

Abstract
Acute insult to the myocardium is associated with substantial loss of cardiomyocytes during the process of myocardial infarction. In this setting, apoptosis (programmed cell death) and necrosis may operate on a continuum. Because the latter is characterized by the loss of sarcolemmal integrity, we propose that an appropriately labeled tracer directed at a ubiquitously present intracellular moiety would allow non-invasive definition of cardiomyocyte necrosis. A trivalent arsenic peptide, GSAO (4-(N-(S-glutathionylacetyl)amino)phenylarsonous acid), is capable of binding to intracellular dithiol molecules such as HSP90 and filamin-A. Since GSAO is membrane impermeable and dithiol molecules abundantly present intracellularly, we propose that myocardial localization would represent sarcolemmal disruption or necrotic cell death. In rabbit and mouse models of myocardial infarction and post-infarct heart failure, we employed In-111-labelled GSAO for noninvasive radionuclide molecular imaging. 111In-GSAO uptake was observed within the regions of apoptosis seeking agent- 99mTc-Annexin A5 uptake, suggesting the colocalization of apoptotic and necrotic cell death processes.
Cell death plays a central role in various cardiovascular diseases. Two morphologically distinct modes of cell death - apoptosis (a programmed process characterized by enzymatic degradation and clean removal of the cell) and necrosis (an uncontrolled process characterized by cell swelling, membrane rupture and spill of its contents) have been reported to contribute to the myocardial tissue loss. It is being increasingly realized that apoptosis and necrosis, rather than being entirely independent, may operate on a continuum, at least in response to noxious stimuli1,2,3.
Numerous strategies have been proposed for the detection of cell death early after onset of ischemia within the time window amenable to intervention. Most experience for the recognition of apoptosis has been obtained with single photon emission computed tomography (SPECT) imaging using 99mTc-Annexin A5 (AA5). 99mTc-AA5 targets externalized phosphatidylserine (PS) on membranes of cells with active apoptotic signaling4 and the clinical feasibility of 99mTc-AA5 imaging has been demonstrated in the setting of myocardial infarction5,6, transplant rejection7 and heart failure8. On the other hand, several radiotracers targeting necrotic cells through membrane disruption have been developed9,10,11. Notably, antimyosin antibody imaging has been successfully employed for the detection of myocardial necrosis associated with myocardial infarction (MI)9, myocarditis9, heart failure12 and cardiac allograft rejection12. However due to technical disadvantages, the necrosis-avid radiotracers have not become popular.
4-(N-(S-glutathionylacetyl)amino)phenylarsonous acid (GSAO) labelled with fluorophores and radionuclides has been used for targeting of cell death in culture, in tumor-bearing mice, and in mice with experimental brain trauma13,14,15. The principle behind GSAO cell death targeting is displayed in figure 1. GSAO is a tripeptide with a trivalent arsenic moiety. The arsenic group binds to dithiols, which are abundantly present in intracellular milieu and virtually absent from the extracellular space16. GSAO cannot reach its intracellular target molecules in intact cells, because it is not able to negotiate across the cell membrane17. Cell membrane γ-glutamyl transferase (GGT) is upregulated during ischemic stress and other situations, splices off GSAO's glutamyl residue and allows cellular entrance of GSAO's metabolite GCAO (4-(N-(S-cysteinylglycylacetyl)amino) phenylarsonous acid)17. However, when radioactive or fluorescent reporter molecules are attached to glutamyl residue of GSAO it is not able to bind to GGT, rendering the molecule membrane impermeable13. After the necrotic process sets in and the sarcolemmal integritiy is lost; and GSAO gains free entry to the intracellular microenvironment. Of numerous dithiol-bearing intracellular targets including filamin A, eukaryotic translation elongation factor 2, and protein disulfide isomerase (PDI), HSP90 is most widely present13. HSP90 comprises approximately 2% of the intracellular protein content and increases by two-threefold in response to acute stress including ischemia18.
Figure 1. Targeting mechanism of GSAO.

GSAO binds to dithiol molecules, which are abundant in the intracellular space and virtually absent in the extracellular space. When labelled with radioactive or fluorescent reporter molecules, GSAO cannot negotiate across the cell membrane of healthy cells (a) but gains free access through disrupted membranes of dying cells (b). Thus, GSAO accumulates in dying cells.
In this study we evaluated feasibility of cell death imaging using 111In-GSAO in mouse and rabbit models of acute MI and a mouse model of chronic MI. In addition, a subgroup of animals, serial SPECT/CT imaging using 99mTc-AA5 and 111In-GSAO was performed to determine the relationship between these two modes of cell death in the setting of myocardial ischemia and reperfusion. Fluorescent GSAO and AA5 were employed in another subgroup of animals for pathological characterization of the mode of cell death. In addition, 111In-GSCA (4-(N-(S-glutathionylacetyl)amino)benzoic acid), which is identical to 111In-GSAO except for replacement of the arsenic group with a carboxylic acid group, was used as a negative control compound in radionuclide experiments.
Results
Fluorescence microscopic characterization of GSAO uptake
Uptake characteristics of GSAO were evaluated and compared with AA5 in mice with acute MI and sham-operated mice. Experimental acute MI was induced by 30-minute coronary ligation followed by 30 minutes of reperfusion. Sham procedure was identical, except that coronary ligation was not performed. Mice with acute MI (n = 3) and sham-operated mice (n = 3) were injected Cy5.5-labelled GSAO and Oregon Green-labeled AA5. Qualitative fluorescent microscopic analysis of heart sections showed that cardiomyocytes of sham-operated mice were positive neither for GSAO nor for AA5. In contrast, in ischemia/reperfusion-injured hearts, cardiomyocytes positive for GSAO and AA5 were observed (Fig. 2a–c). Interestingly, all GSAO-positive cardiomyocytes bound AA5 (Fig. 2a–c), suggesting that necrosis in the reperfused myocardium coexists with apoptotic signaling. Not all AA5 positive cardiomyocytes had taken up GSAO (Fig. 2c).
Figure 2. Characterization of target binding of GSAO.

Tissue sections of acute MI mice, injected fluorescently labeled GSAO and AA5. (a, b) GSAO accumulation (red) was only observed in cells with AA5-positive cell membranes (green). (c) All cells AA5-positive cells were not GSAO positive.
Radiolabelled GSAO imaging in acute myocardial infarction
Acute MI was induced in rabbits and mice by coronary ligation for 40 and 30 minutes, respectively. Reperfusion was achieved by removal of the suture and 111In-GSAO was administered thirty minutes later. Three hours thereafter, in vivo micro-SPECT/micro-CT imaging was performed, animals were sacrificed and hearts were explanted. Next, ex vivo SPECT/CT and planar cardiac imaging were performed. Then, rabbit hearts were cut into ~32 small pieces and mice hearts were sectioned in three short axis slices: basal (remote area), middle (border zone) and apical (infarct). Radiotracer uptake was quantified by γ-counting, and myocardial pieces were histopathologically characterized.
In vivo SPECT/CT imaging in MI rabbits (n = 10) revealed intense uptake of 111In-GSAO in the apical area (Fig. 3a). Ex vivo SPECT/CT and planar imaging (Fig 3b) of explanted hearts confirmed intense apical radiotracer uptake. γ-counting confirmed that the 111In-GSAO uptake in the myocardial infarct was markedly higher than the 111In-GSAO uptake in the remote region (1.10 ± 0.45%ID/g vs 0.03 ± 0.01%ID/g, P = 0.005, Fig. 3c).
Figure 3. 111In-GSAO imaging, quantification and comparison with 99mTc-Annexin A5 and 99mTc-MIBI in rabbits with acute myocardial infarction.

(a) In vivo SPECT (left panels), and fused SPECT/CT images (right panels) in acute MI rabbits revealed intense 111In-GSAO uptake (top) and only modest uptake of radiotracer control compound 111In-GSCA (bottom). (b) Ex vivo planar images confirmed intense 111In-GSAO uptake (top) and modest 111In-GSCA (bottom) uptake. (c) γ-counting of myocardial sections confirmed 111In-GSAO uptake in infarct was higher than in remote area and higher than 111In-GSCA uptake in infarct. Although low, 111In-GSCA uptake in infarct was higher than remote. Difference Between 111In-GSAO and 99mTc-AA5 uptake did not reach significant uptake. Black horizontal lines denote Kruskal-Wallis ANOVAs + Bonferroni correction, and red lines denote Wilcoxon signed rank tests + Bonferroni correction. (d) Serial imaging revealed that 111In-GSAO uptake (top) was predominantly localized in the region of the 99mTc-MIBI perfusion defect (bottom). (e) γ-counting of myocardial sections showed high 111In-GSAO uptake in sections with low MIBI uptake and vice versa. (f) A significant inverse Spearman's correlation between uptake of 111In-GSAO and 99mTc-MIBI was observed. (g) Serial planar imaging in acute MI rabbit demonstrates similar uptake region and higher uptake of 111In-GSAO (top) when compared with 99mTc-AA5 (bottom). (h) γ-counting of myocardial sections showed high 111In-GSAO uptake in sections with high 99mTc-AA5 uptake and vice versa (i). A significant Spearman's correlation between uptake of 111In-GSAO and 99mTc-AA5 was observed.
To study the specificity of 111In-GSAO, the myocardial uptake of negative control compound GSCA was evaluated in acute MI rabbits (n = 5). In vivo (Fig. 3a) and ex vivo SPECT/CT imaging and planar imaging (Fig. 3b) revealed very low cardiac uptake of GSCA; only slightly increased infarct uptake was observed. This was confirmed by γ-counting; when compared with 111In-GSAO, 111In-GSCA uptake in the infarct area was significantly lower (0.07 ± 0.03%ID/g, P = 0.002, Fig 3c). Although low, GSCA uptake in the infarct area was higher than in the remote area (0.03 ± 0.01%ID/g, P = 0.043). This demonstrated that the trivalent arsenical group was necessary for targeting of dying cells. Moreover, myocardial 111In-GSAO uptake in an unmanipulated control animal was similar to 111In-GSAO uptake in the spared myocardium of the MI rabbits (0.01%ID/g vs 0.03 ± 0.01%ID/g), further supporting specificity of GSAO.
To evaluate localization of cardiac 111In-GSAO uptake, three of ten animals receiving 111In-GSAO also received the myocardial perfusion tracer 99mTc-sestamibi (MIBI), immediately after coronary ligation. Serial in vivo SPECT/CT imaging confirmed that 111In-GSAO uptake was localized in the infarct zone as identified by 99mTc-MIBI perfusion defect. Similarly, ex vivo serial SPECT/CT imaging and serial planar imaging (Fig. 3d) revealed uptake of 111In-GSAO in the infarct zone as shown by 99mTc-MIBI perfusion defect. In each animal, γ-Counting of 99mTc-MIBI and 111In-GSAO in myocardial sections confirmed increased uptake of 111In-GSAO in the perfusion defects as sections with low MIBI uptake showed increased 111In-GSAO uptake and vice versa (fig 3e). γ-counting of all myocardial sections from the animals receiving both 111In-GSAO and 99mTc-MIBI revealed an inverse correlation between 111In-GSAO uptake and 99mTc-MIBI uptake (ρ = −0.70, P < 0.01, Fig 3f).
Of ten animals receiving 111In-GSAO, three also received 99mTc-AA5. Serial in vivo SPECT/CT imaging with 111In-GSAO and 99mTc-AA5 revealed similar area of increased uptake; 111In-GSAO uptake was more intense than 99mTc-AA5 uptake. This was confirmed by ex vivo serial SPECT/CT imaging and ex vivo serial planar imaging (Fig. 3g). Quantitative tracer uptake paralleled the imaging data. In each animal, in sections with increased 99mTc-AA5 uptake, 111In-GSAO uptake was also increased (fig. 3h). Using all myocardial sections in all animals receiving 111In-GSAO and 99mTc-AA5, a strong correlation between uptake of the radiotracers was observed (ρ = 0.82, P < 0.01, Fig 3i). Like the fluorescence experiments, this supports that secondary necrosis after apoptotic signaling may be the dominant mode of cell death in myocardial infarction. The 111In-GSAO uptake in the infarct zone was higher than 99mTc-AA5 uptake, although the results did not reach statistical significance, due to the small sample size (1.10 ± 0.45%ID/g vs 0.47 ± 0.23%, P = 0.47). Uptake in the remote area was low for both tracers (0.03 ± 0.01%ID/g vs 0.04 ± 0.02%ID/g, P = NS).
After γ-counting myocardial pieces from infarct, border and remote zones were histologically characterized. Representative examples of the stainings are given in figure 4a. H&E staining revealed hypereosinophilic change and contraction band necrosis in the myocardial sections from the infarct area; the morphological changes were less frequent in the border zone. The remote area was morphologically normal. The rate of apoptosis as shown by terminal deoxyribonucleotide transferase TdT-mediated nick-end labeling (TUNEL) staining (%area positive, %AP) was higher in the infarct (2.83 ± 1.42%AP) and border zone (2.55 ± 1.84%AP) than in the remote zone (0.02 ± 0.0%AP, P < 0.001 for both). The %caspase positive area was higher in the infarct (1.04 ± 0.48%AP) than in the remote zone (0.46 ± 0.48%AP, P = 0.054). 111In-GSAO uptake in the myocardial sections showed a direct correlation with TUNEL (ρ: 0.743, P < 0.001, Fig. 4b) and Caspase-3 (ρ = 0.533, P < 0.001, Fig. 4c) stains, further supporting that necrosis as shown by GSAO is secondary after apoptotic signaling.
Figure 4. Histopathological characterization of 111In-GSAO uptake in rabbit heart after experimental acute myocardial infarction.

H&E staining demonstrates clear signs of tissue damage in infarct and border zone, whereas the remote zone is morphologically normal. TUNEL and Caspase-3 stains demonstrate substantial apoptosis in the infarct zone, whereas positive cells are rarely seen in the remote area. Significant Spearman's Rank correlations between 111In-GSAO uptake and apoptotic signaling as shown by TUNEL (b) and Caspase-3 (c) stains were observed.
The non-target organ distribution of 111In-GSAO and 111In-GSCA demonstrated kidney to be the major organ of radiation burden and urine major route of excretion; all other organs revealed minimum burden for both radiotracers (Supplementary Fig. 1a). Serial blood samples from six animals revealed bi-exponential blood clearance with an initial fast component T1/2α of 5.3 min followed by a slower component T1/2β of 9.7 h; the plateau phase was approached at 15 min (Supplementary Fig. 1b).
In vivo SPECT/CT imaging in mice after acute MI (n = 6) demonstrated high 111In-GSAO uptake; the use of CT allowed precise localization of the radioactivity in the infarcted region of the heart (Fig. 5a,b). Specific apical uptake was confirmed by ex vivo SPECT/CT and planar imaging (Fig. 5c). Imaging experiments revealed absence of specific 111In-GSAO uptake in control animals. 111In-GSAO uptake in the infarct area (2.56 ± 2.75%ID/g) was markedly higher than in remote myocardium (0.48 ± 0.22%ID/g, P = 0.028) and 20-fold higher than apical uptake in unmanipulated control animals (n = 5, 0.13 ± 0.02%ID/g; p = 0.004, Fig. 5d).
Figure 5. In vivo and ex vivo 111In-GSAO imaging and quantification in mice with acute myocardial infarction.
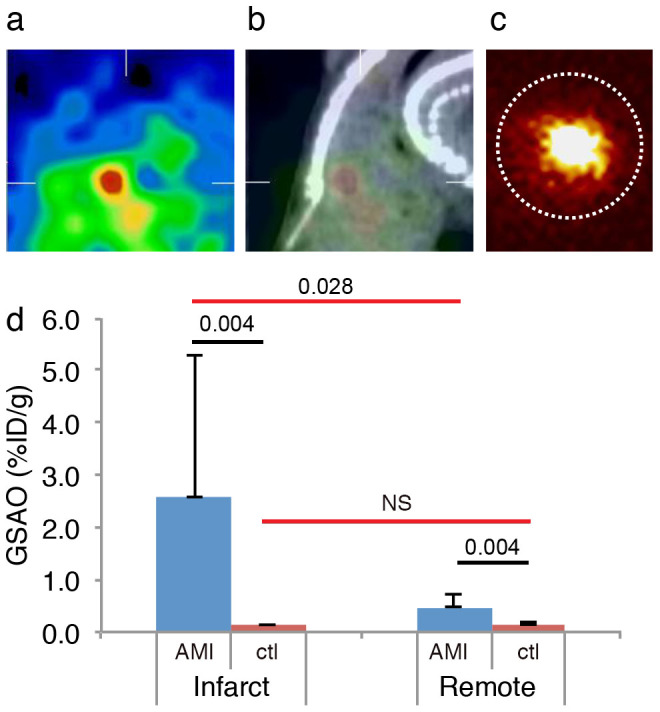
In vivo SPECT (a), and fused SPECT/CT images (b) in acute MI rabbits revealed intense cardiac 111In-GSAO uptake. (c) Ex vivo planar images confirmed intense 111In-GSAO uptake. (d) γ-counting of myocardial sections confirmed 111In-GSAO uptake in infarct (apex) was higher than manyfold higher than in remote area and higher than apical 111In-GSAO uptake in unmanipulated control animals. Black horizontal lines denote Mann-Whitney U tests and red lines denote Wilcoxon signed rank tests.
Radionuclide imaging in a post-MI HF model
111In-GSAO uptake was also evaluated in post-MI HF mouse model, in which the LCA territory was not reperfused. Subgroups underwent imaging experiments at different time points. Also, a disease control group consisting of animals that did not undergo infarction surgery before 111In-GSAO imaging, and a radiotracer control group consisting of animals receiving negative control compound 111In-GSCA imaging at 2 weeks post-MI were used.
111In-GSAO uptake in the HF mice was markedly lower than in AMI mice, and could not be detected by in vivo SPECT/CT imaging. Quantification of radionuclide uptake in short axis slices confirmed that infarct uptake of 111In-GSAO in mice at 2 weeks post-MI (n = 6) was higher than in the five disease control animals that did not undergo infarction surgery (0.42 ± 0.17%ID/g vs 0.13 ± 0.02%ID/g, P = 0.025, Fig 6); remote uptake was not statistically different (0.25 ± 0.12%ID/g vs 0.14 ± 0.04%ID/g, P = 1.0, Fig 6). Moreover, 111In-GSAO uptake at 2w post-MI was significantly higher than uptake of the negative control compound 111In-GSCA in the infarct and remote area (0.04 ± 0.02%ID/g, P < 0.001 and 0.04 ± 0.03%ID/g, P = 0.004, respectively).
Figure 6. Quantification of 111In-GSAO uptake in mice with chronic myocardial infarction.

Γ-counting revealed 111In-GSAO uptake in the infarct area at 2 weeks; uptake was significantly higher than 111In-GSAO uptake in unmanipulated control animals and 111In-GSCA uptake at 2w post-MI in controls. A trend of declining 111In-GSAO uptake over time was observed, but did not reach statistical significance. 111In-GSAO uptake in remote area was higher than 111In-GSCA uptake at 2w post-MI. Results of Kruskal-Wallis ANOVAs + Bonferroni correction are shown.
Moreover, when compared with uptake at 2 weeks post-MI, 111In-GSAO uptake showed a trend of decline at 4 weeks (n = 6, infarct 0.28 ± 0.09%ID/g, P = 1.0; remote 0.22 ± 0.04%ID/g, P = 1.0, Fig 6) and 12 weeks (n = 8, infarct: 0.19 ± 0.07%ID/g, P = 0.332; remote: 0.15% ± 0.1%ID/g, P = 1.0, Fig 6) although differences were not statistically significant.
Discussion
Here we demonstrate cell death imaging in acute myocardial infarction using 111In-GSAO. The specificity of the radiotracer was confirmed by the localization of 111In-GSAO in the infarct zone as shown by 99mTc-MIBI. This was further supported by the lack of uptake of fluorescently labeled GSAO in sham-operated animals outside of the regions directly damaged by the suture. The lack of GSCA uptake in our radionuclide studies confirmed that the trivalent arsenic group on GSAO is responsible for its targeting characteristics15,29,30. The radiotracer showed favorable pharmacokinetic profile with rapid blood clearance and low background uptake in most organs.
In the 1990's evidence of apoptotic signaling in myocardial infarction accumulated19,20,21,22, sparking the discussion over the relative importance of the apoptotic and necrotic forms of cell death. In 2000, 99mTc-AA5 SPECT in patients with acute myocardial infarction revealed intense uptake in the entire region of the perfusion defect5. This observation was provocative as 99mTc-AA5 was believed to identify apoptotic cell death but the infarct area was traditionally expected to be necrotic. It was subsequently proposed that the ischemic insult may be initiated as apoptosis but conclude with secondary necrosis. Ischemic loss of ATP production during myocardial infarction would preclude completion of the energy-dependent apoptosis program23. Restoration of blood flow by reperfusion may either interrupt the apoptotic process to allow cell salvage6,24 or resume the process of apoptosis in critically damaged cells20,22. The latter may even be augmented further by production of radical oxygen species25, or intracellular calcium overload26. These changes may also contribute to secondary necrosis by opening of the mitochondrial permeability transition pore27,28,29,30. It is conceivable that the apoptotic process initiated by noxious stimuli may not follow classical picture of physiologic apoptosis observed during normal turnover of skin or mucosal cells.
The feasibility of cell death imaging using fluorescently labeled and radiolabelled GSAO has recently been demonstrated in tumor-bearing mice and mice with experimental brain trauma13,14,15. GSAO accumulation was characterized by fluorescent microscopy in various cell models of apoptotic and in explanted brains and tumors after in vivo GSAO administration. GSAO was observed intracellularly and colocalized with propidium iodide and Sytox blue, standard markers of membrane disruption, which supported GSAO uptake in secondarily necrotic cells. Similarly, our fluorescent experiments showed that GSAO accumulated intracellularly and only occurred in AA5 + cells, thereby supported the notion that necrosis followed apoptotic signaling in the setting of acute myocardial infarction.
In vivo and ex vivo 111In-GSAO imaging in rabbits and mice with acute myocardial infarction showed intense dual serial imaging with 99mTc-AA5 and 111In-GSAO in rabbits after acute myocardial ischemia and reperfusion revealed the same area of uptake. At a segment level, 111In-GSAO uptake showed the same pattern as 99mTc-AA5 and a strong correlation between uptakes of the two tracers was shown. In addition, 111In-GSAO correlated with presence of Caspase-3 and TUNEL staining, classic markers of apoptosis. This shows that not only does secondary necrosis after apoptotic signaling occur in myocardial ischemia and reperfusion, it may play a dominant role. As in rabbits, high 111In-GSAO uptake was seen in mice with acute myocardial infarction. However, 111In-GSAO uptake in post-myocardial infarction HF model in mice was too low to be detected by micro-SPECT/CT imaging. The most important explanation for this is the lower rate of necrotic cell death in heart failure31,32,33. It is tempting to speculate that the more benign circumstances such as lesser energy depletion and ROS cause lower transition from apoptosis to necrosis in this setting. In fact, the apoptotic process may remain suspended in chronic heart failure34.
A number of imaging tracers for myocardial necrosis have been clinically evaluated. Like 111In-GSAO, most tracers exploited membrane disruption, the hallmark of necrosis as a target. Tc-99 labelled antimyosin antibody was the most widely studied necrosis tracer. Antimyosin antibody imaging has been successfully employed for the detection of myocardial necrosis associated with myocardial infarction9, myocarditis9, heart failure12 and cardiac allograft rejection12. However, because of long circulation time of the radiolabelled antibody, imaging was not feasible for up to 6–12 hours after administration of the agent. 99mTc-pyrophosphate on the other hand, showed maximal myocardial uptake at 24–72 hours after injection, although necrosis imaging at 3 hours post-administration was feasible, maximum myocardial uptake was at 24–72 h. Also, this tracer required residual blood flow, which precluded uptake in the infarct center35. 99mTc-glucarate can be used to necrotic cells in myocardial infarction11,36, and has good imaging characteristics but in the setting of myocardial infarction, use is limited to a clinical window of >9 hours after onset as its target, histone bodies, quickly wash out of the tissue11. Late gadolinium-enhanced MRI has also been used to assess cardiac cell death; the contrast medium accumulates in necrotic cells through the disrupted membranes. However, its specificity is limited, as in all instances of increased extracellular space such as cardiac edema, fibrosis, sarcoidosis and amyloidosis37,38,39. 111In-GSAO appears to have advantages over the previously evaluated cell death imaging techniques. First, 111In-GSAO requires membrane disruption, the hallmark of necrotic or late-apoptotic cell death, for reaching its intracellular targets. Therefore it does not suffer from the lack of specificity of late-gadolinium enhanced MRI. Moreover, its rapid blood clearance results in feasibility of early imaging of cell death using 111In-GSAO. This gives 111In-GSAO an advantage over 99mTc-antimyosin and 99mTc-pyrophosphate. HSP90, the main target of GSAO, functions in complexes. This may allow for targeting over a longer time period, giving 111In-GSAO an edge over 99mTc-glucarate. However, this has to be evaluated in follow-up studies.
Micro-SPECT/CT imaging using 111In-GSAO can be used to visualize necrotic cell death in acute myocardial infarction. The uptake in the HF model was too low to allow imaging. Because the GSAO uptake reflects membrane permeabilization, and occurs predominantly in AA5 positive cells, we propose that secondary necrosis is a dominant mode of cell death in the setting of myocardial ischemia and reperfusion.
Methods
Materials
Cy5.5 GSAO and DTPA-GSAO were prescribed as described previously13 and were a kind gift of Covidien Imaging Solutions (Hazelwood, Missouri, United States). DTPA-GSAO and DTPA-GSCA were labeled with In-111 as described previously13. The other used materials were of standard analytical grade.
Ethical statement
The experimental protocols followed the Guidelines for the Care and Use of Laboratory Animals established by the National Institutes of Health (NIH Publication No. 85–23, revised 1996) and was approved by the Institutional Laboratory Animal Care and Use Committees at the University of California, Irvine and University of Maastricht, Maastricht, The Netherlands.
Experimental myocardial infarction in mice
For fluorescence experiments, C57Bl6/j (Age: 3 months, weight: ~50 g, Jackson Laboratories, Sacramento, CA) and for radionuclide studies Swiss-Webster mice (Age: 3 months, weight ~50 g, Charles River, Wilmington, MA) were used. MI was induced under isoflurane anesthesia (2–3%) using a stereomicroscope (Leica MZ FL III, Leica, Switzerland) as described previously40. Animals were placed on a heating pad in the supine position, intubated under direct laryngoscopy, and mechanically ventilated using a small animal respirator (tidal volume, 1.0 ml; rate, 120 breaths/min, Harvard Apparatus, Holliston, MA). After a minimum thoracotomy, the anterior descending branch of the left coronary artery was ligated with a 6.0-silk suture 3 to 4 mm below the tip of the left atrium. Successful ligation was verified by visual inspection of the LV apex for myocardial blanching, indicating interruption in coronary flow. For induction of acute MI experiments, the suture was removed after 30 minutes to induce reperfusion. The sham operation was identical, but the coronary artery was not ligated. Acute MI and sham animals remained under anesthesia for ensuing radionuclide imaging or fluorescence experiments. For induction of chronic MI, the ligation was not released. In these animals, the chest cavity was closed in layers with 6.0-silk and the skin was closed with 4.0-silk sutures. Thereafter, animals were gradually weaned from the respirator and put back into their cages until imaging procedure.
Experimental acute myocardial infarction in rabbits
Acute experimental MI was induced by the occlusion of left anterior descending coronary artery (LAD) in New Zealand White male rabbits (weight, 3.0–3.5 kg) as described previously24,41. All rabbits were anesthetized with a mixture of ketamine and xylazine (100 mg/ml, 10:1 vol/vol; 2.0 to 3.0 ml subcutaneously). Surgical tracheostomy was performed, and ventilation was maintained with a volume-cycled rodent respirator (Harvard Apparatus, Holliston, MA) provided positive pressure ventilation at 50 mL/cycle and a respiratory rate of 50 cycles/min. After surgical procedure, anesthesia was maintained on 3–4% isoflurane Briefly, the heart was exposed through parasternal thoracotomy, and the pericardium was fenestrated. The LAD was identified and a monofilament suture was placed at the site. The LAD was occluded by tightening the snare created by passing suture through a polyethylene tube. The snare was removed after 40 minutes of occlusion to induce reperfusion. Lead II or III of the electrocardiogram was continuously monitored during the experiments to confirm myocardial ischemia. Animals remained anesthetized for ensuing radionuclide imaging experiments.
Fluorescence experiments in mice
Acute MI mice and sham-operated mice were injected with Cy5.5-labelled GSAO at 5 minutes before ischemia and 1 hour before sacrifice, respectively. All mice received Oregon Green-labeled AA5 10 minutes before sacrifice. Of each fluorescent probe a dosage of 2.5 mg/kg was used. Acute MI animals were sacrificed after 30 m reperfusion, sham mice at 1 hour after sacrifice hearts were snap-frozen in liquid nitrogen, 7 μm thick frozen sections were obtained, dried and mounted in 4,6-diamino-2-phenylindole (DAPI) containing medium and examined with a confocal scanning laser microscope (Bio-Rad) equipped with a krypton/argon mixed gas laser (Ion Laser Technology).
In vivo and ex vivo imaging protocols mice and rabbits
For mice and rabbits, the same imaging procedure was followed. In vivo SPECT imaging was performed at 3 hours after radiotracer administration using a dual-head micro-SPECT γ-camera combined with micro-CT (X-SPECT, Gamma Medica, Inc., Northridge, CA) under isoflurane anesthesia. SPECT images of the heart were acquired in a 64 × 64 matrix at 32 steps at 60 seconds per step with a 247 keV photo peak of 111In with 15% windows using a medium energy parallel-hole collimator. In the animals also undergoing 99mTc-MIBI 99mTc-AA5, this was followed by imaging using the same protocol, at a 140 keV photo peak of 99mTc with 15% windows using a low-energy, high-resolution parallel-hole collimator. Gating was not performed. After SPECT imaging acquisition, a CT scan was acquired using an X-ray tube operating at 50 kVp and 0.6 mA. Images were acquired for 2.5 seconds per view for 256 views in 360° rotation. After transferring to 256 × 256 matrix, the SPECT images and CT studies were fused. After imaging, anesthetized animals were terminated by heart explantation and ex vivo SPECT/CT excised heart was performed using the same protocols but at 45 seconds per step for SPECT. Thereafter, planar 111In-GSAO imaging of the hearts was performed for 15 minutes, followed by 99mTc-MIBI or 99mTc-AA5 planar imaging in GSAO-MIBI and GSAO-AA5 animals. After ex vivo imaging, rabbit hearts were cut into 4 short axis slices and further divided into 31-32 pieces. Mice hearts were cut in 3 short-axis slices. All sections were weighed, and γ-counted in an automatic well-type γ-counter (Perkin Elmer Wallac Inc., Gaithersburg, MD) for calculation of the percent total injected dose per tissue weight (%ID/g) uptake. Tissue samples of the main organs were used for calculation of the %ID/g uptake to evaluate the bio distribution. To correct for the radioactive decay and permit calculation of the concentration of radioactivity as a fraction of the administered dose, aliquots of the injected dose were counted simultaneously. For evaluation of infarct 111In-GSAO uptake in rabbits, the section with the highest 111In-GSAO uptake in the infarct area was used of each animal, and for evaluation of remote 111In-GSAO uptake, the section with the lowest 111In-GSAO uptake in the remote area was selected. In all mice, the apical slice was used for evaluation of infarct uptake, the middle slice for uptake in the border zone and the basal slice for remote uptake.
Pharmacokinetic studies
The pharmacokinetic parameters were derived using the KINFIT module of the MW/PHARM computer program package (Version 3.60, MediWare, Groningen, The Netherlands)42. The data, consisting of the blood concentration of 111In radioactivity or GSAO versus time, were analyzed by non-linear regression analysis using a least-squares weighted simplex algorithm, with data weighted with the reciprocal of the observed value.
Histological, immunohistochemical evaluation
After γ-counting of 111In-GSAO uptake, myocardial pieces of seven rabbits were processed for histopathologic characterization. Of each animal, two pieces from each region (infarct, border zone, remote) were selected.
The myocardial pieces were fixed overnight 4% paraformaldehyde in PBS (pH 7.4 at 4°C), and stored in PBS with 0.02% sodium azide at 4°C until used. The specimens were further processed by dehydration in a graded series of ethanol for paraffin-embedding. The blocks were cut in 5-μm thick sections, floated on a water bath containing deionized water (43°C), and transferred to vectabond (Vector Laboratories Burlingame, California) reagent-treated slides (Vector SP-1800, Vector Laboratories), dried overnight, and stored until ready for use. Sections were deparaffinised by heating (25 minutes at 56°C) and dehydration using xylene and graded series of ethanol. Tissue sections were stained with standard haematoxylin & eosin and Masson's trichrome staining. For immunohistochemical characterization, adjacent sections were incubated with primary antibodies against Caspase-3. After washing with PBS, sections were incubated with a biotinylated secondary antibody. The presence of apoptotic cells was further evaluated using TUNEL staining as described previously. Briefly: exposed DNA fragments were labeled with biotinylated nucleotides (dNTPs) and TdT for 1 h at 37°C after blocking of endogenous perosidase activity using 0.3%hydrogen peroxide and incubation with proteinase K41. For color reactions, sections were incubated with diaminobenzidine. For the assessment of the immunopositive area, stained tissue sections were observed under appropriate magnification (Carl Zeiss, Thornwood, New York), and the images were captured with a high-resolution digital camera (Axiocam, 1,300 × 1,030 pixels, Carl Zeiss) using Axiovision 3.1 software. Digital images were analyzed using Image-Pro Plus 5.0 (Media Cybernetics, Bethesda, Maryland).
Statistical analysis
All results are presented as the mean ± SD. In most cases, the data did not meet the assumptions of parametric tests. For consistency, non-parametric tests were used for all comparisons. Related samples were compared using Wilcoxon Signed Rank test (two groups) or Friedman's Two Way ANOVA followed by post-hoc Wilcoxon Signed-Rank tests and Bonferroni correction for pair-wise significance (more than two groups). Unrelated samples were compared using Mann Whitney U tests (two groups) or Kruskal-Wallis ANOVA followed by Mann-Whitney U tests and Bonferroni Correction for pair-wise significance (more than two groups). When the Kruskal-Wallis test revealed no significant differences among groups, no post-hoc tests were performed and P = NS was reported. Bonferroni correction was also applied to results of multiple Wilcoxon signed rank tests regarding the uptake of radiotracers in infarct vs. remote areas in rabbits (fig 3c). To assess the correlation between 111In-GSAO uptake and 99mTc-AA5 uptake, 99mTc-MIBI uptake and histologic findings (caspase-3 and TUNEL), Spearman's ρs were calculated. P values of <0.05 were considered statistically significant.
Author Contributions
J.N. conceived the study; R.P. developed and produced the targeting agent; M.D. developed the radiolabeling protocols; C.R. conceived the mouse fluorescence experiments; H.Z. conducted the mouse fluorescence experiments; H.Z. and A.P. conducted the mouse imaging experiments; N.T., A.P., T.Y. and T.S. conducted rabbit experiments; J.Z. conducted and analyzed the immunohistochemistry; N.T., H.H., A.P., J.N. performed data analysis; N.T. and H.H. performed statistical analysis; N.T., H.H. and J.N. prepared figures; N.T., H.H. and J.N. wrote the manuscript; J.N. funded the project; H.Z., A.P., R.P., T.Y., J.Z., T.I., R.S., M.D., T.S., A.K., C.R., N.N. and V.F. helped in manuscript writing and editing.
Supplementary Material
Supplementary Figure 1
Acknowledgments
This research was supported by a research grant from the International Research Fund for Subsidy of Kyusyu University School of Medicine Alumni and the Banyu Fellowship Program sponsored by Banyu Life Science Foundation International to N. Tahara, and research grants from the foundation “De Drie Lichten” and the Dutch Heart Association (Dr. E. Dekker student Grant) to H. de Haas. DTPA-GSAO, DTPA-GSCA, Cy5.5 GSAO were supplied by Covidien Imaging Solutions. We thank H.H. Boersma for performing blood clearance excellent statistical support we thank E. Bagiella, supported by National Institutes of Health Grant UL1TR000067.
Footnotes
Dr. Dyszlewski is an employee of Covidien Imaging Solutions, Hazelwood, Missouri. Dr. Panduranghi was an employee of Covidien when the project was being conducted. The other authors claim no conflict of interest.
References
- Konstantinidis K., Whelan R. S. & Kitsis R. N. Mechanisms of cell death in heart disease. Arterioscler Thromb Vasc Biol 32, 1552–1562 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikoletopoulou V., Markaki M., Palikaras K. & Tavernarakis N. Crosstalk between apoptosis, necrosis and autophagy. Biochim Biophys Acta 1833, 3448–3459 (2013). [DOI] [PubMed] [Google Scholar]
- Nicotera P. & Melino G. Regulation of the apoptosis-necrosis switch. Oncogene 23, 2757–2765 (2004). [DOI] [PubMed] [Google Scholar]
- Boersma H. H. et al. Past, present, and future of annexin A5: from protein discovery to clinical applications. J Nucl Med 46, 2035–2050 (2005). [PubMed] [Google Scholar]
- Hofstra L. et al. Visualisation of cell death in vivo in patients with acute myocardial infarction. Lancet 356, 209–212 (2000). [DOI] [PubMed] [Google Scholar]
- Thimister P. W. et al. In vivo detection of cell death in the area at risk in acute myocardial infarction. J Nucl Med 44, 391–396 (2003). [PubMed] [Google Scholar]
- Narula J. et al. Annexin-V imaging for noninvasive detection of cardiac allograft rejection. Nat Med 7, 1347–1352 (2001). [DOI] [PubMed] [Google Scholar]
- Kietselaer B. L. et al. Noninvasive detection of programmed cell loss with 99mTc-labeled annexin A5 in heart failure. J Nucl Med 48, 562–567 (2007). [DOI] [PubMed] [Google Scholar]
- Khaw B. A. & Narula J. Non-invasive detection of myocyte necrosis in myocarditis and dilated cardiomyopathy with radiolabelled antimyosin. Eur Heart J 16 Suppl O, 119–123 (1995). [DOI] [PubMed] [Google Scholar]
- Corbett J. R. et al. 99mTc-pyrophosphate imaging in patients with acute myocardial infarction: comparison of planar imaging with single-photon tomography with and without blood pool overlay. Circulation 69, 1120–1128 (1984). [DOI] [PubMed] [Google Scholar]
- Mariani G. et al. Detection of acute myocardial infarction by 99mTc-labeled D-glucaric acid imaging in patients with acute chest pain. J Nucl Med 40, 1832–1839 (1999). [PubMed] [Google Scholar]
- Narula J. et al. Antimyosin uptake and myofibrillarlysis in dilated cardiomyopathy. J Nucl Cardiol 2, 470–477 (1995). [DOI] [PubMed] [Google Scholar]
- Park D. et al. Noninvasive imaging of cell death using an Hsp90 ligand. J Am Chem Soc 133, 2832–2835 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park D. et al. Optical imaging of treatment-related tumor cell death using a heat shock protein-90 alkylator. Mol Pharm 10, 3882–3891 (2013). [DOI] [PubMed] [Google Scholar]
- Xie B. W. et al. Optical imaging of cell death in traumatic brain injury using a heat shock protein-90 alkylator. Cell Death Dis 4, e473 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donoghue N., Yam P. T., Jiang X. M. & Hogg P. J. Presence of closely spaced protein thiols on the surface of mammalian cells. Protein Sci 9, 2436–2445 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dilda P. J., Ramsay E. E., Corti A., Pompella A. & Hogg P. J. Metabolism of the tumor angiogenesis inhibitor 4-(N-(S-Glutathionylacetyl)amino)phenylarsonous acid. J Biol Chem 283, 35428–35434 (2008). [DOI] [PubMed] [Google Scholar]
- Goetz M. P., Toft D. O., Ames M. M. & Erlichman C. The Hsp90 chaperone complex as a novel target for cancer therapy. Ann Oncol 14, 1169–1176 (2003). [DOI] [PubMed] [Google Scholar]
- Scarabelli T. M. et al. Clinical implications of apoptosis in ischemic myocardium. Curr Probl Cardiol 31, 181–264 (2006). [DOI] [PubMed] [Google Scholar]
- Fliss H. & Gattinger D. Apoptosis in ischemic and reperfused rat myocardium. Circ Res 79, 949–956 (1996). [DOI] [PubMed] [Google Scholar]
- Freude B. et al. Apoptosis is initiated by myocardial ischemia and executed during reperfusion. J Mol Cell Cardiol 32, 197–208 (2000). [DOI] [PubMed] [Google Scholar]
- Gottlieb R. A., Burleson K. O., Kloner R. A., Babior B. M. & Engler R. L. Reperfusion injury induces apoptosis in rabbit cardiomyocytes. J Clin Invest 94, 1621–1628 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narula J. & Strauss H. W. Invited commentary: P.S.* I love you: implications of phosphatidyl serine (PS) reversal in acute ischemic syndromes. J Nucl Med 44, 397–399 (2003). [PubMed] [Google Scholar]
- Kenis H. et al. Annexin A5 uptake in ischemic myocardium: demonstration of reversible phosphatidylserine externalization and feasibility of radionuclide imaging. J Nucl Med 51, 259–267 (2010). [DOI] [PubMed] [Google Scholar]
- Li C. & Jackson R. M. Reactive species mechanisms of cellular hypoxia-reoxygenation injury. Am J Physiol Cell Physiol 282, C227–241 (2002). [DOI] [PubMed] [Google Scholar]
- Orrenius S., Zhivotovsky B. & Nicotera P. Regulation of cell death: the calcium-apoptosis link. Nat Rev Mol Cell Biol 4, 552–565 (2003). [DOI] [PubMed] [Google Scholar]
- Halestrap A. P. Calcium, mitochondria and reperfusion injury: a pore way to die. Biochem Soc Trans 34, 232–237 (2006). [DOI] [PubMed] [Google Scholar]
- Halestrap A. Biochemistry: a pore way to die. Nature 434, 578–579 (2005). [DOI] [PubMed] [Google Scholar]
- Halestrap A. P., Gillespie J. P., O'Toole A. & Doran E. Mitochondria and cell death: a pore way to die? Symp Soc Exp Biol 52, 65–80 (2000). [PubMed] [Google Scholar]
- Arbustini E. & Narula J. Cyclosporin a in reperfusion injury: not opening to cell death knocking at the door? Ann Thorac Surg 89, 1349–1351 (2010). [DOI] [PubMed] [Google Scholar]
- Yue T. L. et al. Possible involvement of stress-activated protein kinase signaling pathway and Fas receptor expression in prevention of ischemia/reperfusion-induced cardiomyocyte apoptosis by carvedilol. Circ Res 82, 166–174 (1998). [DOI] [PubMed] [Google Scholar]
- Condorelli G. et al. Increased cardiomyocyte apoptosis and changes in proapoptotic and antiapoptotic genes bax and bcl-2 during left ventricular adaptations to chronic pressure overload in the rat. Circulation 99, 3071–3078 (1999). [DOI] [PubMed] [Google Scholar]
- Li Z., Bing O. H., Long X., Robinson K. G. & Lakatta E. G. Increased cardiomyocyte apoptosis during the transition to heart failure in the spontaneously hypertensive rat. Am J Physiol 272, H2313–2319 (1997). [DOI] [PubMed] [Google Scholar]
- Narula J., Haider N., Arbustini E. & Chandrashekhar Y. Mechanisms of disease: apoptosis in heart failure--seeing hope in death. Nat Clin Pract Cardiovasc Med 3, 681–688 (2006). [DOI] [PubMed] [Google Scholar]
- De Saint-Hubert M., Prinsen K., Mortelmans L., Verbruggen A. & Mottaghy F. M. Molecular imaging of cell death. Methods (San Diego, Calif.) 48, 178–187 (2009). [DOI] [PubMed] [Google Scholar]
- Narula J., Petrov A., Pak K. Y., Lister B. C. & Khaw B. A. Very early noninvasive detection of acute experimental nonreperfused myocardial infarction with 99mTc-labeled glucarate. Circulation 95, 1577–1584 (1997). [DOI] [PubMed] [Google Scholar]
- Perugini E. et al. Non-invasive evaluation of the myocardial substrate of cardiac amyloidosis by gadolinium cardiac magnetic resonance. Heart 92, 343–349 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada T. et al. Diagnosis of cardiac sarcoidosis and evaluation of the effects of steroid therapy by gadolinium-DTPA-enhanced magnetic resonance imaging. Am J Med 110, 520–527 (2001). [DOI] [PubMed] [Google Scholar]
- de Haas H. J., Arbustini E., Fuster V., Kramer C. M. & Narula J. Molecular imaging of the cardiac extracellular matrix. Circ Res 114, 903–915 (2014). [DOI] [PubMed] [Google Scholar]
- van den Borne S. W. et al. Molecular imaging of interstitial alterations in remodeling myocardium after myocardial infarction. J Am Coll Cardiol 52, 2017–2028 (2008). [DOI] [PubMed] [Google Scholar]
- Kolodgie F. D. et al. Targeting of apoptotic macrophages and experimental atheroma with radiolabeled annexin V: a technique with potential for noninvasive imaging of vulnerable plaque. Circulation 108, 3134–3139 (2003). [DOI] [PubMed] [Google Scholar]
- Proost J. H. & Meijer D. K. MW/Pharm, an integrated software package for drug dosage regimen calculation and therapeutic drug monitoring. Comput Biol Med 22, 155–163 (1992). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1