

Abstract
Mutations in Kinesin proteins (Kifs) are linked to various neurological diseases, but the specific and redundant functions of the vertebrate Kifs are incompletely understood. For example, Kif5A, but not other Kinesin-1 heavy-chain family members, is implicated in Charcot-Marie-Tooth disease (CMT) and Hereditary Spastic Paraplegia (HSP), but the mechanism of its involvement in the progressive axonal degeneration characteristic of these diseases is not well understood. We report that zebrafish kif5Aa mutants exhibit hyperexcitability, peripheral polyneuropathy, and axonal degeneration reminiscent of CMT and HSP. Strikingly, although kif5 genes are thought to act largely redundantly in other contexts, and zebrafish peripheral neurons express five kif5 genes, kif5Aa mutant peripheral sensory axons lack mitochondria and degenerate. We show that this Kif5Aa-specific function is cell autonomous and is mediated by its C-terminal tail, as only Kif5Aa and chimeric motors containing the Kif5Aa C-tail can rescue deficits. Finally, concurrent loss of the kinesin-3, kif1b, or its adaptor kbp, exacerbates axonal degeneration via a nonmitochondrial cargo common to Kif5Aa. Our results shed light on Kinesin complexity and reveal determinants of specific Kif5A functions in mitochondrial transport, adaptor binding, and axonal maintenance.
Keywords: axon degeneration, CMT, HSP, Kinesin, mitochondria, sensory neuron
Introduction
Kinesin superfamily proteins (Kifs) are microtubule-based molecular motors essential for the intracellular transport of various cargos, including organelles, proteins, and RNAs (Hirokawa et al., 2009). The extent of functional redundancy between the 45 known mammalian Kifs is not known (Miki et al., 2001). However, disrupting single kif genes can cause abnormalities in both mice and humans, suggesting unique roles for some Kifs (Hirokawa et al., 2010).
Kif5s, which dimerize to form Kinesin-1, are critical in neurons. Comprised of the ubiquitously expressed Kif5B and the neuron-specific Kif5A and Kif5C, the Kif5 family mediates the transport of diverse cargo (Hirokawa et al., 2010). kif5A (Xia et al., 2003) and kif5C (Kanai et al., 2000) knock-out (KO) mice exhibit neuronal loss and dysfunction, whereas kif5B (Tanaka et al., 1998) KO mice die during embryonic development. Furthermore, Kif5A mutations in humans cause SPG10, a form of Hereditary Spastic Paraplegia (HSP; Reid et al., 2002; Fichera et al., 2004). SPG10 patients display progressive paralysis and spasticity of the limbs, characteristic of HSP, but can also have complex symptoms including peripheral polyneuropathy, blindness, deafness, and cognitive and behavioral changes (Blair et al., 2006; Schüle et al., 2008; Goizet et al., 2009; Crimella et al., 2012). Kif5A mutations have also been associated with Charcot-Marie-Tooth Type 2 (CMT2; Crimella et al., 2012), an axonal peripheral neuropathy characterized by progressive loss of peripheral sensation and muscle wasting (Vallat et al., 2013). The underlying pathologic mechanisms and why the closely related Kif5B and Kif5C do not compensate for Kif5A mutations in these diseases are unclear.
The integral role of mitochondrial transport in neuronal viability and function can be seen in the myriad of neurodegenerative diseases (NDs) associated with dysregulated transport (Sheng and Cai, 2012). Kif5s are key effectors implicated in anterograde mitochondrial transport mainly via an adaptor complex known in Drosophila as the Kif5-Milton-Miro complex (Schwarz, 2013). Milton directly binds Kif5 and Mitochondrial Rho GTPase (Miro), an outer mitochondrial membrane protein that links Kinesin-1 and mitochondria. Mammals have two Milton homologs, Trak1 and Trak2 (trafficking protein kinesin-binding), and two Miro homologs, Miro1 and Miro2. Recent biochemical studies have identified two distinct binding sites for Trak1 and Trak2 to Kif5 (Randall et al., 2013), and Trak1 and Trak2 have been proposed to differentially sort mitochondria (van Spronsen et al., 2013). With three Kif5s, two Traks, and two Miros, redundancy is possible, although the numerous homologs also raise the interesting possibility that different Kif5-Trak-Miro combinations may support unique functions essential to the complex vertebrate nervous system.
Here we assess the in vivo function of kif5A in zebrafish. We find that mutation of kif5Aa, but not its duplicate gene kif5Ab, leads to larval lethality and sensorimotor deficits similar to human patients. Furthermore, we demonstrate that peripheral axons have defective mitochondrial transport and degenerate in kif5Aa mutants. Surprisingly, no other Kif5 can substitute for Kif5Aa, and we show that the C-terminal tail of Kif5Aa mediates its unique, essential function. Moreover, Kif1b, another motor that has been implicated in mitochondrial transport (Nangaku et al., 1994; Wozniak et al., 2005) cannot suppress the deficits of kif5Aa mutant sensory neurons, although kif5Aa and kif1b cooperate to promote peripheral sensory neuron maintenance. Our genetic analyses reveal that Kif5A fulfills a unique role in axonal transport of mitochondria, which is essential for axonal maintenance.
Materials and Methods
Animals and fish stocks
The kif5Aasa7168 and kif5Absa7055 mutations were obtained from the Sanger Zebrafish Mutation Project (Kettleborough et al., 2013) and crossed into Tg(huC:Kaede) (Sato et al., 2006) and Tg(huC:GCaMP5G) (Ahrens et al., 2013) lines, kindly provided by Alex Schier (Harvard University, Cambridge, MA). Embryos were obtained from natural pairwise matings and were reared according to standard procedures (Westerfield, 1995). Because sex is not determined by a single chromosome in zebrafish, it is not possible to predict what the sex of the larvae will be before sexual differentiation at 1.5–2 months of age. The stages of zebrafish development analyzed are before sex determination and differentiation in zebrafish; therefore, larvae that will develop as females, as well as those that will develop as males were analyzed. All procedures and experimental protocols were in accordance with NIH guidelines and approved by the Institutional Review Boards of Albert Einstein College of Medicine and Stanford University School of Medicine.
Genotyping
Genomic DNA was extracted from adult fins or single larvae using standard procedures (Westerfield, 1995). The region surrounding the kif5Aasa7168 mutation was amplified with primers sa7168-F, sa7168-R1, and sa7168-R2. Subsequent restriction enzyme (RE) digestion with DraIII led to a smaller fragment for the mutant allele by dCAPS (Neff et al., 1998). Two reverse primers were used due to a polymorphism within the mutant intron. The region surrounding the kif5Absa7055 mutation was amplified with primers sa7055-F and sa7055-R. Subsequent RE digestion with Mva12691 yielded a smaller fragment for the mutant allele. Primer sequences are in Table 1. kif1bst43 (Lyons et al., 2009) and kbpst23 (Lyons et al., 2008) were genotyped as previously described.
Table 1.
Primer sequences (5′ to 3′)
| Genotyping | |
| sa7168-F | TGGAGAAACGTCTTCGTTCTACG |
| sa7168-R1 | GTGTGTGAATGTGAATGCAGTGCACAGTGT |
| sa7168-R2 | GTGTGTGAATGTGAATGCAGTGCACAGCGT |
| sa7055-F | AGACCGAGCAGAAACTCTGTGG |
| sa7055-R | CCGTAAACCTTCCTGTGGAGAA |
| RT-PCR | |
| kif5Aaex21-F | GAGCAGTCCAAACAGGATCTCA |
| kif5Aaex25-R | GAATCGGGCATTGCTGTAAGAC |
| kif5Abex1-F | TGATATCAGCGCAGAATGCAATA |
| kif5Abex8-R | TTCCTTCAGCCAGAGCTGAGAT |
| ef1α-F | AGCCTGGTATGGTTGTGACCTTCG |
| ef1α-R | CCAAGTTGTTTTCCTTTCCTGCG |
| 3′ RACE | |
| kif5AbRACE-F1 | AAGAGTCGGGAACATGAGAAAA |
| kif5AbRACE-F2 | GGAAGCTGTTCGTTCAAGACCT |
| Full-length cDNAs | |
| kif5AaFL-F | ATGACGGACGCCGCCGCGGAG |
| kif5AaFL-R | TTAACTGGCTGCAGTCTCCTGCTGCATGATGT |
| kif5AbFL-F | CCTCTGTTGCGTTTCTTGTGCT |
| kif5AbFL-R | CCGTTAGTAATGTGGAGTTTCTGTG |
| kif5BaFL-F | ATGGCGGACCCGGCGGAGT |
| kif5BaFL-R | TCAGCTCTTCTCTTGTTTAGTGC |
| kif5BbFL-F | CAGCTGAAGATGGCGGACC |
| kif5BbFL-R | AATATTGAACCCTCCCGTGCTG |
| kif5CFL-F | ATCAAACCAGGATCATACACGA |
| kif5CFL-R | TTTTCACTTGCTCCTGTGATGG |
| kif1b-α/β-FL-F | ATGTCTGGCGCCTCAGTGAAA |
| kif1b-α-FL-R | GTACATGAACCTATAGCCCTGC |
| kif1b-β-FL-R | TGGTACGCTGTAAAGGTAGACA |
| Other cloning | |
| kozakCherry-F | GCCACCATGGTGAGCAAGGGCGAGGA |
| kif5AaΔTailSTOP-R | TTATTACTTCTGGGTGTTAGACCCCC |
| kif5AaTail-F | CAGAAGCAGAAGATTTCCTTTCTT |
| kif5AaΔTailNOSTOP-R | CTTCTGGGTGTTAGACCCCC |
| EcoNI-kif5AaTail-F | CTAGTACCTCGAGGAGGCGCTGAAGGATGCCAAACAGGG |
| pcr8-PvuI-R | TCTCTTGCTTTTGTCAGCAAGAT |
| BsaBI-kif5AaTail-F | CTAGTAGATAAGAATCAGGCTCTGGAGT |
Molecular analysis of kif5Aasa7168 and kif5Absa7055 mutations
To determine the effect of the mutations on the kif5Aa and kif5Ab mRNA transcripts, total RNA was extracted from 6 d postfertilization (dpf) larvae from AB WT, kif5Aawt/sa7168, and kif5Absa7055/sa7055 intercrosses using TRIzol Reagent (15596-026, Invitrogen). kif5Aasa7168/sa7168 mutant larvae were sorted from siblings based on phenotype. cDNA synthesis was performed using SuperScript III First-Strand Synthesis System (18080-051, Invitrogen) according to the manufacturer's instructions. The absorbance at 260 nm and concentration of cDNA was determined using a Nanodrop ND-1000 Spectrophotometer (0.25 μg/reaction was used). Exons 21–25 of kif5Aa were amplified using primers kif5Aaex21-F and kif5Aaex25-R. Exons 1–8 of kif5Ab were amplified using primers kif5Abex1-F and kif5Abex8-R. ef1α was amplified as a loading control using primers ef1α-F and ef1α-R. PCRs were performed using Taq polymerase and an Eppendorf Mastercycler with the following conditions: 95οC for 2 min followed by 40 cycles of 95οC for 30 s, 59οC for 30 s, and 72οC for 1 min, and a final extension of 72οC for 10 min. PCR products were gel extracted (28704, Qiagen) and either sequenced directly or cloned into pGEM-T (A3600, Promega) and sequenced (Macrogen). Primer sequences are in Table 1.
DNA rescue constructs and cloning
Zebrafish kif5Ba cDNA was received from Masahiko Hibi (Nojima et al., 2010).
Full-length zebrafish cDNAs.
Because the 3′ region of kif5Ab was not fully annotated, we performed 3′RACE (18373-019, Invitrogen) using cDNA prepared, as above, from adult brain. Nested PCR was performed by first amplifying with kif5AbRACE-F1 and the abridged universal amplification primer (AUAP), and then kif5AbRACE-F2 and the AUAP. The fragment was cloned into pGEM-T and sequenced. WT cDNAs were amplified from cDNA, prepared as above, from 6 dpf larvae from AB WT intercrosses. Full-length (FL) kif5Aa, kif5Ab, kif5Bb, kif5C, kif1b-α, and kif1b-β were amplified with the primers in Table 1. FL-kif5Ba was PCR-amplified from pcs2-HA-Kif5Ba (Nojima et al., 2010). FL-kif5Aasa7168 was PCR-amplified from 6 dpf kif5Aasa7168 mutant cDNA using the same primers as for FL-kif5Aa. To flank the cDNAs with attL1 and attL2 sites (Hartley et al., 2000; Walhout et al., 2000) cDNAs were then TOPO cloned into pCR8/GW/TOPO (K250020, Invitrogen). 1xFLAG-tags were added to constructs by PCR. GFP, Cherry, and Myc fusions were made by recombining pCR8 constructs with appropriate pCSDest vectors (Villefranc et al., 2007) following the manufacturer's instructions (11791-020, Invitrogen).
Truncated kif5Aas.
pCR8-Cherry-FLAGKif5AaΔTail, which contains amino acids 1–837 of zKif5Aa, was made by PCR-amplifying cherry-FLAGKif5AaΔTail from pCSDest-Cherry-FLAGKif5Aa using primers kozakCherry-F and kif5AaΔTailSTOP-R and TOPO cloning into pCR8/GW/TOPO. pCR8-Kif5AaTail, which contains amino acids 836–1033 of zKif5Aa, was made by PCR-amplifying kif5AaTail from pCR8-FLAGkif5Aa-FL using primers kif5AaTail-F and kif5AaFL-R and TOPO cloning into pCR8/GW/TOPO. A cherry-Kif5AaTail fusion was made by recombining the pCR8 construct with pCSDest-Cherry. Cherry-Kif5AaTail was then amplified from pCSDest-Cherry-Kif5AaTail using primers kozakCherry-F and kif5AaFL-R and TOPO cloned into pCR8/GW/TOPO.
Chimeric kif5s.
To make FLAG-kif5AbAaTail, which contains amino acids 1–871 of zKif5Ab fused to amino acids 888–1033 of zKif5Aa, the kif5AaTail was first amplified from pCR8-FLAGkif5Aa-FL using primers EcoNI-kif5AaTail-F and pcr8-PvuI-R. This fragment was then digested with EcoNI/PvuI and ligated into the corresponding sites in pCR8-FLAGkif5Ab-FL. To make FLAG-kif5BaAaTail, which contains amino acids 1–742 of zKif5Ba fused to amino acids 747–1033 of zKif5Aa, the kif5AaTail was first amplified from pCR8-FLAGkif5Aa-FL using primers BsaBI-kif5AaTail-F and pcr8-PvuI-R. This fragment was then digested with BsaBI/PvuI and ligated into the corresponding sites in pCR8-FLAGkif5Ba-FL.
UAS/Gal4 vectors.
pBH:huC:Gal4 was derived by cloning huC:Gal4 (Paquet et al., 2009) into a Tol2 vector. Bidirectional UAS:DsRed; UAS:Kif5 rescue constructs were made by recombining pCR8 constructs with pT2d-DEST_pA_DsRed.T4_E1b_UAS_E1b_GW-R1-R2_pA (DsRed-UAS-attR1-attR2; Paquet et al., 2009).
Primers
Table 1 shows all primer sequences used in this study.
Imaging
Confocal.
For fixed samples, larvae were mounted in 1% low-melting agarose (A9414, Sigma-Aldrich) in PBS on a glass coverslip bottomed dish. For live samples, larvae were anesthetized with Tricaine (A5040, Sigma-Aldrich) and mounted in 1% low-melting agarose in 1× embryo medium on a glass coverslip bottomed dish. Unless otherwise stated, all fluorescent images were acquired with a Zeiss 5Live Duoscan line-scanning confocal microscope at room temperature (RT). Images were acquired using 10×/0.45, 20×/0.8, 63×/1.4, and 100×/1.4 lenses, and 405, 488, and 561 nm excitation lasers. Images were processed in ImageJ and represent maximum projection z-stacks unless otherwise stated.
Live whole-mount.
Gross phenotypes were imaged with an Olympus SZ61 dissecting microscope and a high-resolution digital camera (model S97809, Olympus America) and Picture Frame 2.0 or 3.0 software (Optronics).
IF labeling
For whole-mount immunofluorescence (IF), larvae were fixed in 2% trichloroacetic acid (T6399, Sigma-Aldrich) in PBS for 2–3 h at RT or 4% paraformaldehyde overnight at 4°C and permeabilized by placing in acetone at −20°C for 7 min. Anti-acetylated Tubulin (T6793, Sigma-Aldrich) was diluted at 1:1000, Anti-DsRed at 1:250 (632496, Clontech), anti-NF-M RMO44 at 1:50 (13-0500, Life Technologies), and anti-FLAG at 1:500 (F3165, Sigma-Aldrich). AlexaFluor488 and AlexaFluor568 (Invitrogen) secondaries were diluted at 1:500.
PTZ seizure susceptibility assay
The behaviors of 3 dpf larvae were recorded using a JVC camcorder (model GZ-MS230AU). Baseline activity was recorded for 10 min, during which time no seizure-like behaviors were observed. After 10 min, larvae were placed in 2.5 mM pentylenetetrazole (PTZ) and embryos were recorded for 17 min. Videos were scored for seizure-like behavior according to established methods (Baraban et al., 2005, 2007). Stage 2 and 3 seizure-like behaviors were scored for each minute interval, with the majority being Stage 2. Behavioral scoring of individual larvae was conducted while blinded with respect to genotypes.
Touch response assay
Two or 6 dpf embryos from heterozygous intercrosses were placed in individual wells in 1× embryo medium. Embryos were touched on the tail with a probe five times and the number of escape responses was recorded.
Axonal coverage of the fin
Maximal z-projections of α-AcTub immunostaining were thresholded using ImageJ and the percentage area covered signal was calculated with the Analyze Particles feature.
TEM
Transmission electron microscopy (TEM) was performed according to previous protocols (Lyons et al., 2008). Briefly, grids were stained with 50% uranyl acetate/50% ethanol, followed by Sato's lead stain and imaged on a JEOL JEM-1400 transmission electron microscope. Five sibling control nerves and 6 kif5Aasa7168/sa7168 mutant nerves (n = 3 animals each) were analyzed. Diameters of axons were measured using ImageJ.
Mitochondria, synaptic vesicle, and lysosome labeling
To colabel axons and mitochondria, circular pBH-huC:Gal4 and pT2d-EXP_pA_memYFP_E1b_UAS_E1b_mitoCFP_pA (memYFP-UAS-mitoCFP; Plucińska et al., 2012) plasmid DNA and Tol2 Transposase RNA transcribed from pCS2FA-transposase (Kwan et al., 2007) were combined (10–25 ng/μl each). Approximately 1 nl of solution was injected into one-cell stage embryos. Embryos were placed in 0.003% phenylthiourea in 1× embryo medium at 24 h postfertilization (hpf) to block pigmentation and screened for the reporter. To colabel axons and synaptic vesicles, circular pBH-huC:Gal4 and syn:GFP-DSR (Meyer and Smith, 2006) plasmid DNA and Tol2 Transposase RNA were combined (10–25 ng/μl each) and injected as above. To colabel axons and lysosomes, circular Crest3:Gal4; UAS:mCherry (Palanca et al., 2013) and pDEST-5kbneurod-Lamp1-eGFP (Drerup and Nechiporuk, 2013) plasmid DNA and Tol2 Transposase RNA were combined (10–25 ng/μl each) and injected as above.
Mitochondrial, synaptic vesicle and lysosomal distribution and mitochondrial dynamics
Axons and mitochondria were colabeled with mitochondria-targeted CFP (mitoCFP) and membrane-targeted YFP (memYFP) as above. Embryos were sorted for expression in the fin and, for calculating mitochondrial density, 1–2 confocal z-stacks were obtained/embryo. Mitochondria were manually counted; axon length was calculated using the Simple Neurite Tracer plugin in ImageJ. Two hundred to 1000 μm of axon was visualized/embryo. Rescue experiments were performed similarly with a coinjected rescue plasmid. Synaptic vesicle and lysosomal distribution analysis was performed similarly using syn:GFP-DSR and pDEST-5kbneurod-Lamp1-eGFP plasmids to label as described above. To analyze mitochondrial dynamics, fin axons were traced back to their main stem axon near the spinal cord. Time lapses were performed in a single z-plane for 5 min with one image/15 s. Dynamics were analyzed using the ImageJ MTrackJ plugin. Mitochondria that moved >0.5 μm between frames were defined as mobile.
Alignment of zebrafish Kif5 sequences
Cloned FL zebrafish Kif5 amino acid sequences were aligned using ClustalW2 multiple-sequence alignment (Larkin et al., 2007; McWilliam et al., 2013). Protein alignments were generated using Jalview v2 (Waterhouse et al., 2009).
Statistical analysis
Statistical analysis was performed using GraphPad Prism 6. Unless otherwise stated, error bars represent ±SEM and statistical significance was estimated by either two-tailed unpaired Student's t test to compare two populations or one-way ANOVA followed by Tukey's multiple-comparison test to compare more than two populations.
Results
kif5Aa, but not kif5Ab, is essential for nervous system function in zebrafish
Loss of kif5A in mouse is perinatal lethal (Xia et al., 2003), but its physiological role was unclear because kif5A mutant embryos displayed limited pathologic signs. Due to a genome-wide duplication event, zebrafish have two kif5A genes, kif5Aa and kif5Ab, with overlapping expression (Campbell and Marlow, 2013). Here we characterized kif5Aa and kif5Ab mutant alleles, kif5Aasa7168 and kif5Absa7055 respectively (Fig. 1A, and data not shown), found by the Sanger Zebrafish Mutation Project (Kettleborough et al., 2013). Sequencing confirmed that both mutations are single base pair substitutions predicted to disrupt essential splice donor sites in each gene (Fig. 1B, and data not shown). RT-PCR analysis revealed two novel kif5Aa transcripts in kif5Aasa7168 mutant larvae (Fig. 1C). The more abundant transcript included the intron between exons 24 and 25, and the less abundant transcript lacked exon 24 (Fig. 1D). Both transcripts contained nonsense codons truncating the tail domain. In homozygous kif5Absa7055 mutants, use of an alternative splice donor site resulted in retention of 8 base pairs of intronic sequence and a nonsense codon within the motor domain.
Figure 1.
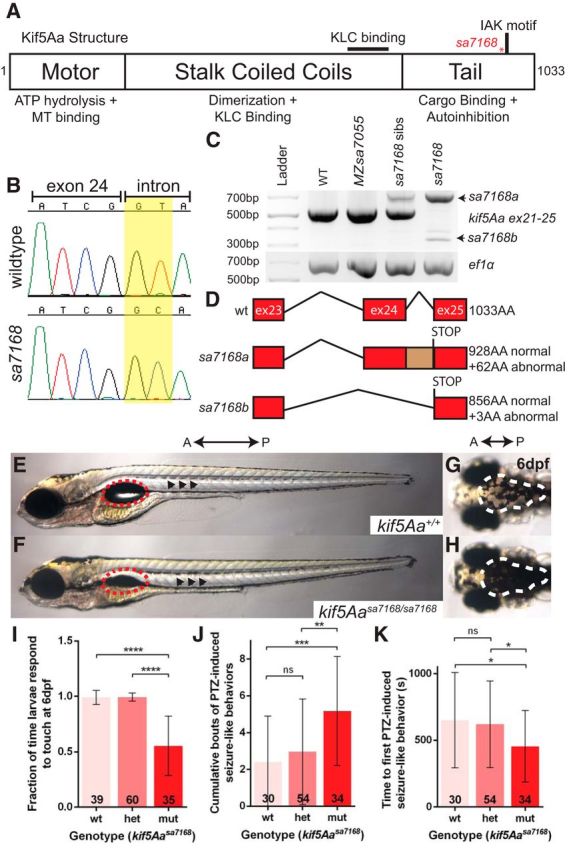
The kif5Aasa7168 mutant allele results in sensorimotor deficits. A, Kif5Aa protein structure. Red asterisk denotes mutation. KLC, Kinesin light chain; MT, microtubule. B, Chromatogram for WT and mutant exon–intron boundaries depicting loss of GT splice donor site in kif5Aasa7168 mutant. C, RT-PCR analysis of kif5Aasa7168 mutant reveals improper splicing of mutant transcripts. D, Novel kif5Aasa7168 mutant transcripts are predicted to lead to premature stop codons and truncated proteins. E, F, Lateral view of 6 dpf larval zebrafish. Red dotted circles denote swim bladder, which is not inflated in kif5Aasa7168 mutants. Black arrowheads denote elevated lateral pigmentation in kif5Aasa7168 mutants. G, H, Dorsal view of 6 dpf larval zebrafish. White dotted line denotes elevated dorsal pigmentation in kif5Aasa7168 mutants. A, Anterior; P, Posterior. I, Touch response of kif5Aasa7168 mutants at 6 dpf. Error bars indicate ±SD; ****p < 0.0001 (one-way ANOVA, Tukey's post-test). J, K, Quantification of PTZ seizure-like induction. J, Cumulative bouts of seizures and (K) seizure latency. Error bars indicate ±SD; *p < 0.05, **p < 0.01, ***p < 0.001 (one-way ANOVA, Tukey's post-test).
To assess the developmental functions of kif5A genes, we analyzed progeny from heterozygous intercrosses for each allele. kif5Aasa7168 homozygous mutants did not inflate their swim bladder by 5 dpf (Fig. 1E,F), and died between 8–12 dpf. Furthermore, kif5Aasa7168 mutants appeared heavily pigmented from 5 dpf onward (Fig. 1E–H), indicating defective background adaptation. Background adaptation involves redistributing melanosomes in melanophores to adapt to ambient light, a process defective in blind mutants. In kif5Aasa7168 mutants, impaired background adaptation is likely due to defective visual processing because the retina appeared properly patterned (data not shown). We also noted a diminished touch response in kif5Aasa7168 mutants at 6 dpf (Fig. 1I). At 2 dpf, however, kif5Aasa7168 mutant touch responses resembled WT and heterozygous siblings, suggesting that the touch response circuit forms normally but becomes compromised in early larvae.
In contrast to kif5Aa mutants, kif5Absa7055 mutants were viable, fertile as adults, and showed no maternal-zygotic deficits. In addition, we observed no enhancement of kif5Aa single mutant phenotypes or additional phenotypes in kif5Aa;kif5Ab compound mutants. Because kif5Aa single and kif5Aa;kif5Ab double-mutants were indistinguishable, we focused on kif5Aasa7168 single mutants, referred to as kif5A mutants hereafter.
kif5A mutations in humans are associated with sensorimotor and cognitive deficits. During free-swimming bouts, WT larvae exhibit synchronous beat-and-glide movements (Buss and Drapeau, 2001). In contrast, kif5A mutants were uncoordinated with asynchronous pectoral fin beating. Reminiscent of motor-circuit deficiencies in human patients with Kif5A mutations, kif5A mutants displayed complex spastic behaviors. From 4 dpf and thereafter, exaggerated and frequent gulping, and jaw and pectoral fin twitching were evident in kif5A mutants, whereas these behaviors were rare in siblings. Furthermore, at 5 dpf and thereafter, kif5A mutants underwent episodes of repetitive trunk contractions followed by periods of inactivity.
Seizures occur with postnatal loss of kif5A in mice (Xia et al., 2003; Nakajima et al., 2012) and in some HSP cases (Finsterer et al., 2012). The abnormal behaviors observed in kif5A mutants are consistent with a hyperexcited state and potential seizures. We tested whether kif5A mutants were predisposed to seizures, using the established PTZ seizure model (Baraban et al., 2005, 2007; Baxendale et al., 2012; Afrikanova et al., 2013) and found that cumulative seizure-like behaviors were significantly increased in GABAA receptor antagonist (PTZ)-treated kif5A mutants compared with siblings (Fig. 1J). Moreover, the first seizure-like behavior occurred earlier in mutants (Fig. 1K). Therefore, kif5A mutants are more prone to PTZ-induced episodes, exhibiting more total events and shorter latency. We detected no defects in motoneuron innervation of somites (Fig. 2C,D) nor overt differences in cranial ganglia or brain axonal architecture (Fig. 2E–J) in kif5A mutants, suggesting that, although they may be present, the neurons controlling motor function are dysfunctional.
Figure 2.
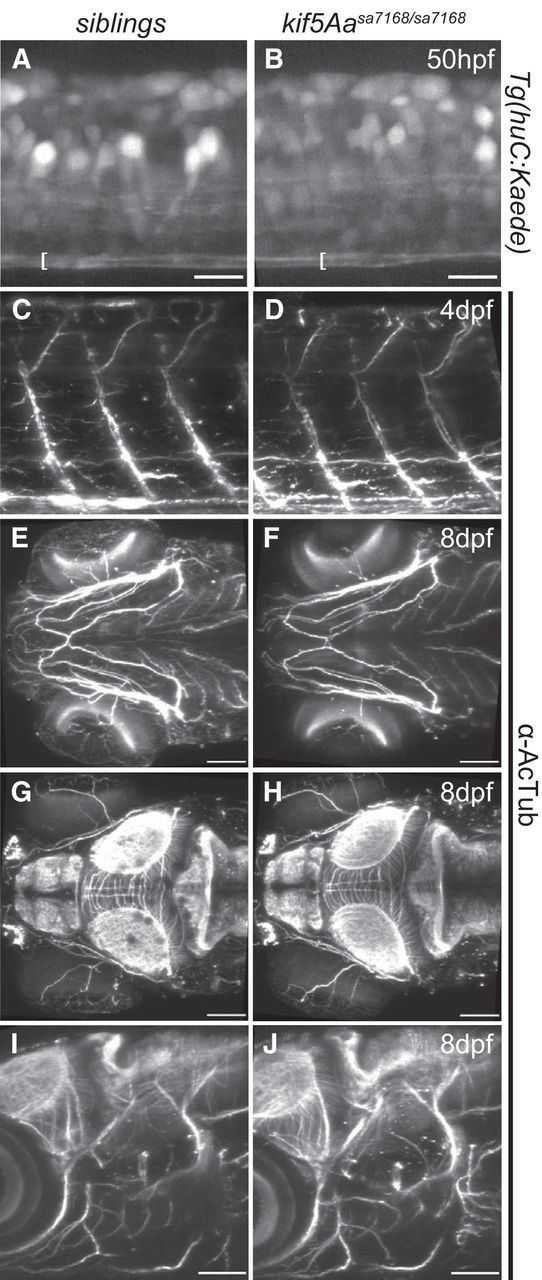
Intact ventral spinal cord axon tracts, cranial axon tracts, and motoneurons in kif5Aasa7168 mutants. A, B, Lateral view of spinal cord at somite 15 in Tg(huc:Kaede) background shows intact ventral axon tracts denoted by the brackets. Scale bar, 15 μm. C, D, Lateral view of larval zebrafish shows intact axonal architecture of motor neurons by α-AcTub. E, F, Ventral view of larval zebrafish showing comparable axonal architecture of jaw stained by α-AcTub. Scale bar, 75 μm. G, H, Dorsal view of larval zebrafish showing intact axonal architecture of brain stained with α-AcTub. Scale bar, 75 μm. I, J, Lateral view. No deficits were apparent in the axonal architecture of brain and cranial nerves stained with α-AcTub. Scale bar, 75 μm. Rostral is to the left in all panels.
Polyneuropathy and axonal degeneration in kif5Aa mutants
Kif5A mutations have been associated with CMT2 (Crimella et al., 2012), which may explain peripheral polyneuropathy among some SPG10 patients (Kawaguchi, 2013). Given the peripheral nerve disruptions in human CMT2 patients, progressive loss of cutaneous sensory innervation might underlie diminished touch reflexes of kif5A mutants. Consistent with this notion, examination of cutaneous axons in the fin revealed reduced fine cutaneous nerve arbors in kif5A mutants (Fig. 3B–D). Reduced innervation was also apparent in another peripheral sensory nervous system component, the posterior lateral line nerve (pLLn). Stretching the length of the larvae, the pLLn is among the longest nerves, and thus, should be especially sensitive to axonal transport deficits. At 8 dpf, the pLLn was thinner and shorter in kif5A mutants than in siblings (Fig. 3E–G), whereas other axonal tracts appeared to be of similar thickness. Furthermore, the pLLn exhibited a “dying back” axonal degeneration phenotype (Coleman, 2005), with axonal swellings and acetylated tubulin (AcTub) accumulations (Fig. 3H,I). Though this phenotype likely reflected axonal degeneration, impaired pLLn outgrowth, as occurs in kif1b (Lyons et al., 2009) and kif1-binding protein (kbp; Lyons et al., 2008) zebrafish mutants, was also possible. Analysis of the pLLn in live Tg(huC:Kaede) transgenic embryos (Sato et al., 2006) at 50 hpf revealed no significant difference in pLLn outgrowth (Fig. 3E); thus, compromised pLLn maintenance was likely. Furthermore, we detected no outgrowth defects in axons in the ventral spinal cord (Fig. 2A,B) as is seen in kif1b zebrafish mutants. To delineate the timeframe of degeneration, we imaged α-AcTub labeled pLLns of 4 dpf larvae. Although the nerve extended the length of the larvae, it was thinner in kif5A mutants (Fig. 3J,K) and had swellings with accumulations of AcTub in pLLn axons (Fig. 3H). Consistent with a role in peripheral sensory axon maintenance as opposed to outgrowth, we saw no difference between kif5A mutants and siblings in axonal coverage of 50 hpf fins (Fig. 3D). In ultrastructural studies, kif5A mutant pLLns had evidence of degenerating axons (Fig. 4B) and significantly fewer large caliber axons, both unmyelinated and myelinated (Fig. 4E–H). Conversely, numbers of small unmyelinated and myelinated axons were similar in kif5A mutants and siblings. These results indicate kif5A is required for maintenance of peripheral axons.
Figure 3.

kif5Aa regulates peripheral sensory neuron maintenance but not outgrowth. A, Diagram of lateral view of larval zebrafish. Blue, pink, and orange boxes denote areas shown in B, C; F, G; and H–M; respectively. B, C, Lateral view of fin showing PCA arbors at 6 dpf. Yellow dotted line marks area of axonal loss. Scale bar, 50 μm. D, Quantification of fin axonal coverage. Error bars indicate ±SEM; ****p < 0.0001 (one-way ANOVA, Tukey's post-test). E, Quantification of pLLn length over time shows pLLn outgrowth (50 hpf), and eventual degeneration (8 dpf). Error bars indicate ±SEM; ***p < 0.001 (one-way ANOVA, Tukey's post-test). F, G, Lateral views of hindbrain and first few somites. Note the thinner pLLn at 8 dpf and other axon tracts. Scale bar, 75 μm. H, I, Higher-magnification of pLLn shows axonal accumulations of AcTub (arrowheads), in kif5Aasa7168 mutants at (F) 4 dpf and (G) 8 dpf, and (G) the truncated pLLn at 8 dpf. Scale bars, 10 μm. J, K, Higher-magnification of pLLn at 4 dpf at somite 15 shows reduced thickness, denoted by blue bracket. Scale bar, 10 μm. L, M, Transgenic fluorescent Ca2+ indicator GCaMP5G shows elevated Ca2+ levels in axonal accumulations of kif5Aasa7168 mutants. Scale bar, 10 μm.
Figure 4.
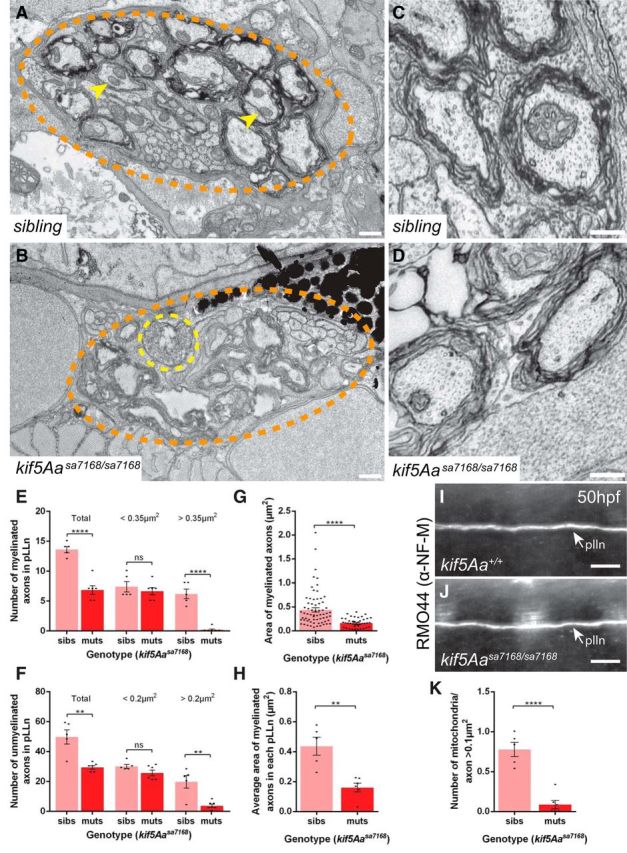
Reduced axon caliber and mitochondria but normal NF-M distribution in kif5Aasa7168 mutant pLLns. A, B, TEM images of cross-sections of the pLLn at somite level 7–8 at 7 dpf. Myelinated axons are evident beneath the basement membrane of (A) WT and (B) kif5Aa mutant axons, which appear smaller, lack mitochondria, and show signs of degeneration. pLLn (orange dotted circle), a degenerating axon (yellow dotted circle), and axonal mitochondria (yellow arrowheads). Scale bar, 0.5 μm. C, D, Higher-magnification TEM images of cross-sections of the pLLn showing intact microtubule and neurofilament networks in (C) WT and (D) kif5Aa mutant axons. E, F, Quantification of total, small diameter, and large diameter (E) unmyelinated and (F) myelinated axons in the pLLn. G, H, Quantification of the (G) area of all myelinated axons in the pLLns and (H) average area of myelinated axons in each pLLn. I, J, Lateral view of pLLn at 50 hpf labeled with α-NF-M antibody RMO44 in (I) WT and (J) kif5Aasa7168 mutants reveals similar NF-M distribution. K, Quantification of mitochondria/axon >0.1 μm2 in the pLLn. For all quantification, nsibs = 3 larvae, 5 nerves; nmuts = 3 larvae, 6 nerves. Error bars indicate ±SEM; **p < 0.01, ****p < 0.0001 (Student's t test).
Extracellular Ca2+ influx into the distal axon occurs during degeneration (Coleman, 2005; Wang et al., 2012). Elevated intracellular Ca2+ can activate Calpain, a serine-threonine protease, leading to cleavage and disruption of structural components like neurofilaments and microtubules (Billger et al., 1988; Johnson et al., 1991). To test whether this occurs in kif5Aa mutant pLLns, we crossed the mutation into the Tg(huC:GCaMP5G) background (Ahrens et al., 2013), which expresses a Ca2+-sensitive GFP variant, GCaMP5G, in neurons (Akerboom et al., 2012). Whereas WT and heterozygotes had weak GCaMP5G fluorescence in the pLLn, kif5A mutants had swellings with strong GCaMP5G signal (Fig. 3L,M), implicating high Ca2+ in degeneration of mutant sensory axons.
kif5Aa mutant pLLn axons and cutaneous nerve arbors lack mitochondria
A conditional kif5A KO mouse associated sensory neuron degeneration with impaired axonal transport of neurofilaments (NF; Xia et al., 2003); however, another conditional kif5A KO mouse showed no NF deficits (Nakajima et al., 2012). Our TEM analysis of the pLLn revealed no obvious defects in either NF or microtubule architecture (Fig. 4C,D), nor did we detect defects in medium NF (NF-M) distribution in zebrafish kif5A mutants (Fig. 4I,J). Therefore, kif5Aasa7168 mutant sensory defects may result from altered transport of another cargo.
Kinesin-I is the key molecular motor thought to transport axonal mitochondria (Tanaka et al., 1998; Kanai et al., 2000; Karle et al., 2012) and defective axonal mitochondrial transport has been linked to NDs (Sheng and Cai, 2012; Hinckelmann et al., 2013). Ultrastructural analysis of pLLn axons showed abundant mitochondria in siblings, and their marked reduction in kif5A mutants (Fig. 4A,B). The total numbers of mitochondria per nerve and per axon >0.1 μm2 were significantly reduced in mutants as compared with siblings (Fig. 4K). This suggested that defective mitochondrial transport might cause pLLn degeneration.
To test whether peripheral cutaneous axon (PCA) arbors were also devoid of mitochondria, we labeled mitochondria and the axons using a huC:Gal4 driver construct to express mosaically a bidirectional UAS plasmid encoding mitoCFP and memYFP (Plucińska et al., 2012) in neurons (Fig. 5A). Mitochondrial density in kif5A mutant PCAs was greatly reduced at 6 dpf (Fig. 5B–H). Because mutant axons were degenerating at this time, mitochondria deficits could be secondary to axonal degeneration, rather than a primary defect. However, at times before and when axonal degeneration and behavioral defects were first apparent, mitochondrial density was significantly reduced, indicating mitochondrial deficiency precedes behavioral abnormalities and axonal degeneration (Fig. 5H). Unlike mitochondria, no difference was observed in the density of synaptic vesicles or lysosomes in mutant and sibling PCAs (Fig. 6). Thus, the kif5A mutation specifically disrupts mitochondria localization.
Figure 5.

Lack of mitochondria in peripheral cutaneous axon arbors and altered transport dynamics in main stem axons. A, Scheme to mosaically label neurons with the mitochondrial reporter. Black box denotes enlarged image, pink box denotes region imaged in B–G, and purple box denotes region imaged in I–M. B–G, Mitochondria are absent from kif5Aasa7168 mutant peripheral cutaneous axon arbors. Arrows denote mitochondria present in sibling axons. Scale bars, 10 μm. H, Decreased mitochondrial density in kif5Aasa7168 mutant axon arbors compared with siblings. Error bars indicate ±SEM; *p < 0.05, ****p < 0.0001 (one-way ANOVA, Tukey's post-test). I, Still images from time-lapses reveal immobile mitochondria (blue arrowheads) in kif5Aasa7168 mutant axons, and anterograde (orange arrowheads) and retrograde (yellow arrowheads) transport in siblings. Scale bars, 5 μm. J, K, Fraction of (J) motile and (K) immotile mitochondria density in kif5Aasa7168 mutant main stem axons. Error bars indicate ±SEM; **p < 0.01 (Student's t test). L, Fraction of mitochondria observed to move at specific run velocities. Anterograde moving mitochondria in kif5Aa mutants move at slower velocities than in siblings; **p < 0.01 (χ2 test). M, Distribution of each mitochondrial run velocity in siblings and kif5Aa mutants. Error bars indicate ±SEM. For time-lapse data in I–M, nsibs = 10 axons, from 10 larvae; nmuts = 9 axons, from 7 larvae.
Figure 6.
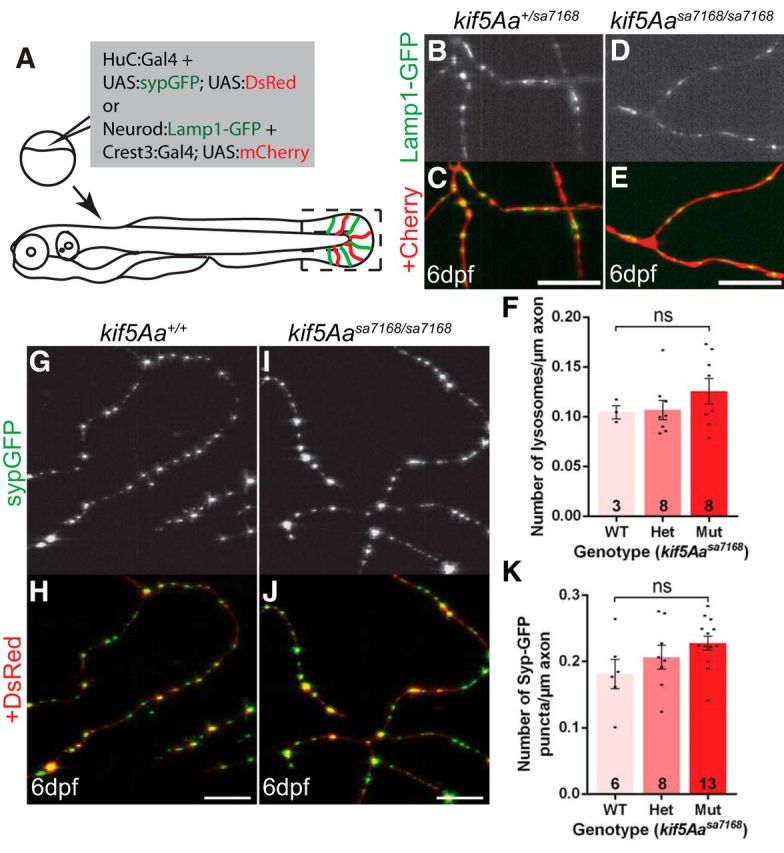
Distributions of lysosomes and presynaptic vesicles are unaffected in peripheral cutaneous axon arbors of kif5Aasa7168 mutants. A, Schematic depicts strategy to mosaically label neurons with the presynaptic vesicle reporter and the lysosomal reporter. Black box denotes region imaged in B–E and G–J. G–K, Presynaptic vesicles are present in both (G, H) WT and (I, J) kif5Aa mutant peripheral cutaneous axon arbors at 6 dpf and there is (K) no difference in density. Scale bars, 10 μm. Error bars indicate ±SEM (one-way ANOVA, Tukey's post-test). B–F, Lysosomes are present in both (B, C) sibling and (D, E) kif5Aa mutant peripheral cutaneous axon arbors at 6 dpf and there is (F) no difference in density. Scale bars, 10 μm. Error bars indicate ±SEM (one-way ANOVA, Tukey's post-test).
Because mitochondria were present in the main stem axons of the PCAs located near the spinal cord (Fig. 5I,K) we performed time-lapse analysis of mitochondrial dynamics in this region. Consistent with loss of a molecular motor required for transport, the fraction of motile mitochondria was reduced in mutants, with no motile mitochondria observed in most cases (Fig. 5I,J). Furthermore, the few mitochondria that moved had altered kinetics with anterograde run velocities on the low end of the normal range (Fig. 5L,M). Surprisingly, the density of immotile mitochondria between siblings and mutants was comparable (Fig. 5K), indicating that localization to the main stem is kif5A independent.
Kif5Aa acts in neurons to localize mitochondria and prevent axonal degeneration
kif5Aa is expressed in neural tissues (Campbell and Marlow, 2013), but it is unclear whether it is limited to neurons. To test whether kif5Aa acts cell autonomously in neurons to prevent degeneration of the pLLn, we mosaically coexpressed full-length FLAG-tagged zebrafish Kif5Aa (FLAG-Kif5Aa) with a DsRed reporter (Paquet et al., 2009) to label the axons (Fig. 7A). FLAG-Kif5Aa caused no defects in sibling pLLn axons (Fig. 7B–D,H). In kif5A mutant pLLns, FLAG-Kif5Aa-expressing axons extended the length of the larvae while the others showed signs of axonal degeneration (Fig. 7E–H). These results indicate that kif5Aa acts cell autonomously in neurons to prevent pLLn axonal degeneration.
Figure 7.

Cell autonomous and hypomorphic activity of Kif5Aasa7168. A, Scheme to mosaically label (B–G) pLLn axons and (I–P) PCAs. Blue dotted box indicates axons imaged in B–G and black dotted box indicates axons imaged in I–P. B–G, OE of FLAG-Kif5Aa has no effect on sibling pLLn axons (B–D) and rescues kif5Aasa7168 mutant axonal degeneration (E–G). White arrows denote axonal accumulations of AcTub present in nonrescued axons. Scale bars, 10 μm. H, Quantification of length of pLLn for FLAG-Kif5Aa OE. Error bars indicate ±SEM; ***p < 0.001 (one-way ANOVA, Tukey's post-test). I–P, OE of FLAG-Kif5Aa has no effect on mitochondrial density of sibling PCAs (I–L) and rescues kif5Aasa7168 density (M–P). Note that in mutants mitochondria appear mainly in OE cells (white arrows) and not in non-OE cells (white arrowheads). Scale bars, 5 μm. Q, Quantification of mitochondrial density relative to nonexpressing sibling control axons for FLAG-Kif5Aa and FLAG-Kif5Aasa7168 OE. Error bars indicate ±SEM; *p < 0.05, ***p < 0.001, ****p < 0.0001 (one-way ANOVA, Tukey's post-test).
To test whether kif5Aa was required in neurons to distribute mitochondria we mosaically coexpressed FLAG-Kif5Aa/DsRed with the mitoCFP/memYFP reporter and examined mitochondrial density in the PCA arbors of the fin (Fig. 7A). As in the pLLn, mitochondrial density was only rescued in kif5A mutant PCAs marked by the DsRed reporter and not in adjacent axons (Fig. 7M–Q). Furthermore, overexpression (OE) of FLAG-Kif5Aa in sibling axons had no effect on mitochondrial density (Fig. 7I–L,Q). These results indicate that kif5Aa acts cell autonomously to distribute mitochondria in PCAs and suggest that its function is tightly regulated.
Using this assay we investigated the nature of the kif5Aasa7168 allele. In WT genotypes, mitochondrial density in cells expressing FLAG-Kif5Aasa7168 and the mitochondrial reporter resembled neighbors expressing only the reporter (Fig. 7Q). Moreover, OE of FLAG-Kif5Aasa7168 suppressed the mitochondrial defect of mutant cells, albeit less so than FLAG-Kif5Aa and with significant variance (Fig. 7Q). These data show that in terms of mitochondrial transport, the kif5Aasa7168 mutation is hypomorphic and not dominant-negative.
Other Kif5s and Kif1b are not sufficient to localize axonal mitochondria in kif5A mutants
Kif5s are thought to act redundantly in mitochondrial transport because Kif5A, Kif5B, and Kif5C can all bind to the known mitochondrial adaptors Trak1 and Trak2 (Brickley et al., 2005; Smith et al., 2006; Brickley and Stephenson, 2011; Chen and Sheng, 2013; Randall et al., 2013; van Spronsen et al., 2013), and very few Kif5 isoform-specific functions are known. All five zebrafish kif5s are expressed in the nervous system (Campbell and Marlow, 2013). Thus, since the mutation did not act antimorphically, the profound axonal mitochondrial deficit in kif5Aasa7168 mutants was surprising. We considered two main explanations for this striking phenotype. First, the other kif5s may simply be less abundant but could substitute if expressed at a higher level. Second, kif5Aa could have a unique function in axonal mitochondria localization that other Kif5 proteins cannot fulfill. Attempts to suppress the mitochondrial deficit of kif5A mutants by expressing the five zebrafish Kif5s in peripheral sensory axons indicate the latter. OE of FLAG-Kif5Aa robustly rescued the mitochondrial density, but no other FLAG-tagged Kif5 rescued mitochondrial density in kif5A mutant PCAs (Fig. 8P), despite robust OE and localization within axons (Fig. 8B–K). Moreover, none of the Kif5s affected mitochondrial density in WT siblings.
Figure 8.
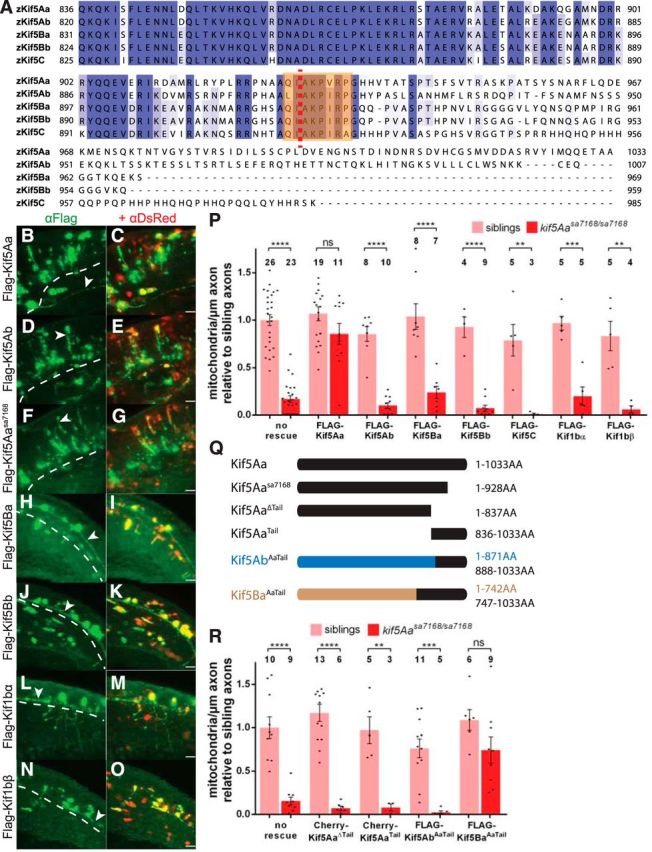
Kif5Aa, but not other motors implicated in mitochondrial transport, restores mitochondria in a tail-dependent fashion. A, Alignment of zebrafish Kif5 C-terminal tail domains reveals significant sequence variation. Orange box denotes conserved IAK motif. Red dotted line denotes site of kif5Aasa7168 mutation. B–O, Lateral views with dorsal to the top and anterior to the left of α-FLAG and α-DsRed staining of embryos injected with FLAG-Kif rescue constructs in the (B–G) hindbrain at 54 hpf and the (H–O) spinal cord at 24 hpf. All FLAG-Kifs are robustly expressed and DsRed expression coincides with FLAG expression. All FLAG-Kifs are present in axons (arrowheads) and cell bodies, as expected. Dotted line denotes boundary of CNS. Scale bars, 25 μm. P, Effects of overexpressing Kif5 family members and Kif1b splice variants on mitochondrial density in WT and kif5Aasa7168 mutant PCAs. Error bars indicate ±SEM; **p < 0.01, ***p < 0.001, ****p < 0.0001 (one-way ANOVA, Tukey's post-test). Q, Kif5 truncations and chimeras used in R. R, OE of Kif5 truncations and chimeras in PCAs variably affects mitochondrial density. Error bars indicate ±SEM; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 (one-way ANOVA, Tukey's post-test).
The Kinesin-3 motor Kif1b has also been implicated in mitochondrial transport in some cell types (Nangaku et al., 1994; Wozniak et al., 2005), though kif1b zebrafish mutants do not display overt mitochondrial deficits (Lyons et al., 2009). kif1b has two main splice forms, kif1bα and kif1bβ (Nangaku et al., 1994; Zhao et al., 2001). Like the other Kif5s, neither OE of Kif1bα nor Kif1bβ affected mitochondrial density in kif5A mutants or siblings (Fig. 8P), despite robust OE and localization to axons (Fig. 8L–O). These results indicate that only Kif5Aa can distribute mitochondria in these sensory axons.
Kif5Aa's C-tail is necessary but not sufficient to distribute mitochondria
Although zebrafish Kif5 proteins are highly similar (Campbell and Marlow, 2013), their C-terminal tails (C-tail) vary, with Kif5Aa and Kif5Ab both having extended, though dissimilar, tails (Fig. 8A). Four points implicate the Kif5Aa C-tail in mitochondrial transport in sensory neurons. (1) The kif5Aasa7168 mutation truncates the C-tail and disrupts axonal localization of mitochondria. (2) Among the Kifs tested, Kif5Aa alone rescued the mitochondrial defect in kif5A mutants. (3) The binding domains of two mitochondrial adaptors, Trak1 and Trak2, reside within the Kif5 C-tail domain (Randall et al., 2013). (4) Mutation of the conserved auto-inhibitory IAK motif (Coy et al., 1999; Friedman and Vale, 1999; Hackney and Stock, 2000; Hackney et al., 2009), disrupted by the kif5Aasa7168 mutation, similarly disrupts mitochondrial transport in Drosophila (Moua et al., 2011). We tested and found that the Kif5Aa C-tail was required to suppress mitochondrial density deficits using a Kif5Aa construct lacking the entire C-tail (Kif5AaΔTail, Fig. 8Q,R). Expression of the Kif5Aa C-tail alone (Kif5AaTail; Fig. 8Q) also did not rescue the defects (Fig. 8R), indicating that the C-tail is necessary but not sufficient. To determine whether the Kif5Aa C-tail could confer rescue activity to other Kif5s, we expressed a chimeric Kif5 with the Kif5Ab motor and stalk domains and the C-tail of Kif5Aa (Kif5AbAaTail; Fig. 8Q). Surprisingly, Kif5AbAaTail did not suppress the mitochondrial deficit. However, because kif5Ab mutants had no phenotype and Kif5Ab did not rescue mitochondrial abundance, it is possible that Kif5Ab lacks a functional motor. Therefore, we expressed another chimeric Kif5 with the Kif5Ba motor and stalk domains and the C-tail of Kif5Aa (Kif5BaAaTail; Fig. 8Q). Kif5BaAaTail suppressed mitochondrial deficits (Fig. 8R) suggesting that the Kif5Aa C-tail confers the capacity to distribute mitochondria to other Kif5s.
Mitochondrial transport-independent role for Kif1b/KBP in peripheral sensory neuron maintenance
Kif1b and its adaptor molecule Kif1-binding protein (KBP) have been implicated in mitochondrial transport in certain cell types (Nangaku et al., 1994; Wozniak et al., 2005). However, to what extent, if at all, this occurs in neurons in vivo is unclear. Zebrafish kif1b (Lyons et al., 2009) and kbp (Lyons et al., 2008) mutants display peripheral nervous system outgrowth deficits, and kbp is additionally implicated in pLLn axonal maintenance. To test whether kif1b also has a role in axonal maintenance and, if so, whether the kif1b/kbp maintenance roles are associated with mitochondrial transport, we analyzed PCA arbors and pLLns of kif5Aasa7168;kif1bst43 (Lyons et al., 2009) and kif5Aasa7168;kbpst23 (Lyons et al., 2008) compound mutants at 6 dpf. Whereas kif1bst43 single mutant PCA arbors were indistinguishable from WT (Figs. 9A,G, 3B), concomitant loss of one copy of kif5Aa in kif1bst43 mutants led to reduced PCA arbors, like kif5Aasa7168 mutants (Figs. 9B,G, 3C). Even more strikingly, PCAs were completely absent in kif5Aasa7168;kif1bst43 homozygous double-mutants (Fig. 9C,G). In addition, the pLLn of kif5Aasa7168;kif1bst43 double-mutants was shorter than kif1bst43 single mutants (Fig. 9I). In contrast to kif1bst43 single mutants, PCAs were reduced in kbpst23 single mutants (Fig. 9D,K); however, as in kif5Aasa7168;kif1bst43 double-mutants, PCAs were completely absent in kif5Aasa7168;kbpst23 double-mutants (Fig. 9F,K). Furthermore, the pLLn of kif5Aasa7168;kbpst23 double-mutants was shorter than kbpst23 single mutants (Fig. 9L). Because both kbp and kif1b contribute to axonal outgrowth, we performed similar experiments at 50 hpf to determine whether these interactions were at the level of maintenance or outgrowth. We observed no axons in the fin of kif5Aasa7168;kbpst23 double-mutants at 6 dpf, but they were indistinguishable from siblings at 50 hpf (Fig. 9K). Furthermore, we observed no enhancement of kbpst23 pLLn outgrowth defects in kif5Aasa7168;kbpst23 double-mutants (Fig. 9L). Similarly, kif5Aasa7168/+;kif1bst43/st43 embryos, which had PCA deficits at 6 dpf, showed no enhancement of outgrowth defects in either the PCA arbors or the pLLn at 50 hpf (Fig. 9H,J), suggesting defects in axonal maintenance rather than outgrowth because kif1b single-mutant PCA arbors appear normal at 6 dpf. To determine whether this maintenance role involved mitochondria as in kif5Aa single mutants, we quantified the number of mitochondria in kif5Aasa7168/+;kif1bst43/st43 and kbpst23/st23 embryos, both of which showed maintenance defects. Surprisingly, mitochondria numbers in the PCAs were not reduced in either genotype (Fig. 9M,N), indicating that reduced arborization is, unlike in kif5Aa single mutants, independent of mitochondrial density. These data indicate that both kif5Aa and kif1b/kbp play roles in peripheral sensory axon maintenance and that there are two independent mechanisms, mitochondrial-dependent and mitochondrial-independent, for maintaining sensory axons.
Figure 9.

kif1b and kbp cooperate with kif5Aa to maintain peripheral sensory axons, independent of mitochondrial distribution. A–C, Effect of kif5Aa dosage on PCAs in kif1bst43 mutants. D–F, Effect of kif5Aa dosage on PCAs in kbpst23 mutants. G, H, Quantification of axonal coverage of the fin in kif5Aa and kif1b genotypes at (G) 6 dpf and (H) 50 hpf. Error bars indicate ±SEM; **p < 0.01, ****p < 0.0001 (one-way ANOVA, Tukey's post-test). I, J, Quantification of pLLn length in kif5Aa and kif1b genotypes at (I) 6 dpf and (J) 50 hpf. Error bars indicate ±SEM; ***p < 0.001, ****p < 0.0001 (one-way ANOVA, Tukey's post-test). K, L, Quantification of (K) axonal coverage of the fin and (L) pLLn length in kif5Aa and kbp genotypes at 50 hpf and 6 dpf. Error bars indicate ±SEM; *p < 0.05, ***p < 0.001, ****p < 0.0001 (one-way ANOVA, Tukey's post-test). M, N, Normal mitochondrial density in PCA arbors of (M) kif1bst43 and kif5Aasa7168/+;kif1bst43/st43 and (N) kbpst23/23 larvae at 6 dpf. Error bars indicate ±SEM (one-way ANOVA, Tukey's post-test).
Discussion
We have shown that zebrafish kif5Aa mutants exhibit hyperexcitability and peripheral polyneuropathy, reminiscent of patients with SPG10 and CMT2. Additionally, OE of Kif5Aa in pLLn axons and peripheral cutaneous axons rescues degeneration and mitochondrial deficits. We show that kif5Aa acts in neurons to maintain mitochondrial density in peripheral sensory axons in a C-tail-dependent manner, and that other Kif5s and Kif1b cannot suppress deficits, providing a mechanism for peripheral sensory axonal degeneration in patients with kif5A mutations. We propose that a distinct Kif5A complex formed via interaction with the Kif5A tail is specifically required for mitochondrial transport in sensory neurons and that distinct Kif5 complexes may mediate mitochondrial transport within discrete neuronal types. Finally, cooperation between Kinesin-1 and Kinesin-3 motors mediates distinct aspects of axonal maintenance, revealing roles for these kinesins in transporting mitochondria and other cargo to maintain diverse axonal architectures and neuronal functions.
Specific role for Kif5Aa C-terminal tail in mitochondrial distribution
Studies of invertebrates have provided a wealth of knowledge about Kinesin-1 functions. However, invertebrates possess only one kif5 gene, khc in Drosophila (Saxton et al., 1991) and unc-116 in Caenorhabditis elegans (Patel et al., 1993), which precludes analysis of Kif5 isoform-specific functions that may be essential to the complexity of the vertebrate nervous system. Due to their similar protein structures and functions in vitro, Kif5A, Kif5B, and Kif5C are thought to act largely redundantly in neurons. However, as evidenced by the distinct kif5A (Xia et al., 2003), kif5B (Tanaka et al., 1998), and kif5C (Kanai et al., 2000) KO mouse phenotypes, and the diverse consequences of human kif5A (Kawaguchi, 2013) and kif5C mutations (Poirier et al., 2013), it is evident that kif5 genes are not fully redundant. Nonetheless, the functional differences among Kif5 proteins have remained largely unknown.
We show that kif5Aa has a unique function in mitochondrial localization, axonal maintenance, and other aspects of sensorimotor function. One simple explanation for these phenotypes and the varied mouse and human phenotypes could be differential expression of kif5s. Strikingly, however, no other Kif5 protein can rescue mitochondrial transport deficits of kif5Aa mutants. In contrast, previous work in mammals shows that Kif5A and Kif5C can suppress mitochondrial deficits in Kif5B knock-outs (Tanaka et al., 1998; Kanai et al., 2000). Because neurons were not examined in those studies, it is possible that Kif5 proteins have redundant roles in some cell types, but not others. Alternatively, Kif5A may substitute for Kif5B or Kif5C but also have unique functions. Supporting this notion, we find that the Kif5Aa C-tail is required for rescue and that it confers the capacity to rescue to Kif5Ba. Similarly, the mouse Kif5A C-tail is required for GABAA receptor transport via the Kif5A specific adaptor GABARAP (Nakajima et al., 2012). It is possible that a similar Kif5Aa specific adaptor may exist for mitochondrial transport as all Kif5s examined have been shown to bind to the known adaptors (Traks and Syntabulin) involved in mitochondrial transport (Smith et al., 2006; Brickley and Stephenson, 2011; Chen and Sheng, 2013; Randall et al., 2013; van Spronsen et al., 2013). An alternative possibility is that the C-tail regulates Trak1/2 or Miro1/2 binding and activity, such that the Kif5Aa-Trak-Miro complex has a distinct activity from other Kif5 complexes. Notably, distinct domains of mammalian Kif5A are used for its binding to Trak1 and Trak2 (Randall et al., 2013). Coupled with a recent report suggesting distinct functions for Trak1 and Trak2 (van Spronsen et al., 2013), and because Kif5A is the only Kif5 with an extended C-tail, the domain by which Kifs interact with Traks, it is plausible that different Kif5 proteins form specific Kif5-Trak-Miro complexes with unique activities required for mitochondrial transport within neuronal subdomains or cell types.
Recent analyses show that transport can be stepwise in fashion, with one motor conducting short-range and another long-range transport steps (Hoerndli et al., 2013). Because mitochondria persist in the main stem axons in kif5Aa mutants, it is possible that other Kif5s facilitate this short-range mitochondrial transport, whereas Kif5Aa is dedicated to long-range transport into axonal arbors. This function could be achieved via specific Kif5 complexes that form according to the cell type and subcellular location.
Kinesins, mitochondrial transport, and axonal maintenance
Mitochondria are crucial for cellular ATP production and regulation of Ca2+ homeostasis (Sheng and Cai, 2012; Schwarz, 2013). Deficits in mitochondrial axonal transport have been associated with several neurodegenerative diseases (Sheng and Cai, 2012), suggesting that axonal mitochondria are critical. In most cases, it is unclear whether the alterations in transport precede or result from pathological changes in the axons. kif5A mutant axons grow normally, have normal distribution of synaptic components and lysosomes, and normal overall structure. Nonetheless, axons of kif5A mutants lack mitochondria and later degenerate. This loss of axonal mitochondria may lead to drastic decreases of local ATP concentrations. Interestingly, mutant pLLns lack both myelinated and unmyelinated large caliber axons; thus, mitochondria may provide ATP required for formation or maintenance of large caliber axons. Furthermore, loss of axonal mitochondria can elevate Ca2+ concentrations. Increased Ca2+ might exacerbate the mitochondrial deficits, as the Ca2+ sensitive Miro releases mitochondria from Kif5 transport machinery in high Ca2+ concentrations (Saotome et al., 2008). Indeed, our data show that kif5A mutant main stem axons have very few motile mitochondria. Our data illustrates a crucial role for axonal mitochondria in maintaining axons and suggests that restoring mitochondrial density or activity may represent a therapeutic avenue in diseases characterized by axonal degeneration, such as HSP or CMT2.
Kif1b and its adaptor molecule KBP have been implicated in mitochondrial transport in certain cell types (Nangaku et al., 1994; Wozniak et al., 2005), and zebrafish kif1b (Lyons et al., 2009), mouse kif1b (Zhao et al., 2001), and zebrafish kbp (Lyons et al., 2008) mutants all display peripheral nervous system deficits. However, whereas zebrafish kif5A mutants display a striking loss of mitochondrial density in axons destined to degenerate, kif5Aasa7168/+;kif1bst43/st43 and kbpst23/23 mutants, both of which show similar PCA reduction to kif5A mutants, do not. The striking lack of cutaneous axon arbors in kif5Aasa7168;kif1bst43 double-mutants indicates that kif5Aa has an additional role in axonal maintenance that is only required when kif1b is lost. Conversely, the maintenance role of kif1b is only necessary when kif5Aa is reduced, because kif1b single-mutant PCAs appear normal. Like kif5Aa;kif1b double-mutants, kif5Aa;kbp double-mutants also lack PCAs. Previous work implicates KBP as a Kif1b binding partner critical for axonal maintenance (Wozniak et al., 2005; Lyons et al., 2008). Our genetic analysis suggests that in addition to Kif5A mediated mitochondria associated axon maintenance, a KBP-dependent mechanism promotes maintenance of peripheral sensory axons by carrying a nonmitochondrial cargo common to Kif1b and Kif5Aa (Fig. 10). Furthermore, because kbpst23/st23 single mutants have deficits in axonal maintenance that are not associated with mitochondria transport, are not observed in kif1bst43 single mutants, and are not worsened by loss of a single copy of kif5Aa, it is likely that KBP serves as the adaptor molecule for both Kif1b and Kif5Aa for this unknown nonmitochondrial cargo. These findings shed new light on the complexity of Kinesin redundancy and demonstrate at least two different means to maintain sensory axons and to prevent degeneration.
Figure 10.

Kif5Aa, Kif1b, and KBP cooperate to maintain peripheral sensory axons. Kif5Aa transports mitochondria into peripheral sensory axons likely via interaction with known Kif5 mitochondrial adaptors, such as TRAK1, and is required for their maintenance. A secondary KBP-dependent mechanism, possibly involving transport of an unknown cargo common to Kif1b and Kif5Aa, further supports maintenance of peripheral sensory axons.
Kif5A and human disease
Mutations in human kif5A can cause both motor and sensory dysfunction characterized as SPG10 (Reid et al., 2002), a form of HSP (Finsterer et al., 2012), or CMT2 (Crimella et al., 2012), depending on the predominant modality affected. kif5A zebrafish mutants display phenotypes consistent with motor and sensory dysfunction characteristic of a mixed model of SPG10 and CMT2.
Similar to SPG10 patients, kif5Aasa7168 mutants display striking motor dysfunction. Generally, HSP-like symptoms have been explained by degeneration of the longest corticospinal tracts (Deluca et al., 2004; Hedera et al., 2005). Similar to the axonal degeneration seen in HSP corticospinal tracts, zebrafish kif5A mutant peripheral sensory axons degenerate, suggesting that specific subsets of central axons that have not been analyzed here may similarly degenerate, and thus contribute to the observed spasticity. Susceptibility of zebrafish kif5A mutants to PTZ-induced seizures indicates potential involvement of GABAA receptors in the pathology. Moreover, conditional postnatal kif5A KO mice, which also develop seizures and motor abnormalities (Xia et al., 2003), associated pathology with impaired GABAA receptor transport (Nakajima et al., 2012). Therefore, hyperactivity due to diminished GABAA receptor transport may be an alternative pathway to spastic behaviors in kif5A mutants and SPG10 patients.
Though the pathogenesis of these disorders is unknown, two proposed mechanisms involve abnormal neurofilament (Xia et al., 2003) or mitochondrial transport caused by kif5A mutations (Karle et al., 2012; Kawaguchi, 2013). We show that mutation of kif5Aa diminishes axonal mitochondria density in peripheral sensory neurons while sparing localization of other known Kif5 cargos examined. Furthermore, loss of mitochondrial density correlates with axonal degeneration and decreased sensory function. Because no defects in Neurofilament-M localization or axon architecture suggestive of loss of neurofilament transport were observed, impaired mitochondrial transport is likely the main defect in sensory neurons of patients with kif5A mutations. Moreover, the interactions between motor mutants point toward other shared cargos that are required for axonal maintenance and may be disrupted in other pathological contexts.
Footnotes
This work was supported by NIH Grants R01GM089979 to F.L.M., R01NS050223 to W.S.T., T32-GM007288 and 1F31NS083258-01A1 to P.D.C., NMSS RG-4756-A-3 to W.S.T., and NCI Cancer Center P30CA013330 to the Analytical Imaging Facility (AIF). We thank members of the Marlow and Talbot labs and L.D. Fricker for helpful discussions, our animal care staff for fish care, the AIF at Einstein for microscopy support, F. Del Bene for discussing data in advance of publication, and J. Saenz for help with PTZ data analysis.
The authors declare no competing financial interests.
References
- Afrikanova T, Serruys AS, Buenafe OE, Clinckers R, Smolders I, de Witte PA, Crawford AD, Esguerra CV. Validation of the zebrafish pentylenetetrazol seizure model: locomotor versus electrographic responses to antiepileptic drugs. PloS One. 2013;8:e54166. doi: 10.1371/journal.pone.0054166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahrens MB, Orger MB, Robson DN, Li JM, Keller PJ. Whole-brain functional imaging at cellular resolution using light-sheet microscopy. Nat Methods. 2013;10:413–420. doi: 10.1038/nmeth.2434. [DOI] [PubMed] [Google Scholar]
- Akerboom J, Chen TW, Wardill TJ, Tian L, Marvin JS, Mutlu S, Calderón NC, Esposti F, Borghuis BG, Sun XR, Gordus A, Orger MB, Portugues R, Engert F, Macklin JJ, Filosa A, Aggarwal A, Kerr RA, Takagi R, Kracun S, et al. Optimization of a GCaMP calcium indicator for neural activity imaging. J Neurosci. 2012;32:13819–13840. doi: 10.1523/JNEUROSCI.2601-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baraban SC, Taylor MR, Castro PA, Baier H. Pentylenetetrazole induced changes in zebrafish behavior, neural activity and c-fos expression. Neuroscience. 2005;131:759–768. doi: 10.1016/j.neuroscience.2004.11.031. [DOI] [PubMed] [Google Scholar]
- Baraban SC, Dinday MT, Castro PA, Chege S, Guyenet S, Taylor MR. A large-scale mutagenesis screen to identify seizure-resistant zebrafish. Epilepsia. 2007;48:1151–1157. doi: 10.1111/j.1528-1167.2007.01075.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxendale S, Holdsworth CJ, Meza Santoscoy PL, Harrison MR, Fox J, Parkin CA, Ingham PW, Cunliffe VT. Identification of compounds with anti-convulsant properties in a zebrafish model of epileptic seizures. Dis Model Mech. 2012;5:773–784. doi: 10.1242/dmm.010090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billger M, Wallin M, Karlsson JO. Proteolysis of tubulin and microtubule-associated proteins 1 and 2 by calpain I and II: difference in sensitivity of assembled and disassembled microtubules. Cell Calcium. 1988;9:33–44. doi: 10.1016/0143-4160(88)90036-X. [DOI] [PubMed] [Google Scholar]
- Blair MA, Ma S, Hedera P. Mutation in KIF5A can also cause adult-onset hereditary spastic paraplegia. Neurogenetics. 2006;7:47–50. doi: 10.1007/s10048-005-0027-8. [DOI] [PubMed] [Google Scholar]
- Brickley K, Stephenson FA. Trafficking kinesin protein (TRAK)-mediated transport of mitochondria in axons of hippocampal neurons. J Biol Chem. 2011;286:18079–18092. doi: 10.1074/jbc.M111.236018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brickley K, Smith MJ, Beck M, Stephenson FA. GRIF-1 and OIP106, members of a novel gene family of coiled-coil domain proteins: association in vivo and in vitro with kinesin. J Biol Chem. 2005;280:14723–14732. doi: 10.1074/jbc.M409095200. [DOI] [PubMed] [Google Scholar]
- Buss RR, Drapeau P. Synaptic drive to motoneurons during fictive swimming in the developing zebrafish. J Neurophysiol. 2001;86:197–210. doi: 10.1152/jn.2001.86.1.197. [DOI] [PubMed] [Google Scholar]
- Campbell PD, Marlow FL. Temporal and tissue specific gene expression patterns of the zebrafish kinesin-1 heavy chain family, kif5s, during development. Gene Expr Patterns. 2013;13:271–279. doi: 10.1016/j.gep.2013.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Sheng ZH. Kinesin-1-syntaphilin coupling mediates activity-dependent regulation of axonal mitochondrial transport. J Cell Biol. 2013;202:351–364. doi: 10.1083/jcb.201302040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman M. Axon degeneration mechanisms: commonality amid diversity. Nat Rev Neurosci. 2005;6:889–898. doi: 10.1038/nrn1788. [DOI] [PubMed] [Google Scholar]
- Coy DL, Hancock WO, Wagenbach M, Howard J. Kinesin's tail domain is an inhibitory regulator of the motor domain. Nat Cell Biol. 1999;1:288–292. doi: 10.1038/13001. [DOI] [PubMed] [Google Scholar]
- Crimella C, Baschirotto C, Arnoldi A, Tonelli A, Tenderini E, Airoldi G, Martinuzzi A, Trabacca A, Losito L, Scarlato M, Benedetti S, Scarpini E, Spinicci G, Bresolin N, Bassi MT. Mutations in the motor and stalk domains of KIF5A in spastic paraplegia type 10 and in axonal Charcot-Marie-Tooth type 2. Clin Genet. 2012;82:157–164. doi: 10.1111/j.1399-0004.2011.01717.x. [DOI] [PubMed] [Google Scholar]
- Deluca GC, Ebers GC, Esiri MM. The extent of axonal loss in the long tracts in hereditary spastic paraplegia. Neuropathol Appl Neurobiol. 2004;30:576–584. doi: 10.1111/j.1365-2990.2004.00587.x. [DOI] [PubMed] [Google Scholar]
- Drerup CM, Nechiporuk AV. JNK-interacting protein 3 mediates the retrograde transport of activated c-Jun N-terminal kinase and lysosomes. PLoS Genet. 2013;9:e1003303. doi: 10.1371/journal.pgen.1003303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fichera M, Lo Giudice M, Falco M, Sturnio M, Amata S, Calabrese O, Bigoni S, Calzolari E, Neri M. Evidence of kinesin heavy chain (KIF5A) involvement in pure hereditary spastic paraplegia. Neurology. 2004;63:1108–1110. doi: 10.1212/01.WNL.0000138731.60693.D2. [DOI] [PubMed] [Google Scholar]
- Finsterer J, Löscher W, Quasthoff S, Wanschitz J, Auer-Grumbach M, Stevanin G. Hereditary spastic paraplegias with autosomal dominant, recessive, X-linked, or maternal trait of inheritance. J Neurol Sci. 2012;318:1–18. doi: 10.1016/j.jns.2012.03.025. [DOI] [PubMed] [Google Scholar]
- Friedman DS, Vale RD. Single-molecule analysis of kinesin motility reveals regulation by the cargo-binding tail domain. Nat Cell Biol. 1999;1:293–297. doi: 10.1038/13008. [DOI] [PubMed] [Google Scholar]
- Goizet C, Boukhris A, Mundwiller E, Tallaksen C, Forlani S, Toutain A, Carriere N, Paquis V, Depienne C, Durr A, Stevanin G, Brice A. Complicated forms of autosomal dominant hereditary spastic paraplegia are frequent in SPG10. Hum Mutat. 2009;30:E376–E385. doi: 10.1002/humu.20920. [DOI] [PubMed] [Google Scholar]
- Hackney DD, Stock MF. Kinesin's IAK tail domain inhibits initial microtubule-stimulated ADP release. Nat Cell Biol. 2000;2:257–260. doi: 10.1038/35010525. [DOI] [PubMed] [Google Scholar]
- Hackney DD, Baek N, Snyder AC. Half-site inhibition of dimeric kinesin head domains by monomeric tail domains. Biochemistry. 2009;48:3448–3456. doi: 10.1021/bi8022575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartley JL, Temple GF, Brasch MA. DNA cloning using in vitro site-specific recombination. Genome Res. 2000;10:1788–1795. doi: 10.1101/gr.143000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedera P, Eldevik OP, Maly P, Rainier S, Fink JK. Spinal cord magnetic resonance imaging in autosomal dominant hereditary spastic paraplegia. Neuroradiology. 2005;47:730–734. doi: 10.1007/s00234-005-1415-3. [DOI] [PubMed] [Google Scholar]
- Hinckelmann MV, Zala D, Saudou F. Releasing the brake: restoring fast axonal transport in neurodegenerative disorders. Trends Cell Biol. 2013;23:634–643. doi: 10.1016/j.tcb.2013.08.007. [DOI] [PubMed] [Google Scholar]
- Hirokawa N, Noda Y, Tanaka Y, Niwa S. Kinesin superfamily motor proteins and intracellular transport. Nat Rev Mol Cell Biol. 2009;10:682–696. doi: 10.1038/nrm2774. [DOI] [PubMed] [Google Scholar]
- Hirokawa N, Niwa S, Tanaka Y. Molecular motors in neurons: transport mechanisms and roles in brain function, development, and disease. Neuron. 2010;68:610–638. doi: 10.1016/j.neuron.2010.09.039. [DOI] [PubMed] [Google Scholar]
- Hoerndli FJ, Maxfield DA, Brockie PJ, Mellem JE, Jensen E, Wang R, Madsen DM, Maricq AV. Kinesin-1 regulates synaptic strength by mediating the delivery, removal, and redistribution of AMPA receptors. Neuron. 2013;80:1421–1437. doi: 10.1016/j.neuron.2013.10.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson GV, Litersky JM, Jope RS. Degradation of microtubule-associated protein 2 and brain spectrin by calpain: a comparative study. J Neurochem. 1991;56:1630–1638. doi: 10.1111/j.1471-4159.1991.tb02061.x. [DOI] [PubMed] [Google Scholar]
- Kanai Y, Okada Y, Tanaka Y, Harada A, Terada S, Hirokawa N. KIF5C, a novel neuronal kinesin enriched in motor neurons. J Neurosci. 2000;20:6374–6384. doi: 10.1523/JNEUROSCI.20-17-06374.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karle KN, Möckel D, Reid E, Schöls L. Axonal transport deficit in a KIF5A−/− mouse model. Neurogenetics. 2012;13:169–179. doi: 10.1007/s10048-012-0324-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi K. Role of kinesin-1 in the pathogenesis of SPG10, a rare form of hereditary spastic paraplegia. Neuroscientist. 2013;19:336–344. doi: 10.1177/1073858412451655. [DOI] [PubMed] [Google Scholar]
- Kettleborough RN, Busch-Nentwich EM, Harvey SA, Dooley CM, de Bruijn E, van Eeden F, Sealy I, White RJ, Herd C, Nijman IJ, Fényes F, Mehroke S, Scahill C, Gibbons R, Wali N, Carruthers S, Hall A, Yen J, Cuppen E, Stemple DL. A systematic genome-wide analysis of zebrafish protein-coding gene function. Nature. 2013;496:494–497. doi: 10.1038/nature11992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan KM, Fujimoto E, Grabher C, Mangum BD, Hardy ME, Campbell DS, Parant JM, Yost HJ, Kanki JP, Chien CB. The Tol2kit: a multisite gateway-based construction kit for Tol2 transposon transgenesis constructs. Dev Dyn. 2007;236:3088–3099. doi: 10.1002/dvdy.21343. [DOI] [PubMed] [Google Scholar]
- Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- Lyons DA, Naylor SG, Mercurio S, Dominguez C, Talbot WS. KBP is essential for axonal structure, outgrowth and maintenance in zebrafish, providing insight into the cellular basis of Goldberg-Shprintzen syndrome. Development. 2008;135:599–608. doi: 10.1242/dev.012377. [DOI] [PubMed] [Google Scholar]
- Lyons DA, Naylor SG, Scholze A, Talbot WS. Kif1b is essential for mRNA localization in oligodendrocytes and development of myelinated axons. Nat Genet. 2009;41:854–858. doi: 10.1038/ng.376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McWilliam H, Li W, Uludag M, Squizzato S, Park YM, Buso N, Cowley AP, Lopez R. Analysis tool web services from the EMBL-EBI. Nucleic Acids Res. 2013;41:W597–W600. doi: 10.1093/nar/gkt376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer MP, Smith SJ. Evidence from in vivo imaging that synaptogenesis guides the growth and branching of axonal arbors by two distinct mechanisms. J Neurosci. 2006;26:3604–3614. doi: 10.1523/JNEUROSCI.0223-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miki H, Setou M, Kaneshiro K, Hirokawa N. All kinesin superfamily protein, KIF, genes in mouse and human. Proc Natl Acad Sci U S A. 2001;98:7004–7011. doi: 10.1073/pnas.111145398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moua P, Fullerton D, Serbus LR, Warrior R, Saxton WM. Kinesin-1 tail autoregulation and microtubule-binding regions function in saltatory transport but not ooplasmic streaming. Development. 2011;138:1087–1092. doi: 10.1242/dev.048645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima K, Yin X, Takei Y, Seog DH, Homma N, Hirokawa N. Molecular motor KIF5A is essential for GABA(A) receptor transport, and KIF5A deletion causes epilepsy. Neuron. 2012;76:945–961. doi: 10.1016/j.neuron.2012.10.012. [DOI] [PubMed] [Google Scholar]
- Nangaku M, Sato-Yoshitake R, Okada Y, Noda Y, Takemura R, Yamazaki H, Hirokawa N. KIF1B, a novel microtubule plus end-directed monomeric motor protein for transport of mitochondria. Cell. 1994;79:1209–1220. doi: 10.1016/0092-8674(94)90012-4. [DOI] [PubMed] [Google Scholar]
- Neff MM, Neff JD, Chory J, Pepper AE. dCAPS, a simple technique for the genetic analysis of single nucleotide polymorphisms: experimental applications in Arabidopsis thaliana genetics. Plant J. 1998;14:387–392. doi: 10.1046/j.1365-313X.1998.00124.x. [DOI] [PubMed] [Google Scholar]
- Nojima H, Rothhämel S, Shimizu T, Kim CH, Yonemura S, Marlow FL, Hibi M. Syntabulin, a motor protein linker, controls dorsal determination. Development. 2010;137:923–933. doi: 10.1242/dev.046425. [DOI] [PubMed] [Google Scholar]
- Palanca AM, Lee SL, Yee LE, Joe-Wong C, Trinh le A, Hiroyasu E, Husain M, Fraser SE, Pellegrini M, Sagasti A. New transgenic reporters identify somatosensory neuron subtypes in larval zebrafish. Dev Neurobiol. 2013;73:152–167. doi: 10.1002/dneu.22049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paquet D, Bhat R, Sydow A, Mandelkow EM, Berg S, Hellberg S, Fälting J, Distel M, Köster RW, Schmid B, Haass C. A zebrafish model of tauopathy allows in vivo imaging of neuronal cell death and drug evaluation. J Clin Invest. 2009;119:1382–1395. doi: 10.1172/JCI37537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel N, Thierry-Mieg D, Mancillas JR. Cloning by insertional mutagenesis of a cDNA encoding Caenorhabditis elegans kinesin heavy chain. Proc Natl Acad Sci U S A. 1993;90:9181–9185. doi: 10.1073/pnas.90.19.9181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plucińska G, Paquet D, Hruscha A, Godinho L, Haass C, Schmid B, Misgeld T. In vivo imaging of disease-related mitochondrial dynamics in a vertebrate model system. J Neurosci. 2012;32:16203–16212. doi: 10.1523/JNEUROSCI.1327-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poirier K, Lebrun N, Broix L, Tian G, Saillour Y, Boscheron C, Parrini E, Valence S, Pierre BS, Oger M, Lacombe D, Geneviève D, Fontana E, Darra F, Cances C, Barth M, Bonneau D, Bernadina BD, N′guyen S, Gitiaux C, et al. Mutations in TUBG1, DYNC1H1, KIF5C and KIF2A cause malformations of cortical development and microcephaly. Nat Genet. 2013;45:639–647. doi: 10.1038/ng.2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randall TS, Moores C, Stephenson FA. Delineation of the TRAK binding regions of the kinesin-1 motor proteins. FEBS Lett. 2013;587:3763–3769. doi: 10.1016/j.febslet.2013.09.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid E, Kloos M, Ashley-Koch A, Hughes L, Bevan S, Svenson IK, Graham FL, Gaskell PC, Dearlove A, Pericak-Vance MA, Rubinsztein DC, Marchuk DA. A kinesin heavy chain (KIF5A) mutation in hereditary spastic paraplegia (SPG10) Am J Hum Genet. 2002;71:1189–1194. doi: 10.1086/344210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saotome M, Safiulina D, Szabadkai G, Das S, Fransson A, Aspenstrom P, Rizzuto R, Hajnóczky G. Bidirectional Ca2+-dependent control of mitochondrial dynamics by the miro GTPase. Proc Natl Acad Sci U S A. 2008;105:20728–20733. doi: 10.1073/pnas.0808953105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T, Takahoko M, Okamoto H. HuC:Kaede, a useful tool to label neural morphologies in networks in vivo. Genesis. 2006;44:136–142. doi: 10.1002/gene.20196. [DOI] [PubMed] [Google Scholar]
- Saxton WM, Hicks J, Goldstein LS, Raff EC. Kinesin heavy chain is essential for viability and neuromuscular functions in Drosophila, but mutants show no defects in mitosis. Cell. 1991;64:1093–1102. doi: 10.1016/0092-8674(91)90264-Y. [DOI] [PubMed] [Google Scholar]
- Schüle R, Kremer BP, Kassubek J, Auer-Grumbach M, Kostic V, Klopstock T, Klimpe S, Otto S, Boesch S, van de Warrenburg BP, Schöls L. SPG10 is a rare cause of spastic paraplegia in European families. J Neurol Neurosurg Psychiatry. 2008;79:584–587. doi: 10.1136/jnnp.2007.137596. [DOI] [PubMed] [Google Scholar]
- Schwarz TL. Mitochondrial trafficking in neurons. Cold Spring Harb Perspect Biol. 2013;5:a011304. doi: 10.1101/cshperspect.a011304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng ZH, Cai Q. Mitochondrial transport in neurons: impact on synaptic homeostasis and neurodegeneration. Nat Rev Neurosci. 2012;13:77–93. doi: 10.1038/nrg3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MJ, Pozo K, Brickley K, Stephenson FA. Mapping the GRIF-1 binding domain of the kinesin, KIF5C, substantiates a role for GRIF-1 as an adaptor protein in the anterograde trafficking of cargoes. J Biol Chem. 2006;281:27216–27228. doi: 10.1074/jbc.M600522200. [DOI] [PubMed] [Google Scholar]
- Tanaka Y, Kanai Y, Okada Y, Nonaka S, Takeda S, Harada A, Hirokawa N. Targeted disruption of mouse conventional kinesin heavy chain, kif5B, results in abnormal perinuclear clustering of mitochondria. Cell. 1998;93:1147–1158. doi: 10.1016/S0092-8674(00)81459-2. [DOI] [PubMed] [Google Scholar]
- Vallat JM, Mathis S, Funalot B. The various Charcot-Marie-tooth diseases. Curr Opin Neurol. 2013;26:473–480. doi: 10.1097/WCO.0b013e328364c04b. [DOI] [PubMed] [Google Scholar]
- van Spronsen M, Mikhaylova M, Lipka J, Schlager MA, van den Heuvel DJ, Kuijpers M, Wulf PS, Keijzer N, Demmers J, Kapitein LC, Jaarsma D, Gerritsen HC, Akhmanova A, Hoogenraad CC. TRAK/Milton motor-adaptor proteins steer mitochondrial trafficking to axons and dendrites. Neuron. 2013;77:485–502. doi: 10.1016/j.neuron.2012.11.027. [DOI] [PubMed] [Google Scholar]
- Villefranc JA, Amigo J, Lawson ND. Gateway compatible vectors for analysis of gene function in the zebrafish. Dev Dyn. 2007;236:3077–3087. doi: 10.1002/dvdy.21354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walhout AJ, Temple GF, Brasch MA, Hartley JL, Lorson MA, van den Heuvel S, Vidal M. GATEWAY recombinational cloning: application to the cloning of large numbers of open reading frames or ORFeomes. Methods Enzymol. 2000;328:575–592. doi: 10.1016/S0076-6879(00)28419-X. [DOI] [PubMed] [Google Scholar]
- Wang JT, Medress ZA, Barres BA. Axon degeneration: molecular mechanisms of a self-destruction pathway. J Cell Biol. 2012;196:7–18. doi: 10.1083/jcb.201108111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse AM, Procter JB, Martin DM, Clamp M, Barton GJ. Jalview version 2: a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009;25:1189–1191. doi: 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerfield M. The zebrafish book: a guide for the laboratory use of Zebrafish (Danio rerio) Ed 3. Eugene, OR: University of Oregon; 1995. [Google Scholar]
- Wozniak MJ, Melzer M, Dorner C, Haring HU, Lammers R. The novel protein KBP regulates mitochondria localization by interaction with a kinesin-like protein. BMC Cell Biol. 2005;6:35. doi: 10.1186/1471-2121-6-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia CH, Roberts EA, Her LS, Liu X, Williams DS, Cleveland DW, Goldstein LS. Abnormal neurofilament transport caused by targeted disruption of neuronal kinesin heavy chain KIF5A. J Cell Biol. 2003;161:55–66. doi: 10.1083/jcb.200301026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Takita J, Tanaka Y, Setou M, Nakagawa T, Takeda S, Yang HW, Terada S, Nakata T, Takei Y, Saito M, Tsuji S, Hayashi Y, Hirokawa N. Charcot-Marie-Tooth disease type 2A caused by mutation in a microtubule motor KIF1Bbeta. Cell. 2001;105:587–597. doi: 10.1016/S0092-8674(01)00363-4. [DOI] [PubMed] [Google Scholar]