

Abstract
Ca2+/calmodulin-dependent protein kinase II is a synapse-enriched kinase in mammalian brains. This kinase interacts with various synaptic proteins to regulate expression and function of interacting proteins and thereby modulates synaptic transmission. CaMKII and its interacting partners are also believed to play a pivotal role in the pathogenesis of various neurological and neurodegenerative disorders, such as Parkinson's disease (PD). In this study, we found that CaMKIIα binds to dopamine D2 receptors (D2R) in vitro. A distal region in the D2R third intracellular loop harbors CaMKIIα binding. Endogenous CaMKIIα was also found to interact with native D2Rs in rat striatal neurons in which D2Rs are expressed at a high level. In addition, in a rat 6-hydroxydopamine lesioned model of PD, chronic levodopa administration induced characteristic dyskinesia. In parallel, levodopa induced an increase in CaMKIIα-D2R interactions in striatal neurons. Intrastriatal injection of a Tat-fusion and CaMKIIα-D2R interaction-dead peptide (Tat-D2Ri) reversed this increase in the interaction between two proteins. Tat-D2Ri also alleviated dyskinetic behaviors induced by levodopa. These results reveal a new interaction between CaMKIIα and D2Rs in striatal neurons which is sensitive to long-term administration of levodopa in PD rats. Prevention of the response of CaMKIIα-D2R interactions to levodopa can alleviate levodopa-induced dyskinesia.
Parkinson's disease (PD) is primarily a result of selective death of dopaminergic neurons in the substantia nigra and consequential loss of dopamine innervations in the striatum. Dopamine replacement therapy with levodopa is so far the most effective therapy. However, chronic levodopa treatment causes abnormal involuntary movements (AIM) known as levodopa-induced dyskinesia (LID), for which the underlying molecular mechanisms are far from clear. Aberrant dopaminergic transmission is considered as a central element in the pathogenesis of LID. In the striatum, both dopamine D1 receptors and dopamine D2 receptors (D2R) are highly expressed1. D1 receptors are known to be predominantly expressed in the direct pathway, i.e., striatonigral projection neurons, as opposed to the fact that D2Rs are expressed in the indirect pathway, striatopallidal neurons. While D1 receptors have been extensively studied for their participations in LID2,3, the role of D2Rs in LID is poorly understood.
Ca2+/calmodulin-dependent protein kinase II (CaMKII) is ubiquitously expressed in the central nervous system and is particularly enriched at excitatory synapses4. This kinase is actively involved in the regulation of essential neuronal activities, including neurotransmitter synthesis and release, postsynaptic receptor signaling, and long-term synaptic plasticity5,6,7. Available data indicate that CaMKII is linked to the pathogenesis and symptomatology of a variety of mental and neurological illnesses, including learning disorder7, cognitive impairment8, schizophrenia9, ischemic10, Alzheimer disease11, epileptic seizures12 and PD13. Recent evidence suggests a possible role of CaMKII in LID. Pharmacological inhibition of CaMKII with a selective inhibitor KN93 ameliorated dyskinesia in a rat PD model14. However, how CaMKII in striatal neurons responds to and mediates LID is unclear. CaMKIIα has been demonstrated to interact with a number of synaptic receptors, including ionotropic and metabotropic glutamate receptors and dopamine D3 receptors, and by directly interacting with these receptors CaMKII vigorously regulates their subcellular distribution and function15,16,17. It is unknown at present however whether CaMKII interacts with D2Rs and if so whether CaMKII-D2R interactions are sensitive to levodopa and contribute to the development of LID.
In this study, we thus examined the relationship between CaMKII and D2Rs in vitro and in vivo. We first investigated whether recombinant CaMKIIα proteins bind to purified D2Rs in vitro and whether their binding is direct. We then mapped a confined binding sequence from a CaMKIIα binding region on the intracellular domain of D2Rs. To determine if native CaMKIIα and D2Rs interact with each other, we carried out coimmunoprecipitation with adult rat striatal lysates. Finally, we tested CaMKIIα-D2R interactions in a rat PD model. We analyzed the changes in CaMKIIα-D2R interactions in striatal neurons in response to chronic levodopa therapy. We also carried out behavioral experiments to determine the role of CaMKIIα-D2R interactions in levodopa-induced dyskinetic behaviors.
Results
CaMKIIα binds to D2Rs
D2Rs contain a characteristically long IL3, usually providing a site for protein-protein interactions18,19. To determine whether CaMKIIα interacts with IL3, we prepared a GST-fusion protein containing IL3 (GST-D2R-IL3) based on the long form of D2Rs as this form is preferentially involved in postsynaptic D2R signaling. We then used GST-D2R-IL3 as immobilized baits to precipitate endogenous CaMKIIα from rat striatal lysates in pull-down assays. As shown in Fig. 1A, GST-D2R-IL3 precipitated CaMKIIα, while GST alone did not. Two other GST-fusion proteins containing IL2 (GST-D2R-IL2) and CT (GST-D2R-CT) were also tested together. They showed no ability to precipitate CaMKIIα (Fig. 1A). These results demonstrate the existence of interactions between CaMKIIα and D2R-IL3. To determine whether CaMKIIα directly interacts with D2Rs, we carried out in vitro binding assays with purified proteins. Purified CaMKIIα was found to bind to the immobilized GST-D2R-IL3 (Fig. 1B). CaMKIIα did not bind to GST alone, GST-D2R-IL2, and GST-D2R-CT (Fig. 1B). Thus, CaMKIIα has the ability to directly bind to D2Rs through the D2R IL3 domain.
Figure 1. CaMKIIα interactions with D2Rs.
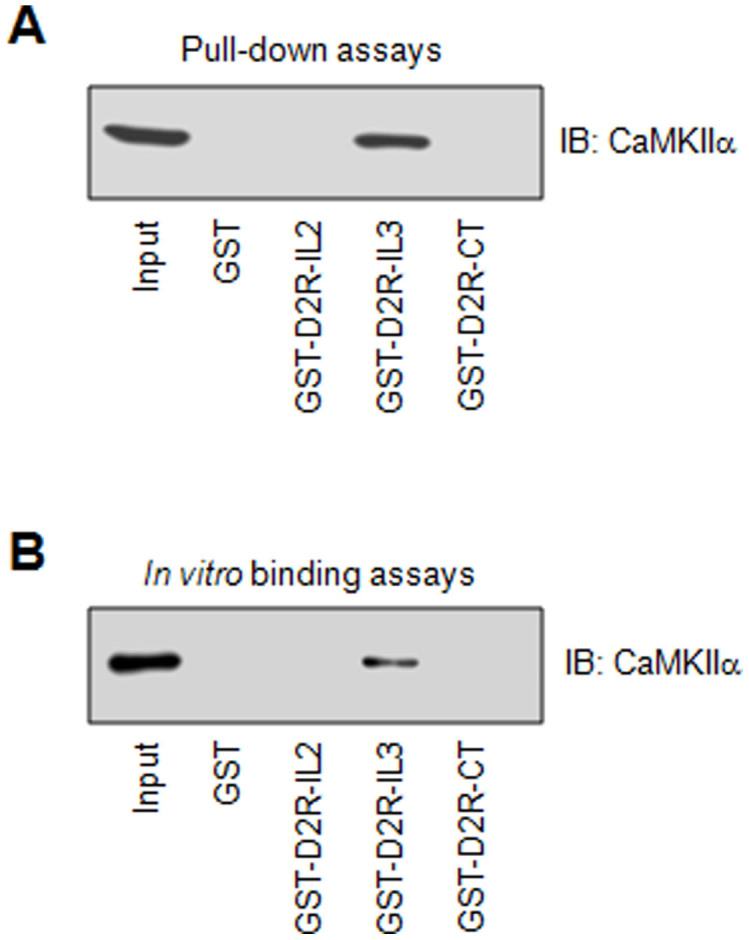
(A) Pull-down assays showing that GST-D2R-IL3 pulled down CaMKIIα from rat striatal lysates. (B) In vitro binding assays showing that GST-D2R-IL3 precipitated purified recombinant CaMKIIα. Note that GST alone and GST fusion proteins containing IL2 (GST-D2R-IL2) and CT (GST-D2R-CT) did not interact with D2Rs. Precipitated proteins from pull-down assays and in vitro binding assays were visualized with immunoblots (IB) using the specific antibodies indicated.
CaMKIIα binds to the distal region of D2R IL3
To identify an accurate CaMKIIα binding site in D2R IL3, we synthesized an N-terminal region of IL3, GST-D2R-IL3(K211-M270), and a CT region of IL3, GST-D2R-IL3(G242-Q374). GST-D2R-IL3(K211-M270) did not precipitate CaMKIIα in binding assays (Fig. 2A). In contrast, GST-D2R-IL3(G242-Q374) precipitated the kinase to an extent similar to full length IL3. Thus, the CT region of D2R-IL3 contains a site that harbors CaMKIIα binding. To further narrow down the sequence required for the CaMKIIα binding, we synthesized a series of truncated IL3 fragments. A short distal IL3 fragment, GST-D2R-IL3(Q345-Q374), strongly precipitated CaMKIIα (Fig. 2B). Other two fragments proximal to Q345-Q374, i.e., GST-D2R-IL3(M281-P310) and GST-D2R-IL3(P308-K340), did not precipitate the kinase. Thus, the C-terminal 30 amino acids (Q345-Q374) of D2R-IL3 contain a core binding motif (Fig. 2C). Notably, a GST protein containing the corresponding region of IL3 in dopamine D3 receptors (D3R), GST-D3R-IL3(K346-Q375), failed to precipitate CaMKIIα (Fig. 2B).
Figure 2. The CaMKIIα binding motif in D2R-IL3.

(A) Binding of CaMKIIα to GST-fusion proteins derived from D2R-IL3. (B) Binding of CaMKIIα to GST-fusion proteins derived from D2R-IL3 and D3R-IL3. Note that Q345-Q374 fragments showed the ability to bind to CaMKIIα. (C) Amino acid sequence of D2R-IL(Q345-Q374). In vitro binding assays were carried out with purified recombinant proteins. Bound CaMKIIα was visualized by immunoblots (IB) of the eluted proteins using a specific antibody.
CaMKIIα interacts with D2Rs in striatal neurons
CaMKIIα and D2Rs are densely expressed in striatal neurons. To determine whether native CaMKIIα and D2R interact with each other in these neurons, coimmunoprecipitation was performed using synaptosomal samples extracted from the adult rat striatum. In proteins immunoprecipitated by anti-D2R antibodies, we readily detected immunoreactivity of CaMKIIα (Fig. 3A). In a reverse coimmunoprecipitation assay, we also observed a D2R band in the CaMKIIα precipitates (Fig. 3B). Full-length blots were presented in Supplementary Figure1. Thus, endogenous CaMKIIα and D2R proteins in striatal neurons form complexes in normal animals with extracorporeal detection.
Figure 3. Interactions of CaMKIIα with D2Rs in adult rat striatal neurons.
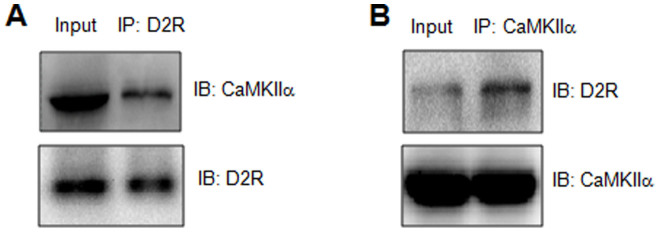
(A) Coimmunoprecipitation of endogenous D2Rs and CaMKIIα using an anti-D2R antibody as the immunoprecipitating antibody. (B) Coimmunoprecipitation of endogenous D2Rs and CaMKIIα using an anti-CaMKIIα antibody as the immunoprecipitating antibody. Coimmunoprecipitated proteins were visualized by immunoblots (IB) of the eluted proteins using antibodies indicated. Full-length blots were presented in Supplementary Figure1.
Effects of chronic levodopa administration on CaMKIIα-D2R interactions
In this study, we investigated the effect of chronic levodopa injection on CaMKIIα-D2R interactions in striatal neurons of 6-OHDA lesioned PD rats. We found that chronic levodopa administration (25 mg/kg, i.p.; twice daily for 22 days) produced an increase in an amount of D2Rs in CaMKIIα precipitates as compared to control PD rats (Fig. 4A and 4B). This indicates an increased rate of CaMKIIα-D2R interactions in striatal neurons following chronic administration of levodopa.
Figure 4. Levodopa-induced CaMKIIα-D2R interactions in the striatum.

(A) Coimmunoprecipitation of CaMKIIα and D2R using rat striatal lysates. Note that chronic administration of levodopa (25 mg/kg, i.p., twice daily for 22 days) increased D2R levels in CaMKIIα precipitates. Intrastriatal injection of Tat-D2Ri rather than Tat-D2Rc (1 μg) reduced the level of D2Rs in CaMKIIα precipitates. (B) A graph illustrating the effect of levodopa on CaMKIIα-D2R interactions and the effect of Tat-fusion peptides on levodopa-induced CaMKIIα-D2R interactions. Full-length blots were presented in Supplementary Figure2. **p < 0.01 versus PD control; ##p < 0.01 versus saline+levodopa.
Based on the CaMKIIα binding region in D2R-IL3 identified above, we synthesized a Tat-fusion interfering peptide (Tat-D2Ri) that contains a Tat cell membrane transduction domain (YGRKKRRQRRR) and a core region of CaMKIIα binding motif (TMSRRKLSQQKEK) on D2R-IL3 (Fig. 4A). The arginine-enriched Tat domain renders cell permeability20 and the CaMKIIα binding motif may compete with endogenous D2Rs for CaMKIIα binding. Indeed, Tat-D2Ri locally injected into the rat striatum (1 μg/o.5 μl, 1 h prior to levodopa) at the final day of chronic levodopa treatment (25 mg/kg, i.p.; twice daily for 22 days) reduced the CaMKIIα-D2R interaction in the injected site (Fig. 4A and 4B). In contrast to Tat-D2Ri, Tat-D2Rc (a sequence-scrambled control) did not alter the increased CaMKIIα-D2R complex formation. Full-length blots were presented in Supplementary Figure2. These results validated the efficacy and selectivity of Tat-D2Ri in disrupting CaMKIIα-D2R interactions in striatal neurons of adult rat brains in vivo. A single intrastriatal injection of the peptide was able to reverse the elevation of CaMKIIα-D2R interactions induced by chronic levodopa administration.
Effects of Tat-fusion peptides on behavioral responses to levodopa
A series of neurobehavioral experiments were carried out to test the effect of Tat-fusion interaction-dead peptides on behavioral activities induced by levodopa. Chronic administration of levodopa induced a gradual increase in ALO AIM scores in PD rats as tested at 2, 7, 14, and 21 days after levodopa administration (Fig. 5A), indicating a development of LID. Interestingly, at day 22, intrastriatal infusion of Tat-D2Ri (1 μg/0.5 μl) alleviated these dyskinetic behavioral changes (Fig. 5B).In contrast, intrastriatal infusion of Tat-D2Rc or saline had no effect on dyskinetic behaviors. Chronic levodopa injection also shorterned the duration of response (Fig. 5C and Fig. 5D). Tat-D2Ri but not Tat-D2Rc significantly reversed this reduction. Detailed data were displayed within tables in supplementary information (Table 1 and Table 2).
Figure 5. Effects of Tat-D2Ri and Tat-D2Rc on dyskinetic behaviors induced by levodopa.

(A) Effects of chronic levodopa administration on ALO AIM scores. (day2 vs day21, lid+saline group: p = 0.03; lid+Ri group: p = 0.00; lid+Rc group: p = 0.03) (B) Effects of Tat-fusion peptides on ALO AIM scores at day 22. (lid+saline vs lid+Ri: p = 0.049; lid+Ri vs lid+Rc: p = 0.025; lid+Rc vs lid+saline p = 1.000) (C) Effects of chronic levodopa administration on the duration of response. (day2 vs day21, saline group: p = 0.00; lid+Ri group: p = 0.00; lid+Rc group: p = 0.00) (D) Effects of Tat-fusion peptides on the duration of response at day 22. (lid+saline vs lid+Ri: p = 0.027; lid+Ri vs lid+Rc: p = 0.044; lid+Rc vs lid+saline p = 1.000). Note that chronic levodopa administration increased ALO AIM scores and reduced the duration of rotation. Tat-D2Ri can significantly reverse these behavioral changes. Levodopa was given to 6-OHDA lesioned PD rats at 25 mg/kg (i.p.; twice daily for 22 days). At day 22, Tat-D2Ri and Tat-D2Rc (1 μg) was infused bilaterally into the striatum 1 h before levodopa. Behavioral tests were conducted at 2, 7, 14, 21, and 22 days. p < 0.05 versus: #day2 of the same group; * lid+saline group on the same day.
Discussion
In this study, we investigated the possible interaction between CaMKIIα and D2Rs. We found that CaMKIIα bound to D2Rs in vitro. This binding appeared to be direct since a specific region in the D2R-IL3 domain formed a binding site for CaMKIIα. Two native proteins were also found to interact with each other in neurons. Based on the coimmunoprecipitation data, CaMKIIα was found to form complexes with D2Rs in adult rat striatal neurons. We then set forth to investigate whether the CaMKIIα-D2R interaction in striatal neurons responds to levodopa therapy in a rat 6-OHDA lesioned PD model. We found that chronic levodopa administration induced a significant increase in the CaMKIIα-D2R interaction. A cell permeable Tat-D2Ri peptide injected into the striatum blocked this increase. Tat-D2Ri peptides also reduced the dyskinetic behavior induced by levodopa. These results indicate that D2Rs are a new interacting partner of CaMKIIα in striatal neurons. This newly identified CaMKIIα-D2R interaction may play a role in LID.
An important finding in this work is the interaction between CaMKIIα and D2Rs. CaMKIIα was found to bind to an intracellular domain of D2Rs. Specifically, CaMKIIα bound to IL3 but not IL2 and CT regions. A small distal region in the C-terminal IL3 forms a site accepting CaMKIIα. Dopamine D3 receptors have also been found to be bound by CaMKIIα15. However, interestingly, CaMKIIα binds to the N-terminal region of D3R IL3 as opposed to its binding to the C-terminal region of D2R IL3. At present, we have not carried out experiments to fully define the functional role of CaMKIIα-D2R interactions in regulating D2R expression and function. As a G protein-coupled receptor, D2Rs are couple to Gαi proteins, through which D2Rs inhibit adenylyl cyclase21 and reduce the downstream cAMP formation22. IL3 is a known region that couples D2Rs to Gαi proteins23. Within IL3, the C-terminal membrane-proximal part participates in the interaction with Gαi21. Noticeably, the CaMKIIα binding motif we identified in D2R-IL3 partially overlaps with the Gαi interaction domain. Thus, CaMKIIα is assumed to have an impact on the D2R-Gαi-adenylyl cyclase signaling pathway. This topic will be investigated in the future.
The basal ganglia circuitry contains a D1 receptor-bearing direct pathway and a D2R-bearing indirect pathway24,25. An essential pathophysiological characteristic of LID is the presence of hypoactivity of the indirect pathway and hyperactivity of the direct pathway26,27. Although D2Rs are considered to be important to LID, direct evidence is incomplete in illustrating the role of D2Rs. Thus, in this study, we attempted to investigate the role of D2Rs in terms of its interactions with CaMKIIα. The important findings obtained in the study include that 1) chronic levodopa administration increased the CaMKIIα-D2R interaction in the striatum of PD rats, 2) This increase was disrupted by an interaction-dead peptide (Tat-D2Ri) but not by a control peptide, and 3) more importantly, Tat-D2Ri showed the ability to reduce dyskinetic behavioral responses to levodopa. These results collectively indicate that the CaMKIIα-D2R interaction in striatal neurons is an important pathway to LID. This interaction was upregulated as a plastic response to long-term levodopa therapy, which may contribute to dyskinetic behavior seen in LID rats. Co-administration of partial D2R agonists with levodopa attenuated levodopa-induced abnormal behavioral response28. Thus, if CaMKIIα exerts an inhibitory regulation of D2Rs, Tat-D2Ri, by removing the CaMKIIα influence, can disinhibit D2Rs and achieve the same effect as that induced by D2R agonists. Consistent with this model, intrastriatal injection of CaMKII inhibitors improved motor performance and synaptic plasticity in the form of long-term potentiation at corticostriatal synapses13. Moreover, intrastriatal infusion of the CaMKII inhibitor KN93 ameliorated dyskinesia in 6-OHDA-lesioned PD rats14,29.
Methods
The protocols of this study were reviewed and approved by the Ethical Committee of the Medical School of Shanghai Jiaotong University. The methods were carried out in accordance with the approved guidelines and regulations. Animals were purchased from the Sippr-BK Ltd (Shanghai, China), Shanghai, fed and regulated in experimental animal center of Xinhua Hospital affiliated to the Medical School of Shanghai Jiaotong University (Shanghai, China).
Expression and purification of glutatione S-transferase (GST)-fusion proteins
cDNA fragments encoding intracellular domains, such as intracellular loops (IL) and C-termini (CT), were generated by polymerase chain reaction amplification from full-length cDNA clones. These fragments include D2R-IL2(D131-R151), D2R-CT(N431-C444), D2R-IL3(K211-Q374), D2R-IL3(K211-M270), D2R-IL3(G242-Q374), D2R-IL3(M281-P310), D2R-IL3(P308-K340), D2R-IL3(Q345-Q374), and D3R-IL3(K346-Q375). These fragments were subcloned into BamHI-EcoRI sites of the pGEX4T-3 plasmid (Amersham Biosciences, Arlington Heights, IL) or SpeI-XhoI sites of the pET-41a (+) plasmid (Novagen, Madison, WI). To confirm appropriate splice fusion, all constructs were sequenced. GST-fusion proteins were expressed in E. coli BL21 cells (Amersham) and purified from bacterial lysates as described by the manufacturer. His-tagged full-length CaMKIIα (M1-H478) was expressed and purified via a baculovirus/Sf9 insect cell expression system.
Affinity purification (pull-down) assay
Pull-down assays were performed with solubilized striatal extracts (50–100 μg of protein) diluted in 1X phosphate buffered saline (PBS)/1% Triton X-100 and incubated with 50% (v/v) slurry of glutathione-sepharose 4B beads (Amersham). Assay solutions were saturated with GST alone or the indicated GST-fusion protein (5–10 μg) for 2–3 h at 4°C. After beads were washed four times with 1X PBS/1% Triton X-100, bound proteins were eluted with 2X lithium dodecyl sulfate (LDS) loading buffer, resolved by SDS-PAGE, and immunoblotted with a specific antibody.
In vitro binding assay
To conduct binding assays, GST-fusion proteins (1–5 μg) were digested with 0.2 NIH unit of thrombin (Amersham) for 2 h at room temperature. The reaction was stopped by phenylmethylsulfonyl fluoride (10 μM). GST was removed by glutathione sepharose (Amersham). The supernatant was equilibrated to binding buffer (200 mM NaCl, 0.2% Triton X-100, 0.1 mg/ml BSA, and 50 mM Tris, pH, 7.5) with 0.5 mM CaCl2 and 1 μM CaM. Binding reactions started after adding GST-fusion proteins and remained for 2–3 h at 4°C. To precipitate GST-fusion proteins, 10% glutathione sepharose was added. The precipitate was washed three times with binding buffer. Bound proteins were eluted with 2X LDS loading buffer, resolved by SDS-PAGE, and immunoblotted with a specific antibody.
Animals and 6-OHDA lesions
Animal experimental design was seen in supplementary figure3. Adult female rats (Sprague Dawley, 180–220 g, Sippr-BK Ltd, Shanghai, China) were used in this study. All procedures were carried out in accordance with guidelines of the National Institutes of Health for the care and use of laboratory animals. Rats were deeply anesthetized by 7% chloral hydrate (0.5 ml/100 g, v/w) and mounted in a stereotaxic apparatus (Narishge, Japan) equipped with a rat adaptor. Animals received unilateral injections of 6-OHDA (8 μg) or saline (sham lesion) into the right medial forebrain bundle (MFB) at the coordinates (mm): AP = −4.4 mm from bregma; ML = −1.2 mm from midline; DV = −7.8 from the dura surface; Tooth bar = −2.4 mm. A volume of 4 μl was injected over 4 min and the Hamilton syringe was kept in place for an additional 5 min before being retracted slowly. At 3 weeks after surgery, rats were tested with a subcutaneous injection of apomorphine at 0.05 mg/kg (WOKO, Japan). Contralateral turns were counted for 30 min after an interval of 10 min. Only those rats displaying rotational asymmetry of >6 turns/min were considered as PD rats and were used for the following neurochemical and behavioral experiments.
Drug treatment
Validated PD rats received vehicle or levodopa injection for 22 days. Vehicle or levodopa methylester (Sigma, St. Louis, MO) was given intraperitoneally (i.p.) at 25 mg/kg in combination with benserazide (6.25 mg/kg). Animals were treated with levodopa and benserazide twice daily (6 h apart). At the final day (day 22), levodopa-treated and dyskinetic rats were randomly divided into 3 groups. These rats received intrastriatal administration of Tat-D2Ri, Tat-D2Rc, or saline. One hour after fully recovery from anesthesia, rats were treated with levodopa and benserazide. For intrastriatal injection, rats were anesthestized by 7% chloral hydrate (0.5 ml/100 g, v/w). A volume of 0.5 μl Tat-D2Ri (1 μg) or Tat-D2Rc (1 μg) or saline was injected at the coordinates: AP = 0.5 mm from bregma; ML = −2.5 mm from midline; and DV = −4.2 from the dura surface. A microsyringe was kept in place for an additional 5 min before being retracted slowly. Rats were left on a warm plate after surgery to avoid hypothermia until recovery.
Behavioral test
The evaluation of AIM was performed according to the rat dyskinesia scale30. On testing days, rats were placed individually in transparent plastic cages 10 min before drug treatment. As described previously31, rat abnormal involuntary movements (AIM) were classified into four subtypes: axial AIM, i.e. dystonic posturing or choreiform twisting of the neck and upper body towards the side contralateral to the lesion; limb AIM, i.e. abnormal, purposeless movements of the forelimb and digits contralateral to the lesion; orolingual AIM, i.e. empty jaw movements and contralateral tongue protrusion; and locomotive AIM, i.e. increased locomotion with contralateral side bias. Each of these subtypes was scored on a severity scale from 0 to 4. During a period of 180 min following levodopa treatment, three subtypes of AIM were assessed as axial, limb, orolingual every 20 min (60 sec monitoring period for each). The ALO AIM were tested at 2, 7, 14 and 21 days during levodopa treatment. At day 22, rats were intrastriatal injected with Tat-D2Ri, Tat-D2Rc or saline. One hour after fully revived from anesthesia, levodopa was administrated and behavioral assessments were then carried out. The response duration was also recorded which was defined previously32. The duration of the rotational response was measured as the time between the first 5 min interval when turning exceeded 20% of the peak rate and the first interval when turning fell below 20% of the peak rate. The peak intensity of rotation was measured as the peak number of contralateral turns in any 5 min interval.
Coimmunoprecipitation and immunoblot
Rat striatal tissue was dissected on ice and homogenized by sonication in an immunoprecipitation lysis buffer (Beyortime, China) plus a protease inhibitor cocktail (Roche Diagnostics, Swiss). To obtain P2 pellets, the homogenate was centrifuged at 800 g for 10 min at 4°C. The supernatant was then centrifuged at 11,000 g for 30 min at 4°C. The pellet was resuspended in the lysis buffer and used for coimmunoprecipitation. Samples were incubated with a rabbit antibody against D2Rs (Millipore) or CaMKIIα (Santa Cruz) overnight at 4°C. The complex was precipitated with protein G agarose beads or protein A agarose beads by gentle rocking for 3 h at 4°C. Samples were suspended in a buffer containing 0.5% SDS and boiled for 5 min. Samples were loaded on 5–10% SDS gels. After electrophoresis, proteins were transferred to a polyvinylidene difluoride membrane (Millipore). Membranes were blocked in 5% nonfat milk for 1 h at room temperature and incubated with a rabbit primary antibody against D2R (Millipore) or a mouse primary antibody against CaMKIIα (Santa Cruz) overnight at 4°C. Membranes were washed and incubated in horseradish peroxidase conjugated secondary antibodies (1:1000) for 1 h at room temperature. Immunoblots were developed with the enhanced electrochemiluminescence reagent (GE Healthcare) and captured by a BIO-RAD molecular imager.
Data analysis
The O.D. value of blots bands were measured by image labtm software and normalized with PD control. SPSS17.0 and Graphpad prism5 were used for stastics and Graphics. Results of behavioral tests are presented as means ± SD. The data were evaluated using a one-way ANOVA followed by a Bonferroni's comparison of groups using least-squares-adjusted means. Probability levels of <0.05 were considered statistically significant.
Author Contributions
L.Z.G. have contributed to the conception and design of the study. Z.S.F. performed the experiments, wrote the main manuscript test and prepared figures 3–5. W.Q. performed the experiments, prepared figures 1 and 2, and revised the article. X.C.L. participated in the statistical analysis. All authors have reviewed the manuscript.
Supplementary Material
Supplementary information
Acknowledgments
The study was supported by the Projects of National Science Foundation of China (No.81171203, 81171204 and 81200871), and Projects of the Shanghai Committee of Science and Technology, China (No.11nm0503300 and 12XD1403800). We thank Dr. Xianyu Liu and Dr. Limin Mao for support and assistances.
References
- Le Moine C. & Bloch B. D1 and D2 dopamine receptor gene expression in the rat striatum: sensitive cRNA probes demonstrate prominent segregation of D1 and D2 mRNAs in distinct neuronal populations of the dorsal and ventral striatum. J Comp Neurol 355, 418–426 (1995). [DOI] [PubMed] [Google Scholar]
- Berthet A. & Bezard E. Dopamine receptors and L-dopa-induced dyskinesia. Parkinsonism Relat Disord 15 Suppl 4, S8–12 (2009). [DOI] [PubMed] [Google Scholar]
- Aubert I. et al. Increased D1 dopamine receptor signaling in levodopa-induced dyskinesia. Ann Neurol 57, 17–26 (2005). [DOI] [PubMed] [Google Scholar]
- Kelly P. T., McGuinness T. L. & Greengard P. Evidence that the major postsynaptic density protein is a component of a Ca2+/calmodulin-dependent protein kinase. Proc Natl Acad Sci U S A 81, 945–949 (1984). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giese K. P., Fedorov N. B., Filipkowski R. K. & Silva A. J. Autophosphorylation at Thr286 of the alpha calcium-calmodulin kinase II in LTP and learning. Science 279, 870–873 (1998). [DOI] [PubMed] [Google Scholar]
- Taha S., Hanover J. L., Silva A. J. & Stryker M. P. Autophosphorylation of alphaCaMKII is required for ocular dominance plasticity. Neuron 36, 483–491 (2002). [DOI] [PubMed] [Google Scholar]
- Silva A. J., Paylor R., Wehner J. M. & Tonegawa S. Impaired spatial learning in alpha-calcium-calmodulin kinase II mutant mice. Science 257, 206–211 (1992). [DOI] [PubMed] [Google Scholar]
- Yabuki Y. et al. Aberrant CaMKII activity in the medial prefrontal cortex is associated with cognitive dysfunction in ADHD model rats. Brain res 1557, 90–100 (2014). [DOI] [PubMed] [Google Scholar]
- Novak G. & Seeman P. Hyperactive mice show elevated D2(High) receptors, a model for schizophrenia: Calcium/calmodulin-dependent kinase II alpha knockouts. Synapse 64, 794–800 (2010). [DOI] [PubMed] [Google Scholar]
- Waxham M. N., Grotta J. C., Silva A. J., Strong R. & Aronowski J. Ischemia-induced neuronal damage: a role for calcium/calmodulin-dependent protein kinase II. J Cereb Blood Flow Metab 16, 1–6 (1996). [DOI] [PubMed] [Google Scholar]
- Min D. et al. The alterations of Ca2+/calmodulin/CaMKII/CaV1.2 signaling in experimental models of Alzheimer's disease and vascular dementia. Neurosci Lett 538, 60–65 (2013). [DOI] [PubMed] [Google Scholar]
- Xu X. et al. Abnormal changes in voltage-gated sodium channels Na(V)1.1, Na(V)1.2, Na(V)1.3, Na(V)1.6 and in calmodulin/calmodulin-dependent protein kinase II, within the brains of spontaneously epileptic rats and tremor rats. Brain Res Bull 96, 1–9 (2013). [DOI] [PubMed] [Google Scholar]
- Picconi B. et al. Abnormal Ca2+-calmodulin-dependent protein kinase II function mediates synaptic and motor deficits in experimental parkinsonism. J Neurosci 24, 5283–5291 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X., Wu N., Song L. & Liu Z. Intrastriatal injections of KN-93 ameliorates levodopa-induced dyskinesia in a rat model of Parkinson's disease. Neuropsychiatr Dis Treat 9, 1213–1220 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X. Y. et al. Activity-dependent modulation of limbic dopamine D3 receptors by CaMKII. Neuron 61, 425–438 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao L. M., Jin D. Z., Xue B., Chu X. P. & Wang J. Q. Phosphorylation and regulation of glutamate receptors by CaMKII. Sheng li xue bao 66, 365–372 (2014). [PMC free article] [PubMed] [Google Scholar]
- Jin D. Z. et al. Phosphorylation and feedback regulation of metabotropic glutamate receptor 1 by calcium/calmodulin-dependent protein kinase II. J Neurosci 33, 3402–3412 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borroto-Escuela D. O. et al. Characterization of the A2AR-D2R interface: focus on the role of the C-terminal tail and the transmembrane helices. Biochem Biophys Res Commun 402, 801–807 (2010). [DOI] [PubMed] [Google Scholar]
- Borroto-Escuela D. O. et al. Dopamine D2 and 5-hydroxytryptamine 5-HT((2)A) receptors assemble into functionally interacting heteromers. Biochem Biophys Res Commun 401, 605–610 (2010). [DOI] [PubMed] [Google Scholar]
- Schwarze S. R., Ho A., Vocero-Akbani A. & Dowdy S. F. In vivo protein transduction: delivery of a biologically active protein into the mouse. Science 285, 1569–1572 (1999). [DOI] [PubMed] [Google Scholar]
- Malek D., Munch G. & Palm D. Two sites in the third inner loop of the dopamine D2 receptor are involved in functional G protein-mediated coupling to adenylate cyclase. FEBS lett 325, 215–219 (1993). [DOI] [PubMed] [Google Scholar]
- Park S. K. et al. Par-4 links dopamine signaling and depression. Cell 122, 275–287 (2005). [DOI] [PubMed] [Google Scholar]
- Ilani T. et al. Coupling of dopamine receptors to G proteins: studies with chimeric D2/D3 dopamine receptors. Cell Mol Neurobiol 22, 47–56 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuen J. A., Chen M., Gloss B. & Calakos N. Drd1a-tdTomato BAC transgenic mice for simultaneous visualization of medium spiny neurons in the direct and indirect pathways of the basal ganglia. J Neurosci 28, 2681–2685 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valjent E. et al. Regulation of a protein phosphatase cascade allows convergent dopamine and glutamate signals to activate ERK in the striatum. Proc Natl Acad Sci U S A 102, 491–496 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rascol O. et al. Cortical motor overactivation in parkinsonian patients with L-dopa-induced peak-dose dyskinesia. Brain 121 (Pt 3), 527–533 (1998). [DOI] [PubMed] [Google Scholar]
- Fisone G. & Bezard E. Molecular mechanisms of l-DOPA-induced dyskinesia. Int Rev Neurobiol 98, 95–122 (2011). [DOI] [PubMed] [Google Scholar]
- Kalda A., Herm L., Rinken A., Zharkovsky A. & Chen J. F. Co-administration of the partial dopamine D2 agonist terguride with L-dopa attenuates L-dopa-induced locomotor sensitization in hemiparkinsonian mice. Behav Brain Res 202, 232–237 (2009). [DOI] [PubMed] [Google Scholar]
- Ba M. et al. Changes in subcellular distribution and phosphorylation of GluR1 in lesioned striatum of 6-hydroxydopamine-lesioned and l-dopa-treated rats. Neurochem Res 31, 1337–1347 (2006). [DOI] [PubMed] [Google Scholar]
- Winkler C., Kirik D., Bjorklund A. & Cenci M. A. L-DOPA-induced dyskinesia in the intrastriatal 6-hydroxydopamine model of parkinson's disease: relation to motor and cellular parameters of nigrostriatal function. Neurobiol Dis 10, 165–186 (2002). [DOI] [PubMed] [Google Scholar]
- Lundblad M. et al. Pharmacological validation of behavioural measures of akinesia and dyskinesia in a rat model of Parkinson's disease. Eur J Neurosci 15, 120–132 (2002). [DOI] [PubMed] [Google Scholar]
- Kong M., Ba M., Song L. & Liu Z. Comparative effects of acute or chronic administration of levodopa to 6-OHDA-lesioned rats on the expression and phosphorylation of N-methyl-D-aspartate receptor NR1 subunits in the striatum. Neurochem Res 34, 1513–1521 (2009). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information