

Abstract
Two siblings, an 11-year-old boy and a 7-year-old girl presented with bilateral symmetrical ptosis and limited eye movements. Having already been reviewed on a number of occasions by a variety of specialists in multiple hospital settings a diagnosis of their ocular motility disorder had remained elusive. We describe their cases, outline the differential diagnosis and review the investigations performed which were influential in finally making a diagnosis.
Background
Two siblings, an 11-year-old boy and a 7-year-old girl, presented to the ophthalmology department. Since birth they had had an abnormal facial appearance with bilateral symmetrical ptosis and profoundly limited ocular movements. They had already been reviewed on a number of occasions in multiple hospital settings. A definitive diagnosis of their ocular motility disorder had not however been made.
Case presentation
Both children were developmentally normal. They were otherwise well with no significant medical history. Neither of their parents showed similar features. They had a sibling without similar ocular abnormalities.
At examination both children had visual acuities of 6/6 bilaterally on a Snellen chart. As can be seen from figure 1 both had bilateral symmetrical non-fatiguable ptosis with compensatory frontalis muscle over action. Additionally the girl had an abnormal head posture, persistently elevating her chin. The boy's ptosis was found to worsen with prolonged attempts at lateral gaze. The eye movements of both children were profoundly limited in all directions of gaze. Facial muscle weakness produced typical myopathic facies. Neurological examination revealed weakness of neck flexion, the deltoid and triceps muscles in both children.
Figure 1.
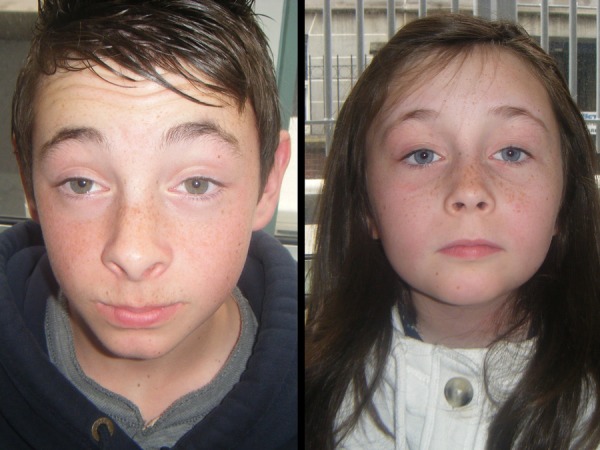
Colour photographs of the two patients described, 11-year-old boy and 7-year-old girl showing their bilateral symmetrical ptosis and the presence of frontalis muscle over-action. Note also how the girl elevates her chin in an attempt to compensate for her blepharoptosis.
Investigations
MRI in each case revealed cystic enlargement of the pineal gland. The pineal cyst at brain MRI of the boy can be easily seen in figure 2. Serum pyruvate and lactate levels were normal in both children. The boy demonstrated a decremental response to repetitive nerve stimulation at suprathreshold electromyography. A dramatic improvement of his ptosis was noted at Tensilon testing.
Figure 2.
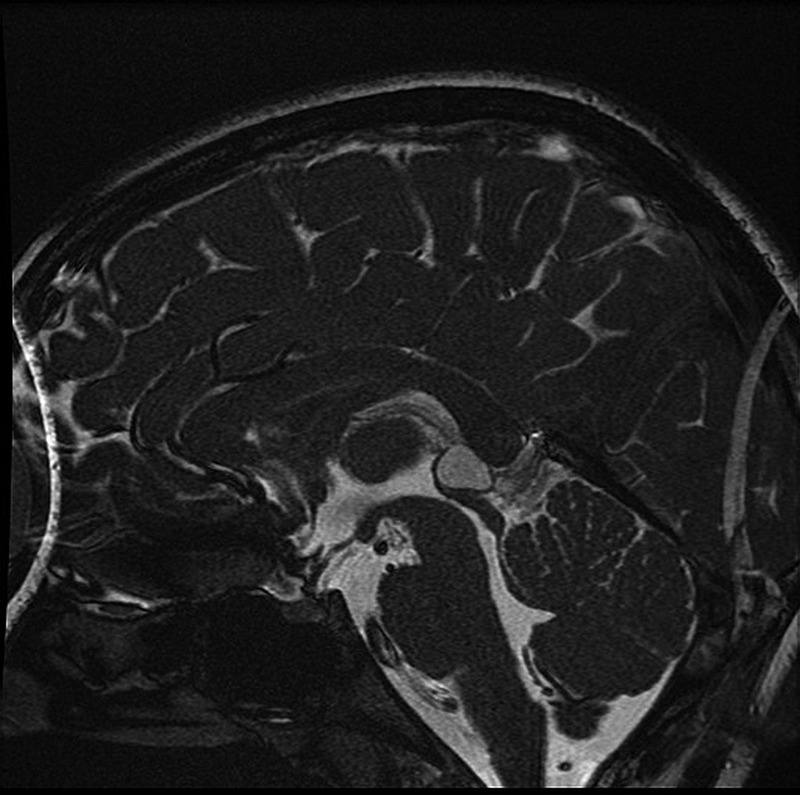
Brain MRI of the boy described here clearly showing a large pineal gland cyst.
Differential diagnosis
The congenital onset of this condition pointed to a genetic or intrauterine aetiology. Common genetic causes of ophthalmoplegia and ptosis include mitochondrial disease and the spectrum of congenital fibrosis of the extraocular muscles (CFEOM).
Chronic progressive external ophthalmoplegia which may be isolated or occur in association with other systemic problems such as with cardiac conduction defects in Kearns-Sayre syndrome causes ophthalmoplegia. The ophthalmolegia of such mitochondrial disorders is however usually progressive. Serum lactate and pyruvate levels are often elevated.
CFEOM invariably presents with ophthlmoparesis but would not be expected to display facial or upper arm weakness.
In Mobiüs syndrome, a congenital aplasia of the lower cranial nerve nuclei, there is weakness of the motor functions of the VI–XII cranial nerves. Bilateral VI nerve palsies reducing horizontal eye movements occur in 50% of those affected. Bilateral VII nerve palsies cause facial muscle weakness and reductions in facial expression. The cases described here however had clear third nerve involvement causing bilateral ptosis.
Pineal gland cysts in children can cause Parinaud's syndrome where defects of up-gaze accompany dilated pupils and light-near dissociation.
Myasthenia gravis is a common cause of ophthalmoplegia but, in the vast majority of presentations it is an acquired condition which usually presents, in late adolescence or adulthood rather than in childhood. Transient neonatal myasthenia gravis can occur as the acetylcholine receptor antibodies that cause autoimmune myasthenia gravis are passed across the placenta from mother to fetus to affect offspring at birth.1
The responses of the boy at electromyography and at tensilon testing are those characteristic of a myasthenic syndrome. Since the clinical presentation was not typical of conventional myasthenia gravis, genetic analysis was performed to test for congenital myasthenia. The finding of genetic mutations in a gene coding for a subunit of the acetylcholine receptor in both the children described here confirmed the diagnosis of congenital myasthenia.
Treatment
Both children were started on the acetylcholinesterase inhibitor pyridostigmine bromide.
Outcome and follow-up
Initially, there was little change in the ocular motility of these children but after more than 18 months of treatment improvements in their eye movements and lid position were apparent. As these rare congenital myasthenic syndromes are present from birth, associated muscular atrophy is inevitable. Improved neuromuscular transmission, as occurs with treatment of these conditions, results, with time, in increased muscle mass, so that a slower response to treatment might be expected in those with congenital myasthenia in comparison to those with conventional myasthenia.
Discussion
The congenital myasthenic syndromes are a group of rare, genetically heterogeneous, inherited disorders of the neuromuscular junction—the connection between a motor axon and skeletal muscle.2 The terminal button of the motor neuron is separated by the synaptic cleft from the motor endplate of the skeletal muscle.3 This ‘postsynaptic’ membrane is lined with acetylcholine receptors to which acetylcholine binds in order to initiate muscle contraction.4 The effect of acetylcholine is terminated by the enzyme acetylcholinesterase which is concentrated within the synaptic cleft.
The majority of the congenital myasthenic syndromes are inherited via autosomal recessive mutations which result in loss of function of neuromuscular transmission.5 6 Autosomal dominant mutations have however also been described.2 These syndromes are divided into those that affect presynaptic, synaptic or postsynaptic function at the neuromuscular junction.7 Presynaptic forms are the rarest affecting an estimated 7–8% of patients while synaptic forms affect approximately 14–15% of patients. Postsynaptic forms are by far the most common and account for 75–80% of these conditions.5
The acetylcholine receptor is a ligand-gated ion channel composed of specific combinations of five subunits (α, ß, γ, ε, δ). The majority of the ‘postsynaptic’ congenital myasthenic syndromes affect one of the acetylcholine receptor subunits.8 In our patients mutations were found in the gene encoding the epsilon subunit of the receptor. The congenital myasthenic syndromes may however also be caused by disorders of the proteins which influence acetylcholine receptor function; rapsyn, plectin, downstream of tyrosine kinase 7 (dok-7) and muscle-specific tyrosine kinase (MuSK).9
The congenital myasthenic syndromes are characterised by fatigable weakness of skeletal muscle for example, limb and extraocular muscles. Cardiac and smooth muscle are not involved. The prevalence of the congenital myasthenic syndromes is estimated to be one-tenth that of myasthenia gravis which is thought to be 25:1 000 000–125:1 000 000.10 Onset is at or shortly after birth or in early childhood. Postchildhood onset of the congenital myasthenic syndromes though described, is extremely rare.11 12 In some individuals, distinctive craniofacial features such as a high-arched palate have been reported. The spectrum of clinical severity varies greatly and both intrafamilial and interfamilial differences are seen.13 The clinical course of these conditions is usually fairly static as with the children described here but in some, sudden, transient but severe exacerbations of weakness and/or episodes of respiratory insufficiency can be triggered by fever, infection or excitement.9 14
The diagnosis of CMS is based on the aforementioned clinical finding of fatigable muscle weakness, a decremental EMG response of the compound muscle action potential on low frequency that is, 2–3 Hz stimulation, absence of antiacetylcholine receptor (AChR) and antibodies to MuSK antibodies in the serum, and lack of improvement of clinical symptoms with immunosuppressive therapy.
Muscular weakness is manifest when a significant number of neuromuscular junctions fail to transmit the motor axon potential to the skeletal muscle fibre muscle fibre. Where neuromuscular transmission is impaired, action potentials though generated by initial stimuli may not be generated by those later in a series resulting in a decremental response at electromyography that is characteristic of the congenital myasthenic syndromes. The absence of serum AChR and anti-MuSK antibodies can help distinguish a CMS from myasthenia gravis and that subset of this disorder caused by anti-MuSK antibodies but will not exclude seronegative types of myasthenia gravis.15 Immunosuppressive therapy forms the main approach to the treatment of autoimmune myasthenia gravis and so the absence of a response to the same suggests an alternative diagnosis. Interestingly, a case of autoimmune myasthenia gravis developing in an individual with CMS has been reported.16
Clinical clues may even point to the particular gene mutated in a CMS.17 18 For example, the clinical features commonly associated with mutations in the gene encoding the epsilon subunit of the acetylcholine receptor include ptosis and ophthalmoplegia as in the cases described here and, additionally, feeding difficulties.19
In an autosomal recessive CMS as described here the parents of an affected child are obligate heterozygotes and therefore each carry one mutant allele. Heterozygotes or ‘carriers’ are asymptomatic. At conception, each child has a 25% chance of being affected, a 50% chance of being an asymptomatic heterozygote carrier and a 25% chance of being unaffected and not a carrier.
To establish the diagnosis in a patient suspected to have a CMS, targeted mutation analysis dependent on the ethnic origin of the patient may be carried out. Subsequent genetic testing is based on the proportion of the CMSs attributed to a mutation in each of the associated genes. Genetic testing for at-risk relatives of an affected individual when the mode of inheritance is autosomal recessive, prenatal diagnosis and preimplantation genetic diagnosis for at-risk pregnancies require prior identification of the disease-causing mutations in the family. The first of these might be most important in new borns or young children who may have a CMS that places them at risk from sudden respiratory failure and in whom treatment to prevent the same could be instituted early, after diagnosis.
Acetylcholinesterase inhibitors which increase the number of acetylcholine receptors activated by each motor nerve impulse and 3, 4-diaminopyridine which increases the amount of acetylcholine released by nerve impulses are the mainstays of treatment in the CMSs. Drugs known to affect neuromuscular transmission and thus exacerbate the symptoms of congenital myasthenia include ciprofloxacin, chloroquine, procaine, lithium, phenytoin, β-blockers, procainamide and quinidine and these should be used with caution in those affected.
Learning points.
The congenital myasthenic syndromes, though not entirely uncommon frequently go undiagnosed as in the cases outlined here. This may be because ophthalmologists are less familiar with the congenital myasthenic syndromes than with conventional ‘autoimmune’ myasthenia gravis or indeed than with other neurological disorders presenting with ocular symptoms and/or signs.20 This report aims to go some way towards increasing the awareness of the congenital myasthenic syndromes within the specialty.
The congenital myasthenic syndromes are also frequently misdiagnosed. They can closely mimic other disorders. In the cases we describe here there were many conflicting factors which made more difficult making the correct diagnosis. A history of variability in symptomatology that is characteristic of myasthenia gravis and is also in keeping with the congenital myasthenic syndromes was absent. The absence too of a relevant family history of congenital myasthenia was initially misleading. At examination, our male patient demonstrated an association of eye and lid movements somewhat suggestive of an aberrant regeneration phenomenon. The pineal gland cysts identified at MRI of both children and which we are now confident were merely incidental findings were also confusing initially. In these cases maintaining a broad differential diagnosis ensured an accurate diagnosis was finally made.
When there is clearly a pathological condition that is not in keeping with commoner conditions then rarer possibilities should be considered as explanatory.
The investigation of individuals with a suspected congenital myasthenia syndrome is complex. Many tests for these disorders are only undertaken in specialist national or international centres and performed following advice from regional or national experts.21 If a diagnosis of congenital myasthenia is suspected then prompt referral for the relevant investigations in the appropriate centre should be made.
The onset and course of disease, treatment efficacy and safety in patients sharing the same genetic defect in the congenital myasthenic syndromes are closely linked to the underlying molecular aetiology. Thus the genetic diagnosis of a congenital myasthenic syndrome enables a rationale for management to be developed. Similarly improved understanding of the molecular biology underlying these syndromes offers the potential for improved therapy and more accurate genetic counselling of affected families.4
The involvement of a multidisciplinary team inclusive of a paediatrician, neurologist, geneticist and ophthalmologist and for the more severe cases a physiotherapist, occupational therapist and speech and language therapist is necessary for the appropriate management of children with the congenital myasthenic syndromes.21
Acknowledgments
The authors would like to thank Gillian O'Mullane, Senior Orthoptist, Temple Street Children's University Hospital, for providing the figures used in this report.
Footnotes
Competing interests: None.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Belasco C, Carbillon L, Louaib D, et al. Neonatal myasthenia gravis. Arch Pediatr 2000;7:263–6 [DOI] [PubMed] [Google Scholar]
- 2.Engel A. Current status of the congenital myasthenic syndromes. Neuromuscul Disord 2012;22:99–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Salpeter M, Loring R. Nicotinic acetylcholine receptors in vertebrate muscle: properties, distribution and neural control. Prog Neurobiol 1985;25:297–325 [DOI] [PubMed] [Google Scholar]
- 4.Nogajski J, Kiernan M, Ouvrier R, et al. Congenital myasthenic syndromes. J Clin Neurosci 2009;16:1–11 [DOI] [PubMed] [Google Scholar]
- 5.Müller J, Mihaylova V, Abicht A, et al. Congenital myasthenic syndromes: spotlight on genetic defects of neuromuscular transmission. Expert Rev Mol Med 2007;9:1–20 [DOI] [PubMed] [Google Scholar]
- 6.Kinali M, Beeson D, Pitt M, et al. Congenital myasthenic syndromes in childhood: diagnostic and management challenges. J Neuroimmunol 2008;201–202:6–12 [DOI] [PubMed] [Google Scholar]
- 7.Palace J, Beeson D. The congenital myasthenic syndromes. J Neuroimmunol 2008;201–202:2–5 [DOI] [PubMed] [Google Scholar]
- 8.Engel A, Shen X, Selcen D, et al. What have we learned from the congenital myasthenic syndromes. J Mol Neurosci 2010;40:143–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barišić N, Chaouch A, Müller J, et al. Genetic heterogeneity and pathophysiological mechanisms in congenital myasthenic syndromes. Eur J Paediatr Neurol 2011;15:189–96 [DOI] [PubMed] [Google Scholar]
- 10.Chaouch A, Beeson D, Hantaï D, et al. 186th ENMC International Workshop: congenital myasthenic syndromes. Neuromuscul Disord 2012;22:566–76 [DOI] [PubMed] [Google Scholar]
- 11.Burke G, Cossins J, Maxwell S, et al. Rapsyn mutations in hereditary myasthenia: distinct early and late onset phenotypes. Neurology 2003;61:826–8 [DOI] [PubMed] [Google Scholar]
- 12.Beeson D, Higuchi O, Palace J, et al. Dok-7 mutations underlie a neuromuscular junction synaptopathy. Science 2006;313:1975–8 [DOI] [PubMed] [Google Scholar]
- 13.Engel A, Sine S. Current understanding of congenital myasthenic syndromes. Curr Opin Pharmacol 2005;5:308–21 [DOI] [PubMed] [Google Scholar]
- 14.Ohno K, Tsujino A, Brengman J, et al. Choline acetyltransferase mutations cause myasthenic syndrome associated with episodic apnoea in humans. Proc Natl Acad Sci USA 2001;98:2017–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hoch W, McConville J, Helms S, et al. Auto-antibodies to the receptor tyrosine kinase MuSK in patients with myasthenia gravis without acetylcholine receptor antibodies. Nat Med 2001;7:365–8 [DOI] [PubMed] [Google Scholar]
- 16.Croxen R, Vincent A, Newsom-Davis J, et al. Myasthenia gravis in a woman with congenital AChR deficiency due to epsilon-subunit mutations. Neurology 2002b;58:1563–5 [DOI] [PubMed] [Google Scholar]
- 17.Cossins J, Burke G, Maxwell S, et al. Diverse molecular mechanisms involved in AChR deficiency due to rapsyn mutations. Brain 2006;129:2773–83 [DOI] [PubMed] [Google Scholar]
- 18.Palace J, Lashley D, Newsom-Davis J, et al. Clinical features of the DOK7 neuromuscular junction synaptopathy. Brain 2007;130:1507–15 [DOI] [PubMed] [Google Scholar]
- 19.Burke G, Cossins J, Maxwell S, et al. Distinct phenotypes of congenital acetylcholine receptor deficiency. Neuromuscul Disord 2004;14:356–64 [DOI] [PubMed] [Google Scholar]
- 20.Engel A. The therapy of congenital myasthenic syndromes. Neurotherapeutics 2007;4:252–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Parr J, Jayawant S. Childhood myasthenia: clinical subtypes and practical management. Dev Med Child Neurol 2007;49:629–35 [DOI] [PubMed] [Google Scholar]