptors excite VTA dopamine neurons by activating an inward current. A, Example VTA neuron that depolarized and started firing in response to the MOP receptor-selective agonist DAMGO (500 nm; application time indicated by horizontal bar above trace). B, Pooled membrane potential data from all quiescent neurons showing a significant depolarization following bath application of the DAMGO (0.5–3 μm). C, Example of a confirmed dopamine neuron excited by DAMGO. Left column, Biocytin fill of recorded neuron (top, red) with post hoc TH immunostaining (middle, green) and merged image (bottom, overlap indicated in yellow). Right column, Current-clamp experiment of this neuron (I = 0) showing response to 500 nm DAMGO. Upper trace, This neuron responded to the MOP receptor agonist DAMGO (500 nm) with a transient increase in firing rate. Lower trace, The concurrent membrane potential in the same neuron illustrating a sustained depolarization following DAMGO application. D, A different example neuron where the MOP receptor-selective antagonist d-Phe-Cys-Tyr-d-Trp-Arg-Pen-Thr-NH2 (CTAP; 100 nm) reversed a DAMGO-induced depolarization. E, Example voltage-clamp (Vm = −70 mV) experiment where DAMGO activated an inward current in the presence of picrotoxin (100 μm), and this current was accompanied by an increase in conductance (blue). F, Another example neuron in which DAMGO was applied twice, first in voltage clamp, resulting in an inward current, and subsequently in current clamp, yielding a depolarization.
Figure 3.

Distribution of significant (p < 0.05) DAMGO-elicited physiological responses in VTA neurons. A, DAMGO-induced changes in membrane potential in neurons not firing spontaneously. B, Among these quiescent neurons, decreases in input resistance (top) were observed in most neurons with significant DAMGO-induced change in membrane potential, including those that were depolarized by DAMGO. A smaller number of neurons showed either an increase (middle) or no change (bottom) in input resistance. C, Distribution of DAMGO-induced increases (top) or decreases (middle) in firing rate in neurons that were firing spontaneously before DAMGO application.
Figure 5.
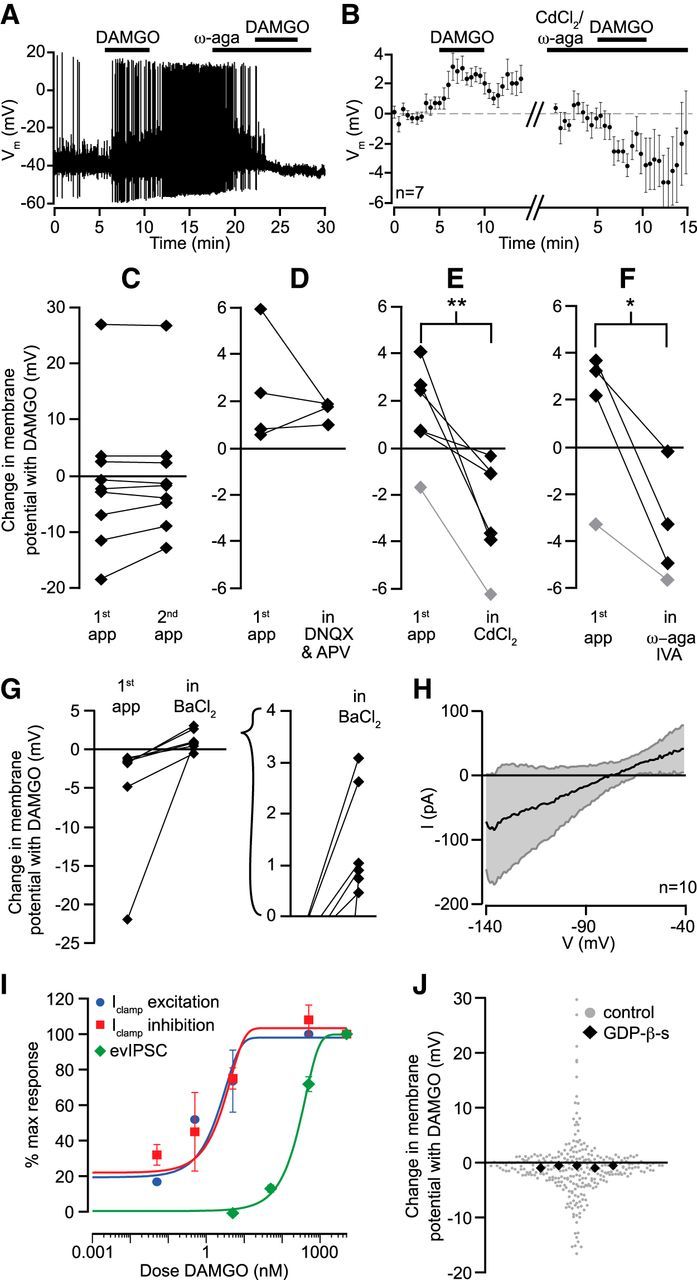
DAMGO excitations require Cav2.1. A, Example neuron where DAMGO (500 nm) application caused a depolarization and increase in AP firing frequency. In the presence of highly selective Cav2.1 (P/Q type) Ca2+ channel blocker ω-agatoxin IVA (100 nm), the same dose of DAMGO caused an inhibition. B, Time course data across seven neurons where either the nonselective Ca2+ channel blocker CdCl2 (100 μm) or ω-agatoxin IVA (100 nm) was used to block a DAMGO-induced depolarization. The neuron in A was excluded from this plot because of the nonlinearity of membrane potential changes when neurons start and stop firing. C, Control data show that repeated applications of DAMGO within the same cell elicited responses of similar magnitudes for both depolarizations and hyperpolarizations. D, In neurons with an initial depolarizing response to DAMGO, a mixture of ionotropic glutamate receptor antagonists did not block DAMGO excitations. E, The nonselective Ca2+ channel blocker CdCl2 (100 μm) completely blocked DAMGO-induced excitations and in most cases unmasked hyperpolarizations. In gray, a small hyperpolarizing effect of DAMGO was made bigger by CdCl2, suggesting an underlying but masked depolarization. F, The highly selective Cav2.1 (P/Q type) Ca2+ channel blocker ω-agatoxin IVA (100 nm) also completely blocked the excitatory effect of DAMGO, unmasking an inhibitory action. In gray, a small hyperpolarizing effect of DAMGO was made bigger by ω-agatoxin IVA. G, In neurons that responded to DAMGO with a hyperpolarization, the K+ channel blocker BaCl2 (500 μm) eliminated the hyperpolarizations, and in most neurons revealed a depolarization (inset). H, In voltage clamp, voltage ramps were used to estimate the reversal potential for the DAMGO effect in neurons that hyperpolarized ≥8 mV in response to DAMGO (n = 10). I, Dose responses were measured for the postsynaptic excitations and inhibitions, as well as for the inhibition of electrically stimulated GABA release (evIPSCs). Dose–response relationships were indistinguishable for postsynaptic DAMGO excitations (blue) and inhibitions (red) but the EC50 value was two orders of magnitude lower than for DAMGO inhibition of evIPSCs (green). n = 4–8 for each concentration. J, Including GDP-β-s (500 μm) in the recording pipette to block G-protein signaling in the recorded cell eliminated all DAMGO responses. GDP-β-s significantly decreased the variation in responses to DAMGO (permutation analysis, p < 0.0005), consistent with a blockade of both excitations and inhibitions.
Figure 2.
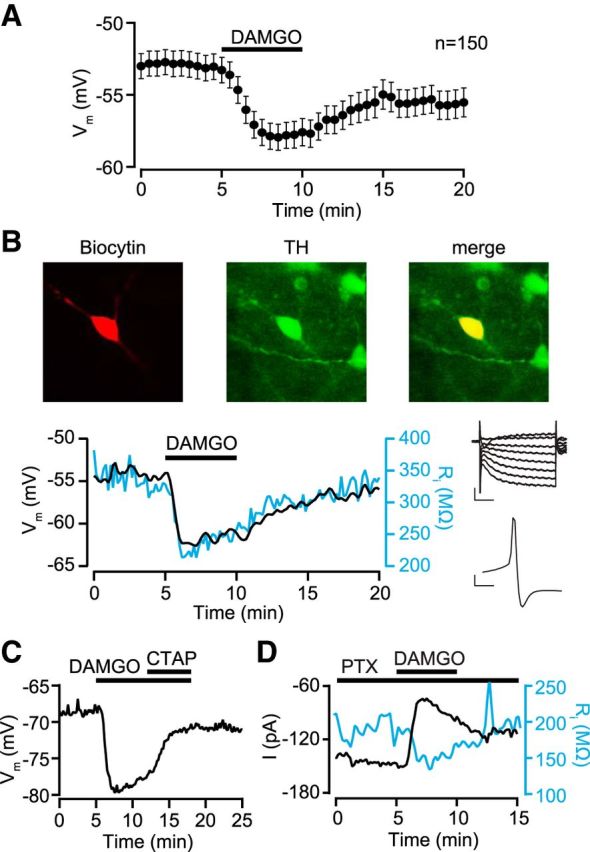
MOP receptors inhibit VTA dopamine neurons by activating an outward current. A, Pooled data from all quiescent neurons that showed a significant hyperpolarization following bath application of the MOP receptor-selective agonist DAMGO. B, Example current-clamp (I = 0) experiment in a neuron filled with biocytin (red, left) and post hoc identified as dopaminergic with immunocytochemistry against TH (green, middle; merged image in yellow on right). DAMGO (500 nm) hyperpolarized this neuron and decreased its input resistance (blue). This neuron was Ih+ and had a short-duration AP (top and bottom insets, respectively). Inset scale bars: top, 100 pA, 50 ms; bottom, 10 mV, 2 ms. C, Example neuron where the MOP receptor-selective antagonist d-Phe-Cys-Tyr-d-Trp-Arg-Pen-Thr-NH2 (CTAP; 100 nm) reversed a DAMGO-induced hyperpolarization. D, Example voltage-clamp (Vm = −70 mV) recording where DAMGO activated an outward current in the presence of picrotoxin (100 μm), and this current was accompanied by an increase in conductance (blue).
Figure 4.
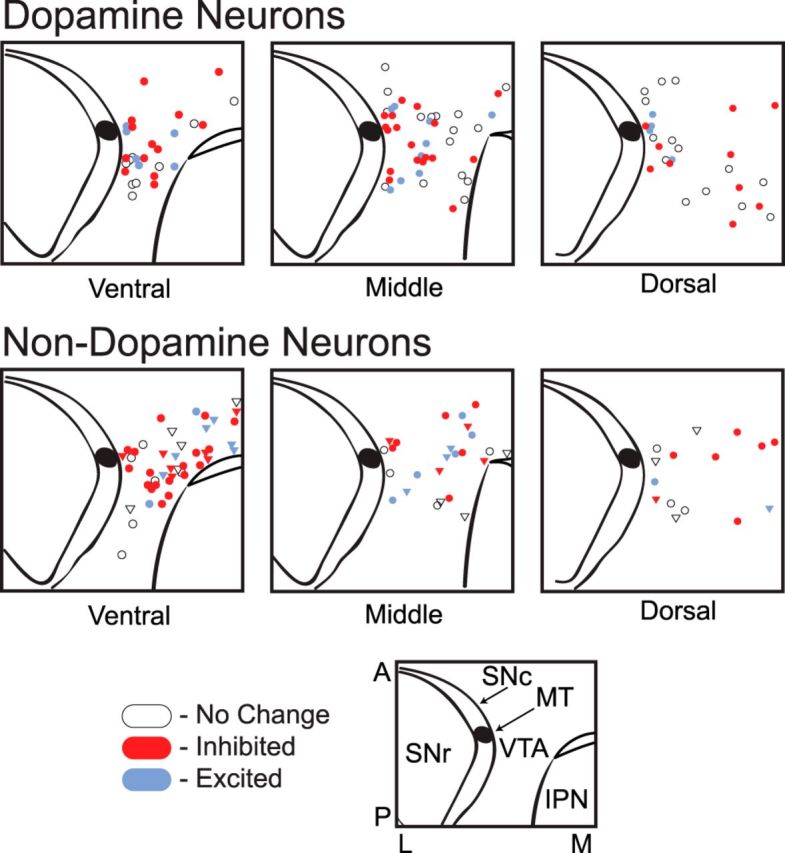
Topographical distribution of sampled neurons within the VTA. Top row, Locations in horizontal slices of cytochemically identified dopamine neurons tested with DAMGO. Bottom row, Identified nondopamine neurons, includes (●) TH− and (▿) Ih− without cytochemistry. Inset, SNc, substantia nigra pars compacta; SNr, substantia nigra pars reticulata; MT, medial terminal nucleus of the accessory optic tract; IPN, interpeduncular nucleus; A, anterior; P, posterior; L, lateral; M, medial.
Excitations are postsynaptic, not disinhibition
One immediate question raised by these data is as follows: are the excitations due to removal of GABA inputs, i.e., disinhibition, as posited in the canonical model? MOP receptor agonists robustly inhibit GABA release onto all VTA neurons in ex vivo recordings (Bonci and Williams, 1997; Margolis et al., 2008), and bath application of the GABAA receptor Cl− channel blocker picrotoxin (100 μm) can depolarize neurons in the slice (5 of 5 neurons, where all neurons had baseline membrane potentials more depolarized than the Cl− equilibrium potential; data not shown) indicating that there is tonic GABA release in VTA slice preparations. However, arguing against the disinhibition model, we observed that most of the DAMGO-induced depolarizations were associated with a concurrent increase in conductance (Fig. 3B). Furthermore, the magnitudes of the excitations were directly and significantly correlated with the increase in conductance (r = 0.66, p = 0.0005). This is consistent with opening a channel, not disinhibition through reduction of an inhibitory input that would decrease membrane conductance through channel closings. To test the GABA disinhibition model directly, responses to DAMGO were measured in voltage clamp (Vm = −60 or −70 mV) in the presence of picrotoxin (100 μm). In 6 of 24 neurons (25%), we observed an inward current in response to DAMGO, a proportion similar to that of excitations observed in control aCSF in current-clamp experiments (Fig. 1E,F). Furthermore, of four neurons where DAMGO induced an inward current in voltage clamp, depolarizations were observed in three when they were tested again with DAMGO after switching into current clamp (Fig. 1F). To provide additional evidence that the MOP receptor excitations were not due to presynaptic inhibition of GABA terminals, we compared the DAMGO dose–response curve for the presynaptic inhibition of GABA release with that for the depolarization. If the two effects were due to activation of MOP receptors on GABA terminals, the dose–response curves for the two actions should overlap. Instead, we found that the EC50 value for DAMGO presynaptic inhibition of GABA release was two orders of magnitude higher than that for the somadendritic hyperpolarizations/depolarizations (Fig. 5I). Thus three independent lines of evidence argue strongly against disinhibition as a major factor in this excitation of VTA neurons: it is associated with a conductance increase, it requires a much lower MOP receptor agonist concentration than presynaptic inhibition of GABA release, and it is insensitive to pharmacological blockade of GABAA receptors.
Many VTA neurons fire spontaneously in the slice and this leads to local GABA, glutamate, and dopamine neuron crosstalk in the VTA (Ford et al., 2009; Omelchenko and Sesack, 2009; Dobi et al., 2010). Because more neurons than expected responded to DAMGO, it is possible that the observed effects in some neurons could be indirect. If the DAMGO effects on the recorded neuron were indirect, i.e., due to a direct receptor action on the firing of other neurons in the slice, the voltage-gated Na+ channel blocker tetrodotoxin (TTX; 500 nm–1 μm) should decrease or eliminate DAMGO responses. In fact, DAMGO-induced hyperpolarizations persisted in the presence of TTX (control response: −9.7 ± 1.9 mV; in TTX: −6.7 ± 2.0 mV; n = 4). Because TTX alone often caused VTA neurons to depolarize, we tested for DAMGO-induced excitations in the presence of TTX in voltage clamp, holding the cells at Vm = −60 mV, further from the reversal potential of a depolarizing conductance, and closer to the basal membrane potential of typical VTA neurons. Under these conditions, 8 of 21 neurons responded to DAMGO with a significant inward current (control: −91 ± 33 pA, n = 3; TTX: −248 ± 131 pA, n = 8). These observations render it unlikely that the inhibitions or excitations produced by DAMGO are indirect.
What MOP receptor-activated conductance(s) depolarize VTA neurons?
Another possibility is that MOP receptor activation increases glutamate release that is independent of AP activity, either by a direct effect on nerve terminals (Velásquez-Martinez et al., 2012) or via an effect on glia (Moussawi et al., 2011). A MOP receptor-induced increase in glutamate release from nerve terminals is unlikely since synaptic experiments have demonstrated that MOP receptor activation inhibits glutamate release onto VTA neurons (Bonci and Malenka, 1999; Margolis et al., 2005). MOP receptors have been observed on microglia and astrocytes (Ruzicka et al., 1995; Dever et al., 2012; Merighi et al., 2013), and activation of microglia can cause glutamate release (Noda et al., 1999). To test whether glutamate is involved in MOP receptor-mediated excitations, we first found a VTA neuron that exhibited an excitation in response to DAMGO, then retested the neuron with DAMGO in the presence of DNQX (10 μm) and d(−)-2-amino-5-phosphonopentanoic acid (AP-V, 50 μm) to block excitatory glutamate receptors. In control experiments where DAMGO was applied twice, the magnitude and direction of membrane potential change was consistent between applications (Fig. 5C). DAMGO effects in the presence of glutamate antagonists were not different from controls (Fig. 5D).
In isolated Purkinje neurons, MOP receptor activation increases a Cav2.1 conductance (Iegorova et al., 2010). Cav2.1 composes ∼40% of voltage-dependent Ca2+ currents in VTA dopamine neurons (Cardozo and Bean, 1995). To test whether a Ca2+ channel is involved in the MOP receptor-induced depolarizations in the VTA, we measured responses to DAMGO in the presence of the Ca2+ channel blocker CdCl2 (100 μm) in neurons that were excited under control conditions. CdCl2 not only prevented excitations, but in 5 of 5 neurons CdCl2 treatment revealed an inhibition in response to DAMGO (Fig. 5E). To test whether Cav2.1 channels were involved, in another set of neurons we retested DAMGO in the presence of the selective blocker ω-agatoxin IVA (100 nm); this also prevented the DAMGO-induced excitations and revealed inhibitions, similar to CdCl2 (Fig. 5A,F). Finally, we also tested whether the excitations depended upon G-protein signaling in the recorded neuron by replacing intracellular GTP with the non-hydrolyzable GDP-β-s (500 μm). None of the five cells tested showed any response to DAMGO after GDP-β-s was delivered into the cell via the recording electrode (Fig. 5J), despite the fact that four of these neurons were tested with and responded to DAMGO in cell-attached configuration before establishing whole-cell access and therefore GDP-β-s delivery, two increasing and two decreasing in firing rate. Consistent with intracellular GDP-β-s eliminating DAMGO signaling, the variability of the differences in Vm between baseline and in DAMGO application in these cells was significantly smaller than across the population of quiescent neurons (permutation analysis of SD, p < 0.0005). These data indicate that excitations require G-protein signaling in the recorded neuron and are consistent with the interpretation that DAMGO causes a direct excitation of a subset of VTA neurons, including dopamine neurons, through G-protein-dependent activation of Cav2.1.
Inhibitions
Our finding that approximately half of all VTA neurons, including 41% of all confirmed dopamine neurons, were inhibited by MOP receptor activation contradicts the assumptions that support the canonical disinhibition model. Postsynaptic MOP receptor-induced inhibitions are typically mediated by the opening of G-protein-activated inwardly rectifying K+ channels (GIRKs). In neurons that were hyperpolarized by DAMGO, we reapplied DAMGO in the presence of the K+ channel blocker BaCl2 (500 μm) to directly test that opening K+ channels mediates the DAMGO-induced inhibitions. Not only did BaCl2 treatment block the inhibitions, in six of seven neurons it uncovered small excitations (Fig. 5G). We further observed that both CdCl2 and ω-agatoxin IVA made DAMGO-induced hyperpolarizations larger (Fig. 5E,F). By applying a voltage ramp in voltage clamp, we determined that the reversal potential for the DAMGO-induced inhibitions is −78 ± 7 mV (n = 8) and correcting for the liquid junction potential estimated at −15 mV yields a reversal potential of −93 mV (Fig. 5H). This is somewhat depolarized from the estimated K+ reversal potential given the aCSF and internal solutions used here (−103 mV), possibly due to the contribution of the excitatory component of MOP receptor signaling in these neurons. The inhibitions are thus most likely an example of the well known opioid receptor activation of a GIRK. It is important to note that although blockade of the DAMGO inhibition by BaCl2 reveals an excitation, intracellular blockade of G-protein signaling with GDP-β-s does not unmask an excitation. This supports the conclusion that both excitations and inhibitions are G-protein mediated and are specific to the recorded neuron. Furthermore, together with the observation that the inhibitions and excitations have the same dose–response curve, this observation provides additional support for the idea that DAMGO concurrently produces both effects directly on the recorded neuron.
MOP receptors are expressed in most VTA dopamine neurons
Together, our observations that the majority (71%) of both dopamine and nondopamine VTA neurons respond to DAMGO (by either excitation or inhibition) indicate that most VTA neurons express the MOP receptor. This was unexpected given that previous studies have reported only modest MOP receptor protein or mRNA expression in the VTA (Mansour et al., 1994, 1995). Furthermore, while some VTA dopamine neurons have previously been reported to express MOP receptor (Garzón and Pickel, 2001) and to be inhibited by MOP receptor activation (Cameron et al., 1997; Margolis et al., 2003; Ford et al., 2006), our proportions of MOP receptor-responsive VTA neurons are higher than predicted. To provide an independent line of support for the conclusion that many VTA neurons express MOP receptor, we measured TH and MOP receptor mRNA from individual VTA neurons with RT-PCR. Of the 21 sampled neurons, 12 (57%) expressed TH mRNA; this proportion is consistent with anatomical findings of proportions of TH+ VTA neurons (Margolis et al., 2006; Nair-Roberts et al., 2008). We found that 10 of 12 neurons that expressed mRNA for TH also expressed MOP receptor mRNA. Among the nine neurons in which TH mRNA was not detectable, MOP receptor mRNA was detected in five. Together with the electrophysiological observations described above, these RT-PCR results provide strong support for the concept that the majority of VTA dopaminergic neurons express MOP receptors.
Discussion
Here we report that a subset of both dopamine and nondopamine VTA neurons are excited by DAMGO. This occurred in the absence of GABAA receptor signaling and required Cav2.1 activity. Consistent with a direct postsynaptic effect, the excitations were associated with an increase in conductance and were blocked by intracellular application of GDP-β-s. A larger proportion of both dopamine and nondopamine neurons were directly inhibited through activation of a GIRK. We confirmed that both excitatory and inhibitory effects of DAMGO occur in VTA neurons from adult rats as well. Blockade of either excitation or inhibition revealed or enhanced the opposite effect, indicating that MOP receptor agonists concurrently activate both GIRKs and Cav2.1 channels in the same neuron.
Direct excitation or disinhibition?
Although excitations of dopamine neurons were frequent, we found no evidence of MOP receptor-mediated disinhibition. This was unexpected because presynaptic inhibition of evoked GABA release by MOP receptor agonists is robust in this preparation and there is significant ongoing tonic GABAA receptor activity as revealed by picrotoxin-induced depolarizations. In other words, even though (1) a reduction of GABA input in this ex vivo preparation does excite VTA dopamine neurons and (2) activation of MOP receptor inhibits GABA neurons and their terminals, inhibition of GABA input is not required for the excitation of VTA neurons by locally acting MOP receptor agonists. However, disinhibition could be significant if the nervous system is intact. There are MOP receptor-sensitive GABAergic terminals in the VTA that arise from neurons in other structures, including the ventral pallidum (Hjelmstad et al., 2013) and the rostromedial tegmental nucleus (Matsui and Williams, 2011), and while these synapses may be very active in vivo, their activity would be minimized in our preparation where their terminals are severed from their cell bodies. Consistent with this idea, spontaneous GABA IPSCs occur at very low rates in VTA neurons in horizontal brain slices (∼1–4 Hz; Margolis et al., 2008; Theile et al., 2008; Xiao and Ye, 2008), and similar rates have been reported for AP-independent miniature IPSCs (Bonci and Williams, 1997; Melis et al., 2002). Furthermore, when measured directly, IPSC frequency is only reduced by ∼30% by TTX (our unpublished observations). Despite the low spontaneous IPSC event rates, there must be a source for the significant tonic GABAA receptor signaling revealed by picrotoxin. One possibility is that there is ambient GABA from a non-neuronal source, such as astrocytes (Jiménez-González et al., 2011), which could explain why it is not significantly regulated by MOP receptors. On the other hand, DAMGO excitations were associated with an increase in conductance. Consequently, although the present results do not rule out a contribution of disinhibition to MOP receptor activation of VTA neurons, they strongly support the conclusion that direct excitation contributes significantly in vivo when MOP agonists are systemically administered.
MOP receptor expression and function is ubiquitous in the VTA
Approximately 20% of VTA neurons, both TH+ and TH−, were excited by MOP receptor activation. Such a Cav2.1-dependent MOP receptor excitatory effect has been reported in cerebellar neurons (Iegorova et al., 2010). Although the Cav2.1-selective blocker ω-agatoxin IVA abolished the excitations, some of the neurons were relatively hyperpolarized at baseline, with membrane potentials at which Cav2.1 channels are reportedly inactive. Consequently, an indirect mechanism could be involved, such as presynaptic MOP receptor enhancement of Ca2+ channel-dependent vesicular release of an excitatory substance. However, MOP receptor activation inhibits release from glutamate terminals in the VTA (Bonci and Malenka, 1999; Margolis et al., 2005), and the excitations we observed were not blocked by glutamate receptor antagonists. Alternatively, MOP receptors might be expressed on glia or at neuronal sites where another depolarizing agent may be released that acts at a G-protein-coupled receptor, such as orexin, substance P, cholecystokinin, or neurotensin. However, the fact that the dose–response curves for the excitations and inhibitions are virtually identical is consistent with a single postsynaptic receptor binding site capable of mediating both excitations and inhibitions. Therefore, the most parsimonious explanation for our data is that these DAMGO-induced excitations are due to postsynaptic MOP receptors on the recorded neurons that open Cav2.1 channels.
We also observed that postsynaptic excitations and inhibitions occurred at much lower concentrations than the presynaptic inhibition of VTA GABA release at terminals. This relationship is consistent with the presynaptic and postsynaptic potency differences reported by Johnson and North for Met-enkephalin in the VTA (1992a). Such differences could be caused by a wide variety of conditions that would affect the affinity of the receptor for the ligand. For instance, the conformation of the receptor may be influenced by the proximity of a G-protein (Kuszak et al., 2009; Malik et al., 2013) or by the local ion concentrations (Wong et al., 1994). There is also evidence that heterodimerization of opioid receptors alters ligand binding (Jordan and Devi, 1999) and the regulator of the G-protein-signaling protein family can alter opioid receptor activation (Georgoussi et al., 2006; Wang et al., 2009). The fact that the postsynaptic receptors are more sensitive to not only DAMGO, as demonstrated here, but also Met-enkephalin raises the possibility that endogenous opioid release acts more potently at postsynaptic than presynaptic sites.
A particularly intriguing observation is that DAMGO-induced excitations were unmasked in most inhibited cells following blockade of K+ currents, and conversely, most excited neurons responded with an inhibition when Ca2+ channels were blocked. The purpose this serves in an individual neuron is not obvious, especially if each receptor is coupled to both downstream ion channels. On the other hand, this observation might make sense if the excitatory and inhibitory actions were initiated by spatially segregated MOP receptors, such that certain dendrites are controlled by MOP receptors coupled to K+ channels, while in other dendrites the MOP receptors are coupled primarily to Ca2+ channels. A similar possibility is that the different MOP receptor effects may segregate according to the source of the nearby synaptic inputs, or may depend upon which other receptors are nearby in the plasma membrane.
While opioid receptors are generally thought of as inhibitory, there is a growing literature supporting the idea that the signaling pathways they employ are actually more varied. In fact, evidence of seven transmembrane domain receptor/G-protein coupled receptor signaling concurrently through several different pathways has been accumulating. In addition to MOP receptors activating Cav2.1 channels in the cerebellum (Iegorova et al., 2010), a variety of stimulatory effects of opioid receptor activation has been reported (Harrison et al., 1998). For example, morphine increases adenylyl cyclase in rat corpus striatum (Puri et al., 1975), and endogenous opioid peptides increase adenylyl cyclase in the rat olfactory bulb in a Gi/o-dependent manner (Onali and Olianas, 1991). In cell culture, there is also evidence that in cells expressing both the MOP receptor and the δ-opioid receptor, DAMGO increases intracellular Ca2+ in a Gi/o-dependent manner (Charles et al., 2003). These and other observations in a variety of brain regions and in different reduced systems lend credence to the interpretation that in the VTA, MOP receptor activation can directly excite neurons by opening a Ca2+ channel.
The high proportion of confirmed dopamine neurons directly inhibited by MOP receptor activation was surprising. At first glance these results differ from the seminal electrophysiological work by Johnson and North (1992b). However, Johnson and North identified “dopamine” neurons as those inhibited by dopamine and not opioids; “GABA” neurons were identified as those insensitive to dopamine and inhibited by opioids. Subsequent work has shown that these criteria are unreliable. However, when we applied the Johnson and North criteria to our neuronal cohort, the electrophysiological properties of the two datasets are strikingly similar (Table 2). However, 28% of our neurons that would have been classified as GABAergic by Johnson and North were TH+. Furthermore, others have shown that some VTA dopamine neurons are inhibited by MOP receptor agonists (Cameron et al., 1997; Ford et al., 2006; but see Chieng et al., 2011). One possible reason that we found more MOP receptor-inhibited dopamine neurons is that our sample size is much larger and includes recordings in the medial VTA, which is not sampled in many ex vivo studies. While we did not observe any topographical sorting of MOP receptor-inhibited dopamine neurons, we excluded substantia nigra pars compacta neurons, which are reportedly MOP receptor insensitive (Lacey et al., 1987). Further, some of the inhibitions were relatively small (Fig. 3) and may have been ignored in previous studies. However, we found that these changes were significant compared with baseline, time locked to drug application, reproducible within cells, and blocked by BaCl2.
Table 2.
Electrophysiological properties of rat VTA neurons sorted according to Johnson and North's 1992 criteriaa
| D2R inhibited; MOP receptor not inhibited | D2R not inhibited; MOP receptor inhibited | |||
|---|---|---|---|---|
| Measure | Johnson and North | Margolis et al. | Johnson and North | Margolis et al. |
| Percentage TH+ | 5 of 8 (63%) | 14 of 19 (74%) | 0 of 1 | 2 of 7 |
| Percentage Ih+b | 100% | 100% (n = 37) | 43% (48 of 60) | 95% (18 of 19) |
| Percentage Ih+ for Ih >20 pA | 97% (36 of 37) | 37% (7 of 19) | ||
| Firing ratec | ||||
| Mean | 1.7 Hz | 1.52 Hz | 3.0 Hz | 3.32 Hz |
| Maximum | 5.8 Hz | 3.24 Hz | 6.75 Hz | |
| Percentage spontaneously active | 148 of 187; 79% | 28 of 37; 76% | 8 of 31; 26% | 10 of 19; 53% |
| Rid | 192 ± 12 | 331 ± 22 | 216 ± 26 | 545 ± 67 |
| Spike threshold (mV)e | −53 | −25.9 ± 0.7 | −63 | −30.5 ± 1.3 |
| Spike duration (ms) | 2.14 ± 0.13 | 1.86 ± 0.20 | ||
aThe seminal ex vivo electrophysiological report of VTA neuron properties by Johnson and North (1992b) concluded that a variety of physiological properties differ between dopamine and GABA neurons. We have not observed such a pattern (Margolis et al., 2006, 2012). However, in Johnson and North's original report, neurons were classified as putative dopamine or putative GABA according to their pharmacological responses to dopamine D2 receptor (D2R) and MOP receptor activation: putative dopamine neurons were inhibited by D2R but not MOP receptor activation, and putative GABA neurons had the converse responses. Here we sort our data for cells tested with both quinpirole and DAMGO for a more appropriate comparison to this previous work. For Ih magnitude, data were categorized as Ih+ or Ih− as described in Materials and Methods. Additionally, since small-magnitude Ih responses were probably difficult to detect with sharp electrodes in the Johnson and North study, we also present the data where only cells with an Ih magnitude >20 pA were counted as positive. Nineteen neurons of our dataset could not be classified by these criteria.
bIn our dataset, Ih magnitude is also significantly larger in principal neurons compared to secondary neurons (p = 0.000003).
cIn our dataset, firing rate is significantly faster in secondary cells (p = 0.0059).
dIn our dataset, Ri is significantly greater in secondary cells (p = 0.00044).
eIn our dataset, the AP threshold is significantly more hyperpolarized in secondary cells (p = 0.0017).
VTA MOP receptors and behavior
The physiologic data are compelling that virtually every VTA neuron is either presynaptically or postsynaptically modulated by MOP receptor activation, and any of these actions may contribute to reward. In fact, even MOP receptor actions at nondopamine neurons can be rewarding, as in naïve rodents where VTA MOP reward is unaffected by dopamine antagonists (Nader and van der Kooy, 1997). The extent of MOP receptor-mediated inhibition of dopamine neurons is also consistent with a growing appreciation of the diversity of VTA dopamine neuron connectivity and function. Particularly noteworthy is evidence that some dopamine neurons are activated by aversive stimuli (Brischoux et al., 2009; Bromberg-Martin et al., 2010). Also, activation of PFC-projecting dopamine neurons can cause conditioned place aversion (Lammel et al., 2012). One possibility is that the MOP receptor-inhibited neurons we report here represent a subpopulation of dopamine neurons that respond to noxious stimulation.
Given the variety of synaptic actions of the MOP receptor in the VTA, it is difficult to predict the relative contribution of GABA terminal inhibition versus somadendritic dopamine neuron excitation to an increase in dopamine neuron firing in an awake behaving rat. This will depend upon a variety of factors, including the activity of specific GABAergic inputs, the membrane potential of the neuron, and the concentration of nonopioid neurotransmitters and modulators. Behaviorally effective systemic doses of morphine do suggest that the more potent postsynaptic effects reported here are significant to behavioral responses: following a typical rodent analgesic dose of morphine in rat (ED50 value, ∼10 mg/kg), brain concentrations of ∼140 ng/g or 0.5 μm are achieved (Patrick et al., 1975). Therefore, a systemic dose of 3 mg/kg morphine, which is sufficient to produce place preference (Shippenberg and Herz, 1987), yields a brain concentration of 150 nm. Assuming equivalent dose responses for morphine and DAMGO, this is a saturating dose for the somadendritic action but is closer to the EC50 value for inhibition of GABA release, raising the possibility that postsynaptic effects dominate in the VTA for systemic opioid administration. Consequently, it is likely that the direct MOP receptor-mediated excitatory effect on VTA dopamine neurons contributes significantly to the rewarding action of MOP receptor agonists. This conclusion is supported by the findings of Zhang et al. (2009) showing that siRNA knockdown of MOP receptors intrinsic to the VTA/substantia nigra region reduces the rewarding effect of systemic MOP receptor agonist administration.
In summary, we report a novel mechanism for neuronal activation by MOP receptor in the VTA, i.e., the opening of somadendritic Cav2.1 channels, an effect seen on both dopamine and nondopamine neurons. MOP receptor-mediated postsynaptic inhibition is more common than excitation even among VTA dopamine neurons. Both postsynaptic excitations and inhibitions are G-protein mediated and both occur concurrently in the same neuron at much lower concentrations of agonist than presynaptic inhibition of GABA release. These observations challenge the canonical disinhibition model and are consistent with the idea that more than one synaptic mechanism can contribute to MOP reward.
Footnotes
This work was supported by National Institute on Drug Abuse Awards R01 DA030529 to E.B.M. and DA008863 to Lakshmi A. Devi, and funds provided by the State of California for medical research on alcohol and substance abuse through the University of California, San Francisco. We thank Mirabel Lim, Joseph Driscoll, Adam Ferris, Peter Fong, Junko Ishikawa, Hagar Lock, and Kelly Pollack for technical support; Antonello Bonci and Lakshmi A. Devi for scientific discussions; and Kevin Bender and Roger Nicoll for feedback on the manuscript.
The authors declare no competing financial interests.
References
- Bonci A, Malenka RC. Properties and plasticity of excitatory synapses on dopaminergic and GABAergic cells in the ventral tegmental area. J Neurosci. 1999;19:3723–3730. doi: 10.1523/JNEUROSCI.19-10-03723.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonci A, Williams JT. Increased probability of GABA release during withdrawal from morphine. J Neurosci. 1997;17:796–803. doi: 10.1523/JNEUROSCI.17-02-00796.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozarth MA, Wise RA. Intracranial self-administration of morphine into the ventral tegmental area in rats. Life Sci. 1981;28:551–555. doi: 10.1016/0024-3205(81)90148-X. [DOI] [PubMed] [Google Scholar]
- Bozarth MA, Wise RA. Anatomically distinct opiate receptor fields mediate reward and physical dependence. Science. 1984;224:516–517. doi: 10.1126/science.6324347. [DOI] [PubMed] [Google Scholar]
- Brischoux F, Chakraborty S, Brierley DI, Ungless MA. Phasic excitation of dopamine neurons in ventral VTA by noxious stimuli. Proc Natl Acad Sci U S A. 2009;106:4894–4899. doi: 10.1073/pnas.0811507106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bromberg-Martin ES, Matsumoto M, Hikosaka O. Dopamine in motivational control: rewarding, aversive, and alerting. Neuron. 2010;68:815–834. doi: 10.1016/j.neuron.2010.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron DL, Wessendorf MW, Williams JT. A subset of ventral tegmental area neurons is inhibited by dopamine, 5-hydroxytryptamine and opioids. Neuroscience. 1997;77:155–166. doi: 10.1016/S0306-4522(96)00444-7. [DOI] [PubMed] [Google Scholar]
- Cardozo DL, Bean BP. Voltage-dependent calcium channels in rat midbrain dopamine neurons: modulation by dopamine and GABAB receptors. J Neurophysiol. 1995;74:1137–1148. doi: 10.1152/jn.1995.74.3.1137. [DOI] [PubMed] [Google Scholar]
- Charles AC, Mostovskaya N, Asas K, Evans CJ, Dankovich ML, Hales TG. Coexpression of delta-opioid receptors with micro receptors in GH3 cells changes the functional response to micro agonists from inhibitory to excitatory. Mol Pharmacol. 2003;63:89–95. doi: 10.1124/mol.63.1.89. [DOI] [PubMed] [Google Scholar]
- Chieng B, Azriel Y, Mohammadi S, Christie MJ. Distinct cellular properties of identified dopaminergic and GABAergic neurons in the mouse ventral tegmental area. J Physiol. 2011;589:3775–3787. doi: 10.1113/jphysiol.2011.210807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dever SM, Xu R, Fitting S, Knapp PE, Hauser KF. Differential expression and HIV-1 regulation of μ-opioid receptor splice variants across human central nervous system cell types. J Neurovirol. 2012;18:181–190. doi: 10.1007/s13365-012-0096-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobi A, Margolis EB, Wang HL, Harvey BK, Morales M. Glutamatergic and nonglutamatergic neurons of the ventral tegmental area establish local synaptic contacts with dopaminergic and nondopaminergic neurons. J Neurosci. 2010;30:218–229. doi: 10.1523/JNEUROSCI.3884-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford CP, Mark GP, Williams JT. Properties and opioid inhibition of mesolimbic dopamine neurons vary according to target location. J Neurosci. 2006;26:2788–2797. doi: 10.1523/JNEUROSCI.4331-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford CP, Phillips PE, Williams JT. The time course of dopamine transmission in the ventral tegmental area. J Neurosci. 2009;29:13344–13352. doi: 10.1523/JNEUROSCI.3546-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garzón M, Pickel VM. Plasmalemmal mu-opioid receptor distribution mainly in nondopaminergic neurons in the rat ventral tegmental area. Synapse. 2001;41:311–328. doi: 10.1002/syn.1088. [DOI] [PubMed] [Google Scholar]
- Georgoussi Z, Leontiadis L, Mazarakou G, Merkouris M, Hyde K, Hamm H. Selective interactions between G protein subunits and RGS4 with the C-terminal domains of the mu- and delta-opioid receptors regulate opioid receptor signaling. Cell Signal. 2006;18:771–782. doi: 10.1016/j.cellsig.2005.07.003. [DOI] [PubMed] [Google Scholar]
- Harrison C, Smart D, Lambert DG. Stimulatory effects of opioids. Br J Anaesth. 1998;81:20–28. doi: 10.1093/bja/81.1.20. [DOI] [PubMed] [Google Scholar]
- Hjelmstad GO, Xia Y, Margolis EB, Fields HL. Opioid modulation of ventral pallidal afferents to ventral tegmental area neurons. J Neurosci. 2013;33:6454–6459. doi: 10.1523/JNEUROSCI.0178-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iegorova O, Fisyunov A, Krishtal O. G-protein-independent modulation of P-type calcium channels by mu-opioids in Purkinje neurons of rat. Neurosci Lett. 2010;480:106–111. doi: 10.1016/j.neulet.2010.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jhou TC, Xu SP, Lee MR, Gallen CL, Ikemoto S. Mapping of reinforcing and analgesic effects of the mu opioid agonist endomorphin-1 in the ventral midbrain of the rat. Psychopharmacology. 2012;224:303–312. doi: 10.1007/s00213-012-2753-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiménez-González C, Pirttimaki T, Cope DW, Parri HR. Non-neuronal, slow GABA signalling in the ventrobasal thalamus targets delta-subunit-containing GABA(A) receptors. Eur J Neurosci. 2011;33:1471–1482. doi: 10.1111/j.1460-9568.2011.07645.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SW, North RA. Opioids excite dopamine neurons by hyperpolarization of local interneurons. J Neurosci. 1992a;12:483–488. doi: 10.1523/JNEUROSCI.12-02-00483.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SW, North RA. Two types of neurone in the rat ventral tegmental area and their synaptic inputs. J Physiol. 1992b;450:455–468. doi: 10.1113/jphysiol.1992.sp019136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan BA, Devi LA. G-protein-coupled receptor heterodimerization modulates receptor function. Nature. 1999;399:697–700. doi: 10.1038/21441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuszak AJ, Pitchiaya S, Anand JP, Mosberg HI, Walter NG, Sunahara RK. Purification and functional reconstitution of monomeric mu-opioid receptors: allosteric modulation of agonist binding by Gi2. J Biol Chem. 2009;284:26732–26741. doi: 10.1074/jbc.M109.026922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacey MG, Mercuri NB, North RA. Dopamine acts on D2 receptors to increase potassium conductance in neurones of the rat substantia nigra zona compacta. J Physiol. 1987;392:397–416. doi: 10.1113/jphysiol.1987.sp016787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammel S, Lim BK, Ran C, Huang KW, Betley MJ, Tye KM, Deisseroth K, Malenka RC. Input-specific control of reward and aversion in the ventral tegmental area. Nature. 2012;491:212–217. doi: 10.1038/nature11527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laviolette SR, Nader K, van der Kooy D. Motivational state determines the functional role of the mesolimbic dopamine system in the mediation of opiate reward processes. Behav Brain Res. 2002;129:17–29. doi: 10.1016/S0166-4328(01)00327-8. [DOI] [PubMed] [Google Scholar]
- Malik RU, Ritt M, DeVree BT, Neubig RR, Sunahara RK, Sivaramakrishnan S. Detection of G protein-selective G protein-coupled receptor (GPCR) conformations in live cells. J Biol Chem. 2013;288:17167–17178. doi: 10.1074/jbc.M113.464065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansour A, Fox CA, Thompson RC, Akil H, Watson SJ. mu-Opioid receptor mRNA expression in the rat CNS: comparison to mu-receptor binding. Brain Res. 1994;643:245–265. doi: 10.1016/0006-8993(94)90031-0. [DOI] [PubMed] [Google Scholar]
- Mansour A, Fox CA, Burke S, Akil H, Watson SJ. Immunohistochemical localization of the cloned mu opioid receptor in the rat CNS. J Chem Neuroanat. 1995;8:283–305. doi: 10.1016/0891-0618(95)00055-C. [DOI] [PubMed] [Google Scholar]
- Margolis EB, Hjelmstad GO, Bonci A, Fields HL. κ-Opioid agonists directly inhibit midbrain dopaminergic neurons. J Neurosci. 2003;23:9981–9986. doi: 10.1523/JNEUROSCI.23-31-09981.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis EB, Hjelmstad GO, Bonci A, Fields HL. Both kappa and mu opioid agonists inhibit glutamatergic input to ventral tegmental area neurons. J Neurophysiol. 2005;93:3086–3093. doi: 10.1152/jn.00855.2004. [DOI] [PubMed] [Google Scholar]
- Margolis EB, Lock H, Hjelmstad GO, Fields HL. The ventral tegmental area revisited: is there an electrophysiological marker for dopaminergic neurons? J Physiol. 2006;577:907–924. doi: 10.1113/jphysiol.2006.117069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis EB, Fields HL, Hjelmstad GO, Mitchell JM. δ-Opioid receptor expression in the ventral tegmental area protects against elevated alcohol consumption. J Neurosci. 2008;28:12672–12681. doi: 10.1523/JNEUROSCI.4569-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis EB, Coker AR, Driscoll JR, Lemaître AI, Fields HL. Reliability in the identification of midbrain dopamine neurons. PLoS One. 2010;5:e15222. doi: 10.1371/journal.pone.0015222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis EB, Toy B, Himmels P, Morales M, Fields HL. Identification of rat ventral tegmental area GABAergic neurons. PLoS One. 2012;7:e42365. doi: 10.1371/journal.pone.0042365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui A, Williams JT. Opioid-sensitive GABA inputs from rostromedial tegmental nucleus synapse onto midbrain dopamine neurons. J Neurosci. 2011;31:17729–17735. doi: 10.1523/JNEUROSCI.4570-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melis M, Camarini R, Ungless MA, Bonci A. Long-lasting potentiation of GABAergic synapses in dopamine neurons after a single in vivo ethanol exposure. J Neurosci. 2002;22:2074–2082. doi: 10.1523/JNEUROSCI.22-06-02074.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merighi S, Gessi S, Varani K, Fazzi D, Stefanelli A, Borea PA. Morphine mediates a proinflammatory phenotype via μ-opioid receptor-PKCε-Akt-ERK1/2 signaling pathway in activated microglial cells. Biochem Pharmacol. 2013;86:487–496. doi: 10.1016/j.bcp.2013.05.027. [DOI] [PubMed] [Google Scholar]
- Moaddab M, Haghparast A, Hassanpour-Ezatti M. Effects of reversible inactivation of the ventral tegmental area on the acquisition and expression of morphine-induced conditioned place preference in the rat. Behav Brain Res. 2009;198:466–471. doi: 10.1016/j.bbr.2008.11.030. [DOI] [PubMed] [Google Scholar]
- Moussawi K, Riegel A, Nair S, Kalivas PW. Extracellular glutamate: functional compartments operate in different concentration ranges. Front Syst Neurosci. 2011;5:94. doi: 10.3389/fnsys.2011.00094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nader K, van der Kooy D. Deprivation state switches the neurobiological substrates mediating opiate reward in the ventral tegmental area. J Neurosci. 1997;17:383–390. doi: 10.1523/JNEUROSCI.17-01-00383.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair-Roberts RG, Chatelain-Badie SD, Benson E, White-Cooper H, Bolam JP, Ungless MA. Stereological estimates of dopaminergic, GABAergic and glutamatergic neurons in the ventral tegmental area, substantia nigra and retrorubral field in the rat. Neuroscience. 2008;152:1024–1031. doi: 10.1016/j.neuroscience.2008.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noda M, Nakanishi H, Akaike N. Glutamate release from microglia via glutamate transporter is enhanced by amyloid-beta peptide. Neuroscience. 1999;92:1465–1474. doi: 10.1016/S0306-4522(99)00036-6. [DOI] [PubMed] [Google Scholar]
- Olmstead MC, Franklin KB. The development of a conditioned place preference to morphine: effects of microinjections into various CNS sites. Behav Neurosci. 1997;111:1324–1334. doi: 10.1037/0735-7044.111.6.1324. [DOI] [PubMed] [Google Scholar]
- Omelchenko N, Sesack SR. Ultrastructural analysis of local collaterals of rat ventral tegmental area neurons: GABA phenotype and synapses onto dopamine and GABA cells. Synapse. 2009;63:895–906. doi: 10.1002/syn.20668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onali P, Olianas MC. Naturally occurring opioid receptor agonists stimulate adenylate cyclase activity in rat olfactory bulb. Mol Pharmacol. 1991;39:436–441. [PubMed] [Google Scholar]
- Patrick GA, Dewey WL, Spaulding TC, Harris LS. Relationship of brain morphine levels to analgesic activity in acutely treated mice and rats and in pellet implanted mice. J Pharmacol Exp Ther. 1975;193:876–883. [PubMed] [Google Scholar]
- Puri SK, Cochin J, Volicer L. Effect of morphine sulfate on adenylate cyclase and phosphodiesterase activities in rat corpus striatum. Life Sci. 1975;16:759–767. doi: 10.1016/0024-3205(75)90352-5. [DOI] [PubMed] [Google Scholar]
- Ruzicka BB, Fox CA, Thompson RC, Meng F, Watson SJ, Akil H. Primary astroglial cultures derived from several rat brain regions differentially express mu, delta and kappa opioid receptor mRNA. Brain Res Mol Brain Res. 1995;34:209–220. doi: 10.1016/0169-328X(95)00165-O. [DOI] [PubMed] [Google Scholar]
- Shippenberg TS, Herz A. Place preference conditioning reveals the involvement of D1-dopamine receptors in the motivational properties of mu- and kappa-opioid agonists. Brain Res. 1987;436:169–172. doi: 10.1016/0006-8993(87)91571-X. [DOI] [PubMed] [Google Scholar]
- Shippenberg TS, Bals-Kubik R, Herz A. Examination of the neurochemical substrates mediating the motivational effects of opioids: role of the mesolimbic dopamine system and D-1 vs. D-2 dopamine receptors. J Pharmacol Exp Ther. 1993;265:53–59. [PubMed] [Google Scholar]
- Steffensen SC, Stobbs SH, Colago EE, Lee RS, Koob GF, Gallegos RA, Henriksen SJ. Contingent and non-contingent effects of heroin on mu-opioid receptor-containing ventral tegmental area GABA neurons. Exp Neurol. 2006;202:139–151. doi: 10.1016/j.expneurol.2006.05.023. [DOI] [PubMed] [Google Scholar]
- Steinberg EE, Keiflin R, Boivin JR, Witten IB, Deisseroth K, Janak PH. A causal link between prediction errors, dopamine neurons and learning. Nat Neurosci. 2013;16:966–973. doi: 10.1038/nn.3413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theile JW, Morikawa H, Gonzales RA, Morrisett RA. Ethanol enhances GABAergic transmission onto dopamine neurons in the ventral tegmental area of the rat. Alcohol Clin Exp Res. 2008;32:1040–1048. doi: 10.1111/j.1530-0277.2008.00665.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai HC, Zhang F, Adamantidis A, Stuber GD, Bonci A, de Lecea L, Deisseroth K. Phasic firing in dopaminergic neurons is sufficient for behavioral conditioning. Science. 2009;324:1080–1084. doi: 10.1126/science.1168878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Zessen R, Phillips JL, Budygin EA, Stuber GD. Activation of VTA GABA neurons disrupts reward consumption. Neuron. 2012;73:1184–1194. doi: 10.1016/j.neuron.2012.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velásquez-Martinez MC, Vázquez-Torres R, Jiménez-Rivera CA. Activation of alpha1-adrenoceptors enhances glutamate release onto ventral tegmental area dopamine cells. Neuroscience. 2012;216:18–30. doi: 10.1016/j.neuroscience.2012.03.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Liu-Chen LY, Traynor JR. Differential modulation of mu- and delta-opioid receptor agonists by endogenous RGS4 protein in SH-SY5Y cells. J Biol Chem. 2009;284:18357–18367. doi: 10.1074/jbc.M109.015453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witten IB, Steinberg EE, Lee SY, Davidson TJ, Zalocusky KA, Brodsky M, Yizhar O, Cho SL, Gong S, Ramakrishnan C, Stuber GD, Tye KM, Janak PH, Deisseroth K. Recombinase-driver rat lines: tools, techniques, and optogenetic application to dopamine-mediated reinforcement. Neuron. 2011;72:721–733. doi: 10.1016/j.neuron.2011.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong CS, Su YF, Watkins WD, Chang KJ. Opioid agonist binding affinity is increased by magnesium in the presence of guanosine diphosphate but decreased by magnesium in the presence of guanyl-5′-yl imidodiphosphate. J Pharmacol Exp Ther. 1994;268:653–661. [PubMed] [Google Scholar]
- Xia Y, Driscoll JR, Wilbrecht L, Margolis EB, Fields HL, Hjelmstad GO. Nucleus accumbens medium spiny neurons target non-dopaminergic neurons in the ventral tegmental area. J Neurosci. 2011;31:7811–7816. doi: 10.1523/JNEUROSCI.1504-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao C, Ye JH. Ethanol dually modulates GABAergic synaptic transmission onto dopaminergic neurons in ventral tegmental area: role of mu-opioid receptors. Neuroscience. 2008;153:240–248. doi: 10.1016/j.neuroscience.2008.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Landthaler M, Schlussman SD, Yuferov V, Ho A, Tuschl T, Kreek MJ. Mu opioid receptor knockdown in the substantia nigra/ventral tegmental area by synthetic small interfering RNA blocks the rewarding and locomotor effects of heroin. Neuroscience. 2009;158:474–483. doi: 10.1016/j.neuroscience.2008.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]