

Introduction
The pharmaceutical industry has entered challenging economic times, and some analysts have questioned the long-term sustainability of its current business model [1]. The average cost of taking a new drug from the chemist’s bench to the pharmacist’s shelf now exceeds $4 billion by some estimates, with almost two-thirds of the costs being related to clinical trials [2]. The economic burden presented by rising development costs will likely be compounded by future reduced market revenues. Improved understanding of disease pathogenesis should allow physicians, perhaps with the aid of genetic testing, to identify potential responder subgroups among larger patient populations (i.e., the rise of “designer drugs”). However, treatment with such drugs will be indicated in smaller percentages of patients with the indicated medical condition so that high volume, blockbuster sales would likely decrease. Society may no longer tolerate rising drug prices, which would be needed to maintain revenues despite reductions in the number of pills sold. Hence, the pharmaceutical business model would strongly benefit from reduced drug development costs, particularly those associated with the conduct of large and prolonged clinical trials. Beyond the operational costs these trials entail, each additional year taken to attain marketing approval represents one year less patent protection during the commercial phase.
From an efficacy standpoint, there are viable strategies that could reduce the size and hence the costs of clinical trials. For example, increased understanding of disease pathogenesis in the context of systems biology should result in the rational design of increasingly effective drugs. More effective new drug candidates, perhaps combined with biomarkers (e.g., genetic testing) to identify the subset of patients most likely to benefit, could demonstrate efficacy in smaller, more cost-effective clinical trials. Novel approaches, such as adaptive trial design [3], may further reduce the clinical trial sample sizes and related costs. However, these efforts will be unrewarded if concern about rare toxicities still demand large and expensive clinical trials to rule out low levels of risk of severe adverse drug events. Indeed, the size of clinical trials for many treatment indications has been increasing in large part due to safety concerns. Troglitazone was approved in 1997 for the treatment of Type 2 diabetes after only 1134 patients were treated with the drug for at least 6 months [4]. Today, approval of a drug for this indication would require many more subjects with longer treatment durations, as well as a large Phase 4 cardiac outcomes study extending well beyond marketing approval. The increasing regulatory demand to detect and quantify the risks of rare or idiosyncratic adverse events in clinical trials is an increasing challenge in drug development today.
Figure 1 shows the major safety reasons for drug withdrawals from the marketplace over three recent decades. Cardiovascular toxicity, the majority accounted for by drug-induced malignant arrhythmias, has represented the major category of adverse events leading to drug withdrawal. There is now a regulatory path to identify the risk of cardiac arrhythmia potential of new drug candidates, including a general requirement for a prolonged QT clinical study. It is therefore expected that there will be a reduction in drug withdrawals due to this problem.
Fig. 1.
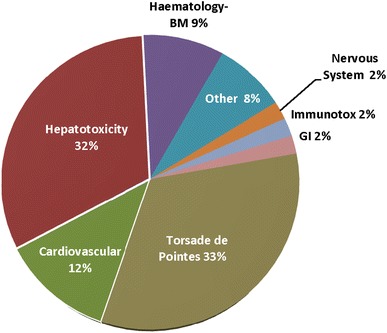
Adverse drug events that have led to withdrawal from the marketplace worldwide between 1975–2007 [22]. GI gastrointestinal, immunotox immunotoxicity, BM bone marrow
There is little reason to have similar optimism regarding rare or idiosyncratic hepatotoxicity. Current efforts to improve liver safety include a push to humanize preclinical screening by using cultured human hepatocytes or humanized mice [5]. The absence of signals in these systems is somewhat reassuring [6] but simple cell culture and current humanized animal models do not mimic human variability in susceptibility to idiosyncratic hepatotoxicity and are therefore of limited value. Until there is a major advance in the understanding of the mechanisms underlying idiosyncratic DILI, it is unlikely that data obtained from any combination of preclinical or early clinical testing will remove regulatory concern about the potential for serious liver toxicity. Accordingly, the Center for Drug Evaluation and Research (CDER) has made great strides towards the performance of comprehensive reviews of clinical trial liver safety databases, when called upon, by expert hepatologists at FDA, most notably Dr. John Senior.
The detection and quantification of liver safety risk from even a very large clinical trial data base is challenging, in large part because the serum biomarkers used to assess liver safety, which have not changed in over four decades, are not ideal [7]. The most sensitive biomarker for hepatocellular injury is serum alanine aminotransferase, but some drugs cause elevations in serum alanine aminotransferase (e.g., heparins, aspirin, and statins) yet have low or absent liver safety risks [8].
A rational approach to the assessment of liver safety in clinical trials was presented in the FDA Guidance Document entitled: “Guidance for Industry Drug-Induced Liver Injury: Premarketing Clinical Evaluation” which was released in 2009 [9]. Most aspects of liver safety assessment were addressed in the document, including frequency of liver chemistry monitoring, normalization of laboratory values to upper limits of the reference range (ULN), and specific heights of liver chemistries for stratification analyses. Among many recommendations, was to consider continued but cautious treatment of subjects manifesting elevations in serum ALT to determine whether they would develop signs of liver dysfunction, particularly elevation in serum total bilirubin >2 X ULN. Subjects with elevations in serum ALT exceeding 3 X ULN and an elevation in serum bilirubin exceeding 2 X ULN (either concomitantly or within one month of the qualifying ALT elevation) were termed “Hy’s Law Cases” if the injury was hepatocellular (no “substantial elevation” in serum alkaline phosphatase) and no other cause but study drug could be identified. A Hy’s Law Case was defined as the gold standard signal for potential of a drug to cause progressive, serious liver injury. However, in a Hy’s Law Case, serum ALT and bilirubin may not be biomarkers of potential for severe liver injury, but rather indicators of severe liver injury. At least one subject in a clinical trial was discontinued due to a drug-induced rise in serum ALT value above 8 X ULN while the serum bilirubin was normal, but went on to develop fatal liver failure [10]. It would therefore be highly desirable to identify biomarker characteristics that can accurately define liver safety of new drug candidates without putting any research subjects at risk.
Goals of the Workshop
The 2009 FDA guidance is a landmark document that represented a major advance in creating a rational and standardized approach to the assessment of liver safety in clinical trials. Nonetheless, there were some potentially important areas not covered in depth in the document including the following four topics:
Essential Data Elements Required to Assess Liver Safety and the Standardization of Data Collection
There are several efforts underway to establish standards for terminology and data collection for regulatory submissions. These include the Clinical Data Interchange Standards Consortium (CDISC), a non-profit consortium developing data standards for clinical study protocols, and the specification and reporting of test results. In addition[11], the Food and Drug Administration Safety and Innovation Act (FDASIA) passed by the Congress in 2012 stipulates that the FDA must establish standardized clinical data terminology for electronic submissions and standardization of drug application data[12]. It is therefore an opportune time to have expert input on what data elements relevant to liver safety should be collected in clinical trials, and what terminology should become universally adopted.
It is clear that the optimal data elements to assess liver safety do not yet exist but are likely to evolve from the study of biospecimens archived from clinical trials. Retrospective analysis of archived DNA from clinical trials has already identified risk alleles for DILI that could in the future be useful as one component of a larger set of key measurements in the diagnosis of DILI but also support risk management through personalized medicine approaches. It is also universally recognized that the serum tests for liver safety currently employed in clinical trials are suboptimal and hence data obtained with these biomarkers is inherently flawed. There are global efforts underway to develop new biomarkers that will hopefully provide more suitable data elements to assess liver safety. However, full appreciation of the potential value and limitations of these biomarkers will require their application to many thousands of specimens obtained in clinical trials of drugs that are both safe and not safe for the liver, as well as in diverse patient populations with varying susceptibility to DILI. Such an effort may only be feasible if the pharmaceutical industry adopts standards for serum collection and archiving.
Liver Safety Data Management
If a liver safety signal is suspected in a clinical trial, companies are now generally required to submit data to CDER in a format that can be readily analyzed by a software program created at CDER termed “evaluation of Drug Induced Serious Hepatotoxicity” or eDISH [13]. This software was developed after publication of the 2009 guidance and is therefore not mentioned in the document. eDISH facilitates analysis of liver safety data in several ways, including graphically displaying the peak serum ALT and bilirubin values obtained in each subject in a clinical trial, and linking the individual subject points with relevant data for that subject, including a visual display to facilitate the interpretation of changes in liver chemistries. The full potential for data visualization methods in liver safety assessment has not been achieved and it only makes sense to have standard or at least compatible approaches within the industry and regulatory bodies. It would also be ideal if the relevant liver safety data for each subject was directly linked to archived biospecimens (e.g., DNA, serum) obtained from that subject. Standardization of these processes across the industry would greatly facilitate future precompetitive efforts to define genetic and non-genetic biomarkers that could revolutionize assessment of liver safety and management of the risk it poses.
Causality Assessment
Assessing causality, especially of serious liver injuries, is obviously a critical determinant in assessing liver safety risk in clinical trials. Although it provides a list of common alternative causes of acute liver injury, the 2009 guidance document [9] does not deal with the causality assessment process. The guidance also does not recommend a causality assessment scale reflecting varying degrees of certainty in causal link.
Liver Safety Assessments in Special Populations
The 2009 guidance does not give specific guidance in the interpretation of potential liver safety signals in patients with pre-existing liver diseases. This has become a particular challenge in treatment trials of patients with chronic viral hepatitis, which have seen a great expansion in recent years [14]. A second area is oncology where liver chemistry abnormalities may reflect involvement of the liver in the malignancy or concomitant hepatotoxic treatments. As more mechanism based and target focused drugs have entered the clinic, and chronic rather than intermittent dosing becomes more common, safety has become an increasing concern [15].
International harmonization of the approach to these four topics would have many advantages, including reduced discrepancies in the interpretation of liver safety data and facilitation of precompetitive sharing of liver safety data and relevant biospecimens.
To address these four areas, an all day workshop was convened in Boston on November 9, 2012. The workshop was jointly sponsored by the Hamner-University of North Carolina Institute for Drug Safety Sciences and the European Innovative Medicines Initiative (IMI). In attendance at the workshop were representatives of regulatory agencies from the U.S. FDA, Health Canada and Japan. In addition, a hepatologist from China frequently involved in liver safety assessments for the CFDA attended. Leading liver safety experts from academia and industry were also in attendance (the attendee list is available as an electronic supplementary material (See ESM 1)). The assigned leaders for the topics above and their colleagues prepared a comprehensive list of focused questions to be addressed in the break out groups as well as possible responses to the questions. These drafts were circulated to confirmed attendees and additional input was gathered prior to the workshop.
Consensus opinions achieved at the workshop were incorporated into summaries which were recirculated among participants of the break out groups and their colleagues for additional feedback. Key issues were also discussed during an evening session at the annual FDA/AASLD/Pharma Hepatotoxicity Conference in April 2013.
Accomplishments
As documented in the following manuscripts, consensus on some issues was achieved by the working group members who discussed essential data elements and standards, data management tools, and causality assessment. Consensus was also reached on related areas, such as the need for archiving of DNA and serum samples in clinical trials. Consensus could not be reached on the approach to assessing liver safety in the special populations, particularly as regards the appropriate reference values for elevations in liver chemistries. Areas that were not resolved included how best to express liver chemistries (e.g., fold upper limits of normal vs. fold baseline), how best to define the baseline reference value, and action levels for liver chemistry abnormalities. It was the consensus that recommendations in this regard should be data based and that the requisite data was not generally available.
The Future
It was clear to all participants that the data necessary to address some important issues were simply not available at the present time. This was most evident regarding special populations, but was also evident in other areas. It was agreed that optimal progress going forward will require access to large amounts of liver safety data across many clinical trials in many different patient populations. It should be noted that there have been increased efforts for public disclosure of clinical trial data [16]. Some potential sources of the relevant data are discussed below:
Increased Efforts for Public Disclosure of Clinical Trial Data
eDISH
CDER has now accumulated extensive liver safety data in the eDISH format from over 100 clinical trials involving over 150,000 patients (Dr. John Senior, personal communication). Such data would represent an unprecedented resource to address important questions like when should treatment modification criteria be changed based on characteristics of the patient population. For example, is the range of ALT values obtained from patients with congestive heart failure similar to those with Parkinson’s disease? These data could also be used to address the relative value of expressing liver chemistries as fold upper limits of normal vs fold baseline. For example, if there exists significant interpatient variation in the hepatocyte content of ALT, the absolute magnitude of random fluctuations in serum ALT during placebo treatment should positively correlate with the subject’s mean value. Such a finding would provide a basis for support of fold baseline as the appropriate unit for ALT expression in clinical trials.
Unfortunately, attendees were told that direct access to the data contained in eDISH format at CDER was probably not possible, even with approval of the individual companies from which the data was obtained. This apparently reflects legal concerns regarding such issues as required assurances of data integrity. With the significant constraints that regulatory authorities have in publically releasing all systematically collected clinical trial data relevant to DILI research, an alternate route is the development of a pre-competitive consortium, which would obtain the data directly from sponsors. This should be technically feasible since sponsors would likely have coalesced, analyzed and submitted the data in eDISH compatible formats as a component of regulatory submissions.
The Drug Induced Liver Injury Network (DILIN)
The National Institutes of Health has supported DILIN since 2003 [17] and will continue to support this network until at least 2018. DILIN has created a registry of subjects who have experienced idiosyncratic DILI to over 200 different marketed drugs, and has collected genomic DNA, serum and urine in addition to extensive phenotypic data from these individuals. Although a few of the cases in DILIN have been from clinical trials of new drug candidates, the majority of cases are due to marketed drugs, including recently approved medications. The continued existence of DILIN underscores the potential importance of standardized liver safety data collection and management in clinical trials, and linkage of the data to archived biospecimens. This is illustrated by the recent experience with lumiracoxib. This drug was withdrawn from worldwide markets in 2007, shortly after its market entry due to several cases of acute liver failure, including liver transplantation. Because the company producing the drug (Novartis) had archived DNA from Phase 3 clinical trials they were able to perform a genome wide association study (GWAS) on just 41 treated subjects who experienced an ALT elevation exceeding 5 X ULN and 176 treated subjects who maintained serum ALT elevations <1.5 X ULN throughout treatment [18]. This analysis identified a risk allele that was also present in the only 3 post-marketing cases of severe liver injury (with jaundice) that agreed to genetic analysis. Based on these data showing a high negative predictive value of serious DILI outcomes in individuals lacking the risk allele, reintroduction of lumiracoxib with a companion genetic test was proposed [19]. There are other recent similar examples of using archived DNA from clinical trials to identify DILI risk factors after severe DILI cases are observed in late clinical development or post marketing [20, 21]. It seems highly likely that in the future DILIN will enroll cases of significant liver injury attributed to newly approved drugs and retrospective analysis of systematically archived DNA from the clinical trials of that drug will provide personalized medicine approaches to risk management. This process will be greatly facilitated by a standardization of data elements, data management tools, and links between the data and archived biospecimens.
The SAFE-T Consortium
This consortium is sponsored in Europe by the Innovative Medicines Initiative and the European Federation of Pharmaceutical Industries and Associations (EFPIA). The goal is to develop and validate improved biomarkers to assess liver, kidney and vascular safety in clinical trials (http://www.imi-safe-t.eu). Regarding liver safety, high throughput assays developed or in development include the liver specific microRNA miR122 and mechanistic biomarkers that detect apoptosis, necrosis and activation of immune responses. These biomarkers could be revolutionary in the assessment of liver safety in clinical trials, potentially enabling accurate assessments from small study populations with more limited durations of study drug exposure. However, establishing the full value of these biomarkers will require their application to literally thousands of serum samples obtained in large clinical trials involving diverse patient populations. Such an effort can only be accomplished on a precompetitive basis and may only be feasible if universal standards for data and biospecimen acquisition and management are adopted at least by the major pharmaceutical companies.
A Liver Safety Research Consortium
As noted above, there was enthusiasm among the workshop participants to continue to work together toward international harmonization of liver safety assessment approaches. There was also consensus that this could only be accomplished by analysis of large amounts of liver safety data already collected across the industry in clinical trials. A major hurdle towards precompetitive efforts to access and analyze these data has been the lack of uniform standards for acquisition of the relevant data elements, data management tools, and the lack of standardized protocols to link phenotypic data with archived biospecimens. Access to existing data will therefore require an organized, sustained effort.
The proposal to create a Liver Safety Research Consortium analogous to the highly successful Cardiac Research Safety Consortium (CSRC) (http://www.cardiac-safety.org) was endorsed by the attendees at the workshop. The CSRC, like the workshop, includes representatives from industry, academia and regulatory agencies. The CSRC is administratively supported by modest contributions from industry using a sliding scale based on gross sales. The CSRC has exerted substantial influence on regulatory policy and standards across the industry through an extensive series of white papers. In addition to coordinating analysis of existing data, the CSRC has initiated de novo studies to fill critical gaps in knowledge. There was broad interest among many attendees at the workshop to promote the development of a Liver Safety Research Consortium which would in a pre-competitive manner gather and analyze clinical trial data relevant to DILI risk.
Summary
The workshop was successful in bringing international experts from industry, academia, and regulatory agencies together to address the need for industry-wide standardization of liver safety data collection, causality assessment, and liver safety data management. It should be noted that the consensus statements contained in the manuscripts are simply that. They are not regulatory policy, but hopefully will stimulate progress toward the lengthy process of updating and revising the 2009 guidance document.
There was near unanimous agreement that future guidelines and policies should be based as much as possible on data that should be available through precompetitive collaboration. Importantly, the workshop established a core international group of concerned experts from academia, industry and regulatory bodies to improve and standardize the approaches to assessing liver safety in clinical trials. An important goal to consider in this effort would be the establishment of a Liver Safety Research Consortium.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
The Innovative Medicines Initiative and the Hamner-University of North Carolina Institute for Drug Safety Sciences sponsored the workshop, part of which is summarized in this article. This article is part of a supplement entitled Liver Safety Assessment in Clinical Drug Development: A Best Practices Workshop report, which was guest edited by Drs. Paul B. Watkins, Michael Merz and Mark I. Avigan. The guest editing by Dr. Avigan does not reflect the position of, nor imply endorsement from, the US Food and Drug Administration or the US Government. Drs. Watkins, Merz and Avigan did not receive any honoraria for guest editing the supplement. All manuscripts were peer reviewed by Dr. Rolf Teschke. Dr. Rolf Teschke has no conflicts of interest to declare and did not receive any honoraria for peer reviewing the supplement; however, he received a free yearly online subscription to the journal Drug Safety.
The Innovative Medicines Initiative (http://www.imi.europa.eu/) is a public-private partnership set up by the European Commission in 2008 to relieve the bottlenecks in drug development and to provide economic stimulus. With a €2 billion commitment, the IMI now has an important portfolio of projects where experts from academia, industry and regulatory bodies collaborate on an unprecedented scale and at a non-competitive level to develop tools and technologies. Drug-induced liver injury has been a focus of several projects including the SAFE-T (Safer and Faster Evidence-based Translation) consortium, which is working on clinical qualification of new biomarkers to better detect and characterize liver toxicity, and MIP-DILI, which is working to determine the optimal preclinical testing to detect potential of liver injury in patients.
The Hamner-University of North Carolina Institute for Drug Safety Sciences (IDSS – http://www.thehamner.org/idss/), located in Research Triangle Park, NC, is dedicated to solving drug safety challenges through a variety of innovative approaches including mouse genetics, mechanistic biomarkers, and culture models derived from induced pluripotent stem cells. Efforts in drug-induced liver injury include the DILI-sim Initiative, a public-private partnership developing computer models to explain and predict drug-induced liver injury.
Conflict of interest
Authors Paul B. Watkins, Michael Merz, Mark I. Avigan, Neil Kaplowitz, Arie Regev and John R. Senior declare no conflicts of interest that are directly relevant to the content of this article.
Footnotes
The views expressed are those of the authors and do not necessarily represent the position of, nor imply endorsement from, the US Food and Drug Administration or the US Government.
References
- 1.Sams-Dodd F. Is poor research the cause of the declining productivity of the pharmaceutical industry? An industry in need of a paradigm shift. Drug Discov Today. 2013;18:211–217. doi: 10.1016/j.drudis.2012.10.010. [DOI] [PubMed] [Google Scholar]
- 2.Herper M. The truly staggering costs of inventing new drugs. In: Forbes. 2012. http://www.forbes.com/forbes/2012/0312/strategies-pharmaceuticals-lilly-stagger-cost-inventing-new-drugs.html. Accessed 1 April 2014.
- 3.Van der Graaf R, Roes KC, Van Delden JJ. Adaptive trials in clinical research: scientific and ethical issues to consider. JAMA. 2012;307:2379–2380. doi: 10.1001/jama.2012.6380. [DOI] [PubMed] [Google Scholar]
- 4.Watkins PB, Whitcomb RW. Hepatic dysfunction associated with troglitazone. N Engl J Med. 1998;338:916–917. doi: 10.1056/NEJM199803263381314. [DOI] [PubMed] [Google Scholar]
- 5.Garcia S, Freitas AA. Humanized mice: current states and perspectives. Immunol Lett. 2012;146:1–7. doi: 10.1016/j.imlet.2012.03.009. [DOI] [PubMed] [Google Scholar]
- 6.Kaplowitz N. Avoiding idiosyncratic DILI: two is better than one. Hepatology. 2013;58:15–17. doi: 10.1002/hep.26295. [DOI] [PubMed] [Google Scholar]
- 7.Senior JR. Alanine aminotransferase: a clinical and regulatory tool for detecting liver injury-past, present, and future. Clin Pharmacol Ther. 2012;92:332–339. doi: 10.1038/clpt.2012.108. [DOI] [PubMed] [Google Scholar]
- 8.Senior JR. Monitoring for hepatotoxicity: what is the predictive value of liver “function” tests? Clin Pharmacol Ther. 2009;85:331–334. doi: 10.1038/clpt.2008.262. [DOI] [PubMed] [Google Scholar]
- 9.FDA. Guidance for industry drug-induced liver injury: premarketing clinical evaluation. In: US FDA guidances (drugs). 2000. http://www.fda.gov/downloads/Drugs/.../Guidances/UCM174090.pdf. Accessed 1 April 2014.
- 10.Watkins PB. Drug safety sciences and the bottleneck in drug development. Clin Pharmacol Ther. 2011;89:788–790. doi: 10.1038/clpt.2011.63. [DOI] [PubMed] [Google Scholar]
- 11.Babre DK. Clinical data interchange standards consortium: a bridge to overcome data standardisation. Perspect Clin Res. 2013;4:115–116. doi: 10.4103/2229-3485.111779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Novack GD. What does the Food and Drug Administration safety and innovation act mean for you? Ocul Surf. 2013;11:206–209. doi: 10.1016/j.jtos.2013.04.002. [DOI] [PubMed] [Google Scholar]
- 13.Watkins PB, Desai M, Berkowitz SD, Peters G, Horsmans Y, Larrey D, et al. Evaluation of drug-induced serious hepatotoxicity (eDISH): application of this data organization approach to phase III clinical trials of rivaroxaban after total hip or knee replacement surgery. Drug Saf. 2011;34:243–252. doi: 10.2165/11586600-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 14.Ilyas JA, Vierling JM. An overview of emerging therapies for the treatment of chronic hepatitis C. Med Clin N Am. 2014;98:17–38. doi: 10.1016/j.mcna.2013.10.011. [DOI] [PubMed] [Google Scholar]
- 15.Sivendran S, Latif A, McBride RB, Stensland KD, Wisnivesky K, Haines L, et al. Adverse event reporting in cancer clinical trial publications. J Clin Oncol. 2014;32:83–89. doi: 10.1200/JCO.2013.52.2219. [DOI] [PubMed] [Google Scholar]
- 16.Mello MM, Francer JK, Wilenzick M, Teden P, Bierer BE, Barnes M. Preparing for responsible sharing of clinical trial data. N Engl J Med. 2013;369:1651–1658. doi: 10.1056/NEJMhle1309073. [DOI] [PubMed] [Google Scholar]
- 17.Hoofnagle JH. Drug-induced liver injury network (DILIN) Hepatology. 2004;40:773. doi: 10.1002/hep.20445. [DOI] [PubMed] [Google Scholar]
- 18.Singer JB, Lewitzky S, Leroy E, Yang F, Zhao X, Klickstein L, et al. A genome-wide study identifies HLA alleles associated with lumiracoxib-related liver injury. Nat Genet. 2010;42:711–714. doi: 10.1038/ng.632. [DOI] [PubMed] [Google Scholar]
- 19.Pharmacogenomics Reporter. Novartis eyeing US market for Prexige launch with companion test in Genomeweb. 2010. http://www.genomeweb.com/dxpgx/novartis-eyeing-us-market-prexige-launch-companion-test Accessed 1 April 2014.
- 20.Spraggs CF, Budde LR, Briley LP, Bing N, Cox CJ, King KS, et al. HLA-DQA1*02:01 is a major risk factor for lapatinib-induced hepatotoxicity in women with advanced breast cancer. J Clin Oncol. 2011;29:667–673. doi: 10.1200/JCO.2010.31.3197. [DOI] [PubMed] [Google Scholar]
- 21.Kindmark A, Jawaid A, Harbron CG, Barratt BJ, Bengtsson OF, Andersson TB, et al. Genome-wide pharmacogenetic investigation of a hepatic adverse event without clinical signs of immunopathology suggests an underlying immune pathogenesis. Pharmacogenomics J. 2008;8:186–195. doi: 10.1038/sj.tpj.6500458. [DOI] [PubMed] [Google Scholar]
- 22.Stevens JL, Baker TK. The future of drug safety testing: expanding the view and narrowing the focus. Drug Discov Today. 2009;14:162–167. doi: 10.1016/j.drudis.2008.11.009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.