

Abstract
Leflunomide is a novel immunomodulatory drug prescribed for treating rheumatoid arthritis. It inhibits the activity of protein tyrosine kinases and dihydroorotate dehydrogenase, a rate-limiting enzyme in the pyrimidine nucleotide synthesis pathway. Here, we report that A77 1726, the active metabolite of leflunomide, inhibited the phosphorylation of ribosomal protein S6 and two other substrates of S6K1, insulin receptor substrate-1 and carbamoyl phosphate synthetase 2, in an A375 melanoma cell line. A77 1726 increased the phosphorylation of AKT, p70 S6 (S6K1), ERK1/2, and MEK through the feedback activation of the IGF-1 receptor–mediated signaling pathway. Invitro kinase assay revealed that leflunomide and A77 1726 inhibited S6K1 activity with IC50 values of approximately 55 and 80 μM, respectively. Exogenous uridine partially blocked A77 1726–induced inhibition of A375 cell proliferation. S6K1 knockdown led to the inhibition of A375 cell proliferation but did not potentiate the antiproliferative effect of A77 1726. A77 1726 stimulated bromodeoxyuridine incorporation in A375 cells but arrested the cell cycle in the S phase, which was reversed by addition of exogenous uridine or by MAP kinase pathway inhibitors but not by rapamycin and LY294002 (a phosphoinositide 3-kinase inhibitor). These observations suggest that A77 1726 accelerates cell cycle entry into the S phase through MAP kinase activation and that pyrimidine nucleotide depletion halts the completion of the cell cycle. Our study identified a novel molecular target of A77 1726 and showed that the inhibition of S6K1 activity was in part responsible for its antiproliferative activity. Our study also provides a novel mechanistic insight into A77 1726–induced cell cycle arrest in the S phase.
Introduction
The phosphoinositide 3-kinase (PI3K) pathway is frequently activated in human cancers and plays essential roles in cell proliferation, apoptosis, protein synthesis, and metabolism. The PI3K pathway is activated through amplification or mutations of the genes encoding protein kinases or deletion of the tumor suppressor phosphatase and tensin homolog [1]. In recent years, extensive efforts in developing the inhibitors of the PI3K pathway as novel therapeutic agents to treat certain types of cancer in which the PI3K pathway is hyperactivated have been thwarted by unacceptable toxicity or poor pharmacokinetics [2], [3]. So far, only everolimus and temsirolimus, two rapamycin analogs that inhibit the mammalian target of rapamycin (mTOR), have been shown to be beneficial in several cancer types [2], [3].
Leflunomide (Arava) is an immunomodulatory drug for the treatment of rheumatoid arthritis. Early studies revealed that A77 1726 has two biochemical activities, the inhibition of tyrosine phosphorylation and inhibition of pyrimidine nucleotide synthesis [4], [5], [6], [7], [8], [9], [10], [11]. The ability of A77 1726 to inhibit the activity of dihydroorotate dehydrogenase (DHO-DHase), a rate-limiting enzyme in pyrimidine nucleotide synthesis, is about 10 to 100 times more potent than its ability to inhibit the activity of protein tyrosine kinases such as p56lck, p59fyn, and PDGF receptor [4], [5], [6], [7], [8]. The inhibition of pyrimidine nucleotide synthesis is thought to be the mechanism of action of leflunomide [12], [13]. White et al. [14] reported that leflunomide inhibits transcriptional elongation of the genes involved in self-renewal of neural progenitor cells through inhibition of DHO-DHase activity. These investigators further demonstrated that leflunomide at low doses cooperates with PLX4720, a B-Raf kinase inhibitor, to effectively inhibit melanoma cell proliferation and tumor growth [14]. Our early studies using a lymphadenopathy and autoimmune disease model in MRL/MpJ-lpr/lpr mice and a tumor xenograft model demonstrated that the immunosuppressive and antitumor activities of leflunomide are largely independent of the pyrimidine nucleotide synthesis pathway [4], [5] since uridine co-administration with leflunomide normalized pyrimidine nucleotide levels in tumor tissues but did not antagonize the antitumor activity of leflunomide in two xenograft models [5]. Those studies suggest that leflunomide may exert its antiproliferative and immunosuppressive activity [4], [5] independent of its inhibitory effect on pyrimidine nucleotide synthesis.
S6K1 is a member of serine/threonine protein kinases A, G, and C family, including AKT and mTOR. S6K1 is one of the predominant effectors of the mTOR complex 1 (mTORC1; Figure 7) [15]. The mTORC1-S6K1 pathway plays an important role in regulating protein synthesis, cell growth, metabolism, and aging [15]. S6K1 is overexpressed or activated in primary liver neoplasms, ovarian cancers, and many other types of malignancy due to the gene mutations in the PI3K pathway [15], [16]. S6K1 gene amplification occurs in 10% of breast cancers and is associated with a poor prognosis [17]. S6K1 serves as a biomarker to predict breast cancer in response to rapamycin [18]. Two recent studies demonstrated that S6K1 phosphorylates carbamoyl phosphate synthetase 2 (CAD), a rate-limiting enzyme involved in pyrimidine nucleotide synthesis, and stimulates its enzymatic activities [19], [20]. There have been considerable efforts in search for the specific inhibitors to target this important player in the mTORC1-S6K1 pathway. Numerous small molecule compounds that inhibit S6K1 alone or both S6K1 and AKT are at the early stage of clinical trials for anticancer therapy [15]. Here, we report that leflunomide and its active metabolite, A77 1726, are the inhibitors of S6K1 and that the inhibition of S6K1 activity contributes to its antiproliferative effect on A375 tumor cells.
Figure 7.
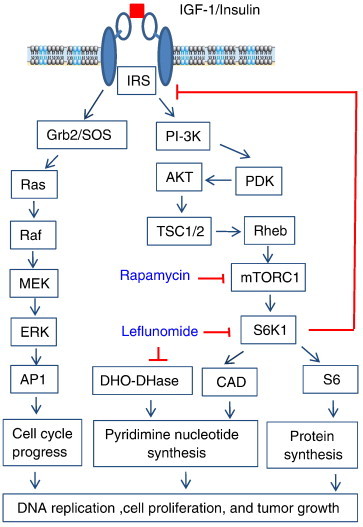
Mechanisms of action of A77 1726 on cell cycle progress and proliferation. A77 1726 inhibits the activity of S6K1, leading to the feedback activation of the PI3K and MAP kinase pathways through IGF-1 receptor. MAP kinase activation accelerates the entry of cell cycle into the S phase. However, due to the depletion of pyrimidine nucleotide pools through the inhibition of DHO-DHase and CAD activity, DNA replication and chromosome duplication cannot be completed, leading to the stall of cell cycle in the S phase. A77 1726 suppresses cell proliferation by inhibiting DNA and protein synthesis. In some types of cancer, A77 1726 may also inhibit cell proliferation by inhibiting the activity of protein tyrosine kinases.
Experimental Procedures
Reagents
Leflunomide and A77 1726 were kindly provided by Cinkate Corporation (Oak Park, IL). PPP was purchased from Santa Cruz Biotechnology, Inc (Santa Cruz, CA). U0126, LY294002, and rapamycin were purchased from Cell Signaling Technology (Danvers, MA). OSI-906 was purchased from Selleckchem.com (Houston, TX). Antibodies against ERK1/2, MEK1/2, Raf-1, p90 RSK, GSKα/β, 4E-BP, PDK1, AKT, mTOR, S6K1, and S6 and their corresponding phosphor antibodies including ERK1/2T202/Y204, MEK1/2S217/S221, Raf-1S338, p90 RSKT356/S360, GSKα/βS21/9, 4E-BPT37/46, PDKS241, AKTS473, AKTT308, mTORS2448, S6K1T389, S6S235/236 insulin receptor substrate-1S1101 (IRS-1S1101), and CADS1859 were purchased from Cell Signaling Technology.
Cell Lines
A375 cells are a melanoma cell line with BRAFV600E mutation and wild-type phosphatase and tensin homolog/PI3KC and p53. MCF-7 cells are an estrogen-positive breast cancer cell line with PI3KC mutation but with wild-type p53. BT-20 cells are a breast cancer cell line with PI3KC mutations (P539R and H1047R) and p53 (K132Q) mutation. A375 and BT-20 cells were cultured in complete Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% FBS, streptomycin (100 μg/ml) and penicillin (100 units/ml), and l-glutamine (2 mM). MCF-7 cells were grown in the complete MEM medium supplemented with 10% FBS, streptomycin and penicillin, l-glutamine, non-essential amino acids (1 ×), Hepes buffer (10 mM), and sodium pyruvate (1 mM). All three cell lines were purchased from American Tissue Culture Collection (Manassas, VA).
Western Blot
Cells seeded in six-well plates were harvested and lysed in NP-40 lysis buffer [50 mM Tris-HCl (pH 8.0), 150 mM NaCl, 1% NP-40, 5 mM EDTA, 10 μg/ml aprotinin, 10 μg/ml leupeptin, and 1 mM PMSF]. After incubation on ice for 30 minutes, the cell lysates were prepared by spinning down at 4°C, 15,000 rpm for 15 minutes. After electrophoresis and transfer to Immobilon membrane, proteins of interest were probed with their specific antibodies, followed by HRP-conjugated goat anti-rabbit or anti-mouse IgG and SuperSignal Western Pico enhanced chemiluminescence substrate (Pierce Chemical Co, Rockford, IL).
In Vitro p70 S6 Kinase Assay
The ability of leflunomide and A77 1726 to inhibit p70 S6 kinase assay was conducted by using an ADP-Glo Kinase assay system (Promega Corporation, Madison, WI). Briefly, A77 1726 or leflunomide diluted in the kinase buffer was mixed with recombinant p70 S6 kinase (100 ng per reaction) and incubated at room temperature for 30 minutes. Peptide substrate of p70 S6 kinase (5 μg per reaction) and ATP (10 μM, final concentration) was added, with a total final volume of 25 μl. After incubation for 1 hour, ADP-Glo reagent (25 μl) was added to each reaction. After incubation for 40 minutes, kinase detection substrate (luciferin; 50 μl per reaction) was added. After incubation for 30 minutes, luciferase activity was measured by reading in a luminescence plate reader. The experiment was conducted in triplicate and repeated once with similar results. The data from one experiment were presented as mean ± SD.
S6K1 Knockdown
S6K1 siRNA ON-TARGETplus SMARTpool was synthesized by Dharmacon (Lafayette, CO) and purchased from Fisher Scientific (Pittsburg, PA). This S6K1 siRNA pool containing three different siRNAs has been previously shown to efficiently suppress S6K1 expression [21], [22]. A scrambled control siRNA was purchased from Invitrogen Life Technologies (Grand Island, NY). A375 cells seeded in a six-well plate were transfected with siRNA using Lipofectamine RNAiMAX (Invitrogen Life Technologies) according to the manufacturer’s instruction. After incubation for 48 hours, the cells were harvested and analyzed for S6K1 expression and for the phosphorylation of S6K1, AKT, S6, and CAD by Western blot.
Cell Proliferation Assay
A375 cells were seeded in 96-well plates at a density of 2000 per well in the absence or presence of indicated concentrations of A77 1726, PLX4720, or uridine (200 μM). After incubation for 72 hours, cell proliferation was monitored by using an ATP-based Cell-Glo assay (Promega Corporation) following the manufacturer’s instruction.
DNA Replication and Cell Cycle Analysis
Upon 60% confluence, A375 cells were treated with vehicle or with indicated concentrations of A77 1726 or indicated inhibitors for 24 hours. Cells were pulsed with 10 μM bromodeoxyuridine (BrdU) for 2 hours. Cells were harvested and denatured with 2 N HCl for 5 minutes at room temperature followed by neutralization with 0.1 M borate buffer (pH 8.5). After washing and blocking with normal mouse serum, the cells were stained with an Alexa Fluor 488–conjugated anti-BrdU monoclonal antibody (BD Biosciences, San Jose, CA), followed by analysis in a Becton Dickson FACScan flow cytometer. Alexa Fluor 488–conjugated mouse IgG was included as a control. For cell cycle analysis, the cells were harvested and fixed in 2 ml of cold 70% ethanol in phosphate-buffered saline (PBS) overnight at 4°C. Fixed cells were then washed three times with PBS and treated with RNaseA (100 μg/ml in 0.5 ml of PBS). After incubation at room temperature for 30 minutes, cells were stained with 2.5 μl of propidium iodide (10 mg/ml) and immediately analyzed for DNA content in a Becton Dickson FACScan flow cytometer.
Statistical Analysis
The differences in A375 cell proliferation between different treatment groups were statistically analyzed by using an unpaired Student's t test. A P value of < .05 was considered statistically significant. All statistics was performed with SigmaPlot 11 software (Systat Software Inc., Richmond, CA).
Results
Feedback Activation of the PI3K and MAP Kinase Pathways by A77 1726
The PI3K and MAP kinase pathways play important roles in cell proliferation, differentiation, and cell cycle progress. Several genes in these two pathways are frequently mutated and have a commanding role in driving tumorigenesis and tumor cell proliferation [2], [3]. We first tested whether A77 1726 affected the MAP kinase and PI3K pathways in an A375 melanoma cell line. A375 cells grown in six-well plates were starved in the media containing 0.5% FBS for 2 hours and then treated with the indicated concentrations of A77 1726 for another 2 hours. A77 1726 strongly induced ERK1/2T202/204 and MEKS217/S221 phosphorylation (Figure 1A) but had no effect on Raf-1 phosphorylation (data not shown) in A375 cells. Of note, A77 1726 at 50 μM always induced MEK phosphorylation more effectively than ERK phosphorylation in multiple experiments. Induction of ERK1/2 phosphorylation was similarly achieved in A77 1726–treated A375 cells cultured in the medium containing 10% FBS (data not shown). A77 1726 strongly induced phosphorylation of AKTS473 and S6K1T389 in a dose-dependent manner but had no or only minimal effect on phosphorylation of PDK1S241, mTORS244, as well as GSKα/βS21/9, p90 RSKT353/356, and 4E-BPT37/46 (data not shown). In contrast, A77 1726 inhibited the phosphorylation of ribosomal protein S6S235/S236 in a dose-dependent manner (Figure 1A). Recent studies have shown that CAD, a rate-limiting enzyme involved in pyrimidine nucleotide synthesis, is phosphorylated by S6K1 at Ser-1859 [19], [20]. As shown in Figure 1A, A77 1726 inhibited CAD phosphorylation in a dose-dependent manner. In addition, S6K1 phosphorylates IRS-1 at Ser-1101 and suppresses IGF-1 receptor–mediated activation of the PI3K pathway [23]. A77 1726 inhibited IRS-1S1101 in a dose-dependent manner (Figure 1A). Inhibition of S6S235/236 phosphorylation was also observed in BT-20 and MCF-7 cells (data not shown). Similar results were obtained with leflunomide (data not shown). The stimulatory effect on AKTS473 and S6K1T389 phosphorylation and the inhibitory effect of A77 1726 on S6S235/236 phosphorylation were very rapid and long-lasting, within a few minutes after exposure to A77 1726 and lasted for up to 48 hours (Figure 1B).
Figure 1.
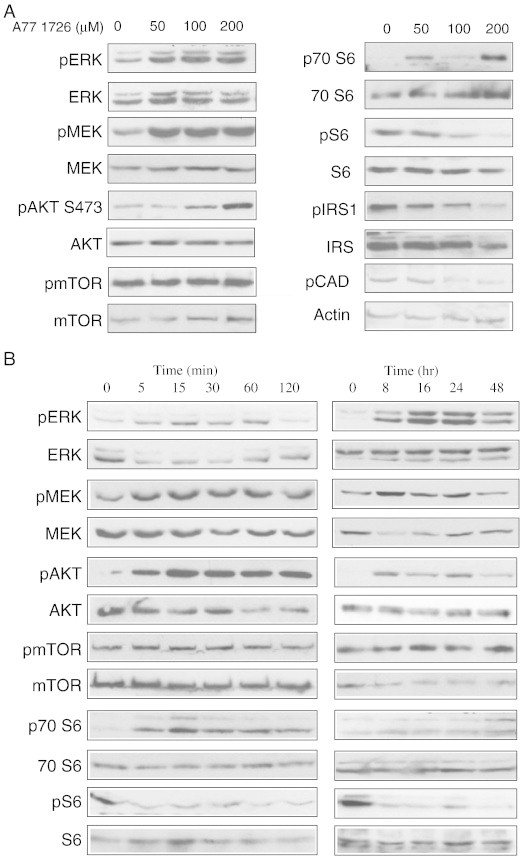
Effect of A77 1726 on the PI3K and MAP kinase pathways. A375 cells seeded in a six-well plate were starved in DMEM containing 0.5% FBS for 2 hours and treated with the indicated concentration of A77 1726 for another 2 hours (A) or treated with A77 1726 (100 μM) for the indicated time (B). Cells were harvested and analyzed for protein phosphorylation by specific antibodies as indicated. Protein loading was monitored by stripping membrane and reprobing with antibodies against non-phosphorylated proteins.
Feedback Activation of the PI3K and MAP Kinase Pathways by A77 1726 Is Independent of Its Inhibitory Effect on Pyrimidine Nucleotide Synthesis
A77 1726 inhibits pyrimidine nucleotide synthesis [12]. To rule out the possibility that the activation of PI3K and MAP kinase pathways was due to pyrimidine nucleotide depletion, we tested whether exogenous uridine affected the phosphorylation of several signaling molecules in the MAP and PI3K pathways. As shown in Figure 2A, uridine (200 μM) did not block A77 1726–induced phosphorylation of ERK1/2T202/Y204, MEK1/2S217/S221, and AKTS473, suggesting that the effect of A77 1726 on the MAP and PI3K pathways is not mediated through its inhibitory effect on pyrimidine nucleotide synthesis.
Figure 2.
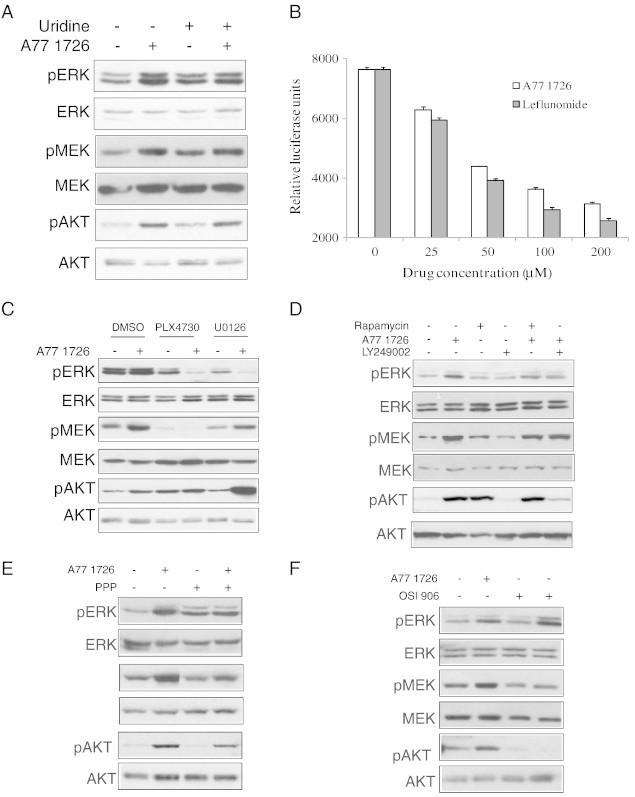
Mechanisms of A77 1726–induced feedback activation of the PI3K and MAP kinase pathways. (A) Inability of uridine to reverse the effect of A77 1726 on the PI3K and MAP kinase pathways. A375 cells seeded in a six-well plate were starved in DMEM containing 05% FBS for 2 hours and treated with the indicated concentration of A77 1726 in the absence or presence of uridine (200 μM) for 2 hours. Phosphorylated and total proteins were analyzed by Western blot as described in Figure 1. (B) In vitro S6K1 kinase assay. Leflunomide and A77 1726 diluted at the final concentrations as indicated were premixed with S6K1 for 30 minutes, followed by the addition of peptide substrate of S6K1 and incubation for 1 hour. S6K1 activity was measured by using an ADP-Glo system. The experiment was repeated with similar results. The data represent the mean ± SD from one experiment in triplicate. (C) The effect of the MAP kinase pathway inhibitors on A77 1726–induced feedback activation of the PI3K and MAP kinase pathways. A375 cells seeded in six-well plates were starved in DMEM containing 0.5% FBS for 2 hours and then pretreated with vehicle (0.1% DMSO) or the inhibitors of the MAP kinase pathway (PLX4720, 1 μM; U0126, 10 μM) for 1 hour. Cells were then treated with A77 1726 (100 μM) or rapamycin (20 nM) as indicated for 2 hours. Cells were harvested and analyzed for protein phosphorylation by specific antibodies as indicated. (D–F) The effect of PI3K, mTOR, and IGF-1 receptor inhibitors on A77 1726–induced feedback activation of the PI3K and MAP kinase pathways. A375 cells seeded in six-well plates were starved in DMEM containing 0.5% FBS for 2 hours and then pretreated with vehicle (0.1% DMSO), LY294002 (10 μM) or rapamycin (20 nM) (D), PPP (1 μM) (E), or OSI-906 (0.2 μM) (F) for 1 hour. Cells were then treated with A77 1726 (100 μM) as indicated for 2 hours. Cells were harvested and analyzed for protein phosphorylation by specific antibodies as indicated. Protein loading was monitored by stripping membrane and reprobing with antibodies against non-phosphorylated proteins.
Validating S6K1 as a Molecular Target of A77 1726
The ability of A77 1726 to inhibit S6, IRS-1, and CAD but to increase AKT and S6K1 phosphorylation strongly suggests that S6K1 is the molecular target of A77 1726. To prove this, we conducted an in vitro kinase assay to determine if leflunomide and A77 1276 directly inhibited S6K1 activity. Indeed, leflunomide and A77 1726 inhibited the activity of recombinant S6K1 in a dose-dependent manner, with IC50 values of approximately 55 and 80 μM, respectively (Figure 2B).
The Effect of the MAP Kinase Pathway Inhibitors on A77 1726–Induced MAP Kinase Pathway Feedback Activation
The inhibitors of the MAP kinase pathway were used to gain the mechanistic insight into how A77 1726 induced MAP kinase pathway activation in A375 cells. PLX4720 (a Raf kinase inhibitor) blocked A77 1726–induced phosphorylation of ERK1/2T202/Y204 and MEK1/2S217/S221. U0126 (a MEK inhibitor) blocked A77 1726–induced ERK1/2T202/Y204 phosphorylation but had no effect on MEK phosphorylation (Figure 2C). Interestingly, inhibition of the MAP kinase pathway by PLX4720 and U0126 enhanced AKTS473 phosphorylation in A77 1726–treated A375 cells.
The Effect of the PI3K Pathway and IGF-1 Receptor Inhibitors on A77 1726–Induced PI3K Pathway Feedback Activation
A prior study demonstrated that rapamycin-mediated feedback activation of the MAP kinase pathway is mediated through PI3K-induced Ras activation in MCF-7 cells [24]. Here, we tested whether PI3K was involved in A77 1726–induced MAP kinase pathway activation. LY294002, a PI3K inhibitor, had little effect on A77 1726–induced phosphorylation of MEK and ERK1/2 but largely blocked A77 1726–induced AKTS473 phosphorylation (Figure 2D). It is well established that S6K1 phosphorylates IRS-1S1101 and suppresses the IGF-1 receptor–mediated activation of the PI3K pathway (Figure 7) [2]. We tested whether inhibition of IGF-1 receptor tyrosine kinase activity led to the suppression of A77 1726–induced PI3K and MAP kinase pathway activation. As shown in Figure 2E, PPP, a specific inhibitor of IGF-1 receptor, had no effect on base levels of ERK1/2T202/Y204, MEK1/2S217/S221, and AKTT473 phosphorylation and largely blocked A77 1726–induced AKTS473 phosphorylation. OSI-906, a second IGF-1 receptor inhibitor, blocked A77 1726–induced AKTS473 and MEK1/2S217/S221 phosphorylation (Figure 2F). PPP and OSI-906 had no or minimal effect on A77 1726–induced ERK1/2T202/Y204 phosphorylation.
Antiproliferative Effect of A77 1726
We next tested whether the antiproliferative activity of A77 1726 on A375 cells was mediated by its inhibitory effect on pyrimidine nucleotide synthesis. A77 1726 at the concentration of 50, 100, and 200 μM inhibited the proliferation of A375 by 39%, 65%, and 88%, respectively (Figure 3A), with an IC50 value of approximately 65 μM. In the presence of exogenous uridine, A77 1726 at the concentration of 50, 100, and 200 μM inhibited the proliferation of A375 by 17%, 24%, 70%, respectively. Thus, uridine partially blocked the inhibitory effect of A77 1726 (Figure 3B). PLX4720 at 1 nM by itself had no effect on untreated or A77 1726–treated A375 cell proliferation (Figure 3A). PLX4720 (100 or 500 nM) by itself inhibited A375 proliferation (Figure 3A). When used in combination with A77 1726 in the absence or presence of uridine, PLX4720 had an additive effect on inhibiting the proliferation of A375 cells (Figure 3).
Figure 3.
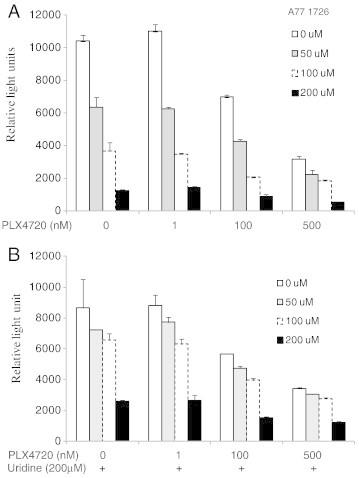
Antiproliferative effect of A77 1726. A375 cells were seeded in 96-well plates (2000 cells per well) and incubated at the indicated concentrations of A77 1726 for 72 hours in the absence or presence of various concentrations of PLX4720 with (A) or without uridine (200 μM) (B). Cell proliferation was analyzed by an ATP-based Cell-Glo assay. Data from one representative of three experiments with similar results were shown.
Effect of S6K1 Knockdown on the PI3K Signaling Pathway and Cell Proliferation
To confirm that inhibition of S6K1 activity contributed to the antiproliferative effect of A77 1726, we tested if S6K1 knockdown led to the suppression of cell proliferation. As shown in Figure 4A, S6K1 transfection effectively suppressed S6K1 expression in A375 cells, leading to decreased S6 and CAD phosphorylation but increased AKTS473 phosphorylation. S6K1 knockdown alone significantly decreased A375 cell proliferation by 20% (Figure 4B). A77 1726 at concentrations of 100 and 200 μM inhibited the proliferation of control siRNA-transfected A375 cells by 23% and 48%, respectively. A77 1726 at concentrations of 100 and 200 μΜ reduced the proliferation of S6K1 siRNA-transfected A375 cells by 9% and 37%, respectively, compared to that in untreated S6K1 siRNA-transfected A375 cells. Combination of S6K1 knockdown and A77 1726 did not achieve an additive antiproliferative effect, suggesting that A77 1726 inhibits A77 1726 proliferation in part by suppressing S6K1 activity.
Figure 4.
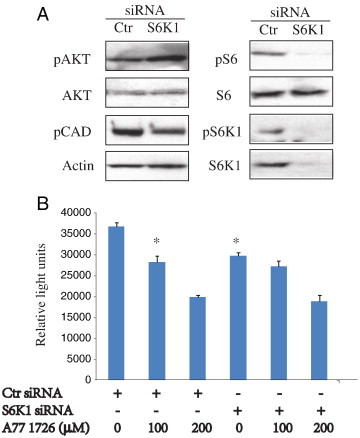
Effect of S6K1 knockdown on feedback PI3K pathway activation and cell proliferation. (A) S6K1 knockdown. A375 cells seeded in a six-well plate were transfected with scrambled or S6K1 siRNA (2.5 nmol each). After incubation for 48 hours, the cells were harvested and analyzed for S6K1 expression and phosphorylation of the indicated proteins by Western blot. (B) The effect of S6K1 knockdown on cell proliferation. A375 cells seeded in a 96-well plate were transfected with a scrambled control siRNA or S6K1 siRNA. After incubation overnight, the cells were incubated in the absence or presence of A77 1726 (100 or 200 μM) for 72 hours and analyzed for cell proliferation by an ATP-based Cell-Glo assay. Data represent the mean ± SD from one of two experiments in triplicate with similar results. *P < .05, compared to control siRNA-transfected A375 cells.
Effect of A77 1726 on Cell Cycle and DNA Synthesis
We next tested whether the antiproliferative effect of A77 1726 was mediated by arresting cell cycle progress. A375 cells were treated with the indicated concentration of A77 1726 and/or uridine for 24 hours and analyzed for cell cycle by propidium iodide staining. As shown in Figure 5A, A77 1726 at 50 μM was sufficient to arrest cell cycle in the S phase. Consistent with this observation, a significantly higher number of cells treated with 50 μM were labeled with BrdU than those treated with A77 1726 at 100 or 200 μM (Figure 6A). Uridine alone had little effect in cell cycle arrest but normalize cell cycle in A375 cells treated with A77 1726 at three different concentrations (Figure 5A). BrdU labeling revealed that uridine blocked the increase of BrdU incorporation in A375 cells mediated by A77 1726 (Figure 6A).
Figure 5.
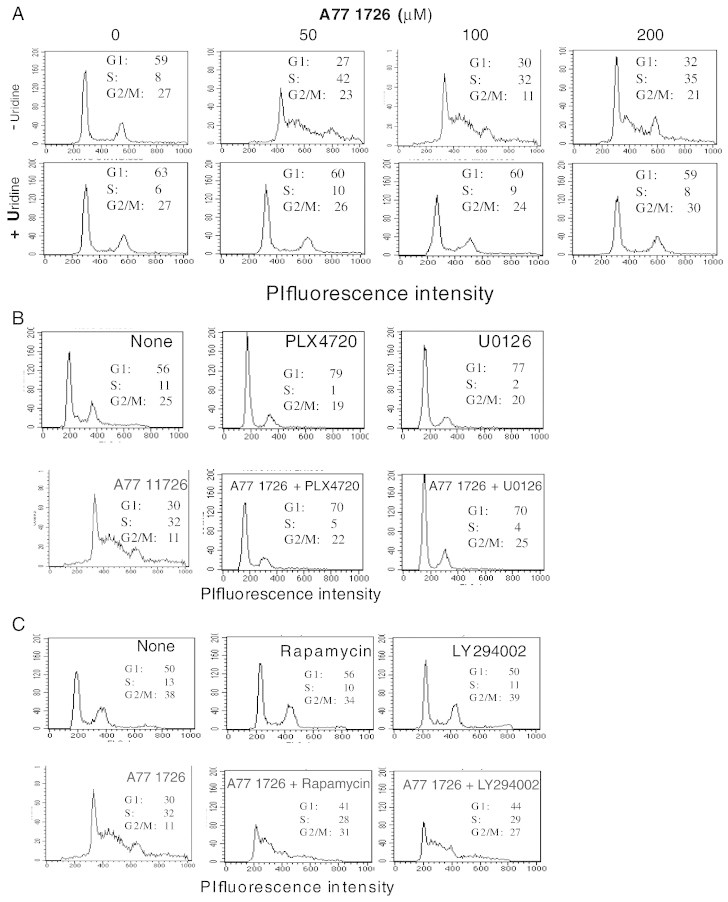
Effect of exogenous uridine and MAP kinase pathway inhibitors on A77 1726–stimulated S phase entry and cell cycle arrest. (A) Ability of uridine to relieve the cell cycle arrest in the S phase. A375 cells grown in six-well plates were treated with the indicated concentration of A771726 with or without uridine (200 μM) for 24 hours. Cell cycle was analyzed in a flow cytometer as described in the Materials and Methods section. (B and C) Effect of the MAP kinase pathway inhibitors (B) and the PI3K pathway inhibitors (C) on A77 1726–mediated cell cycle arrest in the S phase. A375 cells were treated with A771726 (100 μM) in the absence or presence of 0.1% DMSO, PLX 4720 (1 μM), U0126 (10 μM) (B), or rapamycin (20 nM) and LY294002 (10 μM) (C) for 24 hours. Single-cell suspensions were prepared and analyzed for cell cycle in a flow cytometer.
Figure 6.
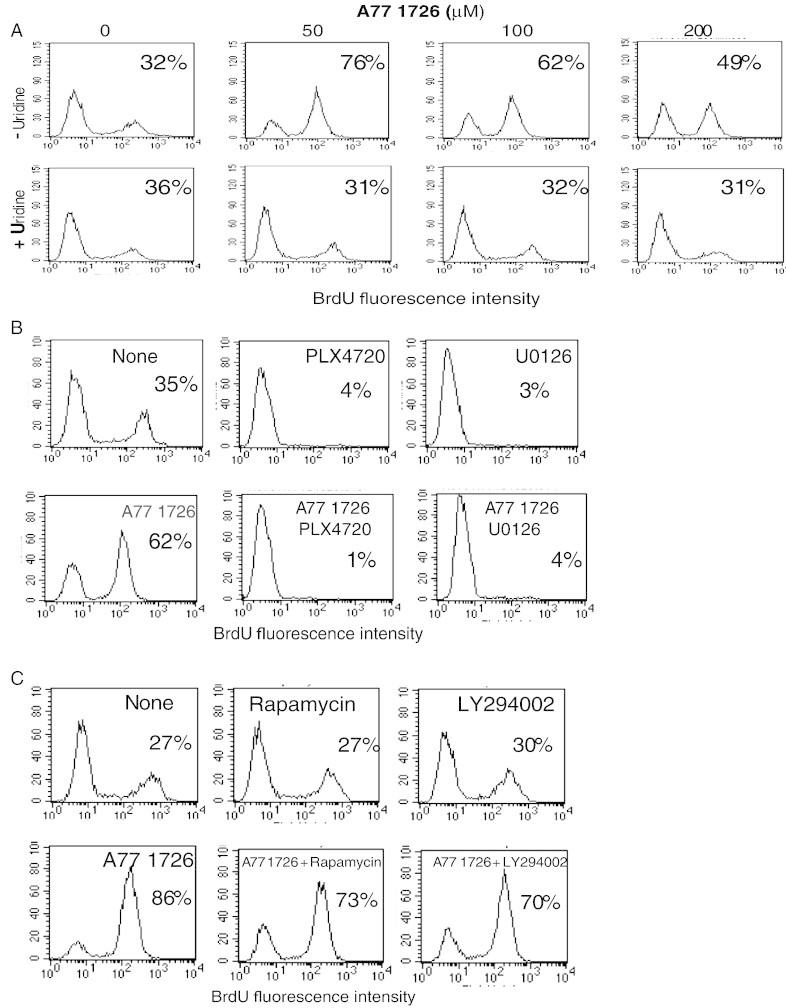
Effect of exogenous uridine and MAP kinase pathway inhibitors on A77 1726–stimulated BrdU incorporation. (A) Effect of uridine to A77 1726–induced DNA synthesis. A375 cells were treated with the indicated concentration of A771726 in the absence or presence of uridine (200 μM) for 22 hours. After pulsing with BrdU (10 μM) for 2 hours, cells were harvested and analyzed for BrdU incorporation by staining with an Alexa Fluor 488–conjugated anti-BrdU monoclonal antibody followed by flow cytometry. (B and C) Effect of the MAP kinase pathway inhibitors (B) and the PI3K pathway inhibitors (C) on A77 1726–stimulated BrdU incorporation. A375 cells were treated with A77 1726 (100 μM) in the presence of 0.1% DMSO, PLX4720 (1 μM), U0126 (10 μM) (B) or rapamycin (20 nM), and LY294002 (10 μM) (C). After incubation for 22 hours, the cells were pulsed with BrdU for 2 hours. Single-cell suspensions were stained for BrdU incorporation and analyzed for cell cycle in a flow cytometer as described in A.
We then tested whether the inhibitors of the MAP kinase pathways affected A77 1726–induced DNA synthesis. As shown in Figure 5B, PLX 4720 (1 μM) and U0126 (10 μM) led to the arrest of the cell cycle in the G1 phase in untreated or A77 1726–treated A375 cells. BrdU labeling revealed that PLX 4720 (1 μM) and U0126 (10 μM) alone were able to completely block DNA replication in A375 cells in the absence or presence of A77 1726 (Figure 6B). These results suggest that A77 1726 treatment led to accelerated DNA synthesis through MAP kinase activation.
Finally, we tested the inhibitors of the PI3K pathway on A77 1726–induced cell cycle progress and DNA replication. Rapamycin or LY249002 alone did not lead to the arrest of cell cycle in the G1 phase (Figure 5C) and was unable to promote cell cycle progress of A77 1726–treated A375 cells. BrdU labeling revealed that LY294002 or rapamycin alone did not significantly affect DNA synthesis but slightly attenuated A77 1726–stimulated DNA synthesis (Figure 6C). These observations suggest that A77 1726–induced cell cycle arrest and DNA synthesis is independent of its effect on the feedback activation of the PI3K pathway.
Discussion
Several prior studies have demonstrated that A77 1726 is capable of inhibiting the PI3K pathway. For example, Baumann et al. [25] reported that the phosphorylation of AKTT308, AKTS473, 4E-BPT37/46, and S6K1T389 is inhibited in H929 and OPM-2 myeloma cell lines after incubation for 24 and 48 hours with A77 1726 (200 μM). Liacini et al. [26] reported that A77 1726 weakly inhibits PDK1 and AKT phosphorylation in a renal CCD1105 cell line and primary human tubular cells, suggesting that A77 1726 may target a kinase upstream of PDK1 or AKT. Sawamukai et al. [27] reported that A77 1726 at 100 and 200 μM inhibits c-Kit ligand-induced PDKS241, AKTT308, and GSK3βS9 in human mast cells. While these studies showed that the PI3K signaling pathway is suppressed by A77 1726, the molecular target of A77 1726 has remained elusive. Our present study demonstrated the ability of A77 1726 to inhibit the phosphorylation of three substrates (S6, CAD, and IRS-1) of S6K1 and the ability of A77 1726 to directly inhibit S6K1 activity in an in vitro kinase assay. Our studies identified S6K1 as a novel molecular target of A77 1726.
IRS-1 is an adaptor protein that interacts with the IGF-1 or insulin receptor and plays a critical role in mediating insulin- and IGF-1–induced activation of the PI3K [28]. S6K1 phosphorylates IRS-1 at Ser-1101 and suppresses PI3K activation. Our study demonstrated the inhibition of IRS-1S1101 phosphorylation by A77 1726 and suggests that A77 1726 induces the feedback activation of the PI3K pathway through the IGF-1 receptor–mediated signaling pathway. Indeed, two inhibitors of the IGF-1 receptor tyrosine kinase, PPP and OSI-906, blocked A77 1726–induced feedback activation of the PI3K pathway. We speculate that A77 1726 does not directly stimulate the IGF-1 receptor tyrosine kinase activity but rather enhances the IGF-1 receptor/IRS-1–mediated PI3K activation. Consistent with this notion, several prior studies suggest that rapamycin induces AKT feedback activation in an IGF-1 receptor–dependent manner [29], [30], [31]. Furthermore, two S6K1 inhibitors induce feedback activation of the PI3K pathway by inhibiting IRS-1 phosphorylation [32], [33].
Though IRS-1 is also required for IGF-1 receptor–mediated activation of the MAP kinase pathway, the underlying mechanisms are less clear [28]. Cook et al. reported that A77 1276 activates Raf-1 kinase in a BON human gastrointestinal carcinoid cell line [34]. Our present study showed that A77 1726 induced ERK1/2 and MEK phosphorylation in A375 cells (Figure 1) but had no effect on Raf-1 phosphorylation (data not shown). PPP and OSI-906 had no effect on the basal level of MEK and ERK phosphorylation. These two inhibitors weakly inhibited A77 1726–induced MEK phosphorylation but had minimal or no effect on ERK phosphorylation. Interestingly, Carracedo et al. [24] reported that rapamycin induces feedback activation of the MAP kinase pathway through Ras. It is likely that A77 1726–induced MEK and ERK phosphorylation may be mediated through the IGF-1 receptor and/or Ras.
The finding that A77 1726 induced the PI3K and MAP kinase pathway feedback activation through the IGF-1 receptor has very important clinical implications. Leflunomide treatment alone may not have strong anticancer effect due to the feedback activation of both PI3K and MAP kinase pathways in some types of cancer. However, leflunomide in combination with an IGF-1 receptor inhibitor or with the PI3K and/or MAP kinase inhibitors may achieve a synergistic effect. Leflunomide in combination with low-dose everolimus leads to the control of Kaposi’s sarcoma in a case report [35]. White et al. [14] reported that leflunomide in combination with PLX4720 achieved a synergistic effect in a melanoma xenograft mouse model.
Prior studies showed that A77 1726 arrests cell cycle progress in the S phase in human gastrointestinal carcinoid [34], prostate, and cutaneous squamous cancer cell lines [36]. Huang et al. [37] suggested that cell cycle arrest in the S phase in K562 cells by A77 1726 is mediated by nucleotide depletion that relies on mutant p53. Our present study demonstrated that A77 1726 stimulated BrdU incorporation but arrested cell cycle arrest in the S phase in wild-type p53 A375 cells. We postulate that activation of the MAP kinase pathway stimulates the cell cycle entry into the S phase. However, depletion of pyrimidine nucleotide pools in A77 1726–treated cells prevents the completion of DNA synthesis and chromosomal duplication (Figure 7), leading to the stall of the cell cycle in the S phase. In support of this notion, MAP kinase pathway inhibitors (U0126 and PLX2720) arrested the cell cycle in the G1 phase in A77 1726–treated A375 cells. It should be noted that A77 1726 arrests cell cycle progress in the G1 phase in lymphocytes [8] and in several myeloma cell lines [25], probably due to the lack of IGF-1 receptor and/or the lack of the MAP kinase activation in these cells.
The antiproliferative activity of A77 1726 in lymphocytes and tumor cells has been well documented. However, the underlying molecular mechanisms are not fully understood. Depletion of pyrimidine nucleotide pools in vitro in cell culture by A77 1726, particular at low concentrations (< 50 μM), is largely responsible for its antiproliferative activity. Uridine was able to partially block the inhibitory effect of A77 1726 used at 50 or 100 μM on cell proliferation but had little effect to reverse the ability of A77 1726 at 200 μM to inhibit cell proliferation. These results suggest that A77 1726 used at high concentrations largely exerts its antiproliferative activity independent of its anti-pyrimidine mechanism. A recent study demonstrated that leflunomide but not A77 1726 functions as an agonist of aryl hydrocarbon receptor to contribute to its antiproliferative activity [38], though underlying mechanisms are still not fully understood. In the present study, we demonstrated that both leflunomide and A77 1726 inhibited S6K1 activity. Since its downstream effector, ribosomal protein S6, plays a critical role in protein synthesis, A77 1726 may exert its antiproliferative activity in part by inhibiting S6 kinase phosphorylation and activation. In support of this notion, S6K1 knockdown significantly inhibited A375 cell proliferation, and S6K1 in combination with A77 1726 did not reach an additive effect.
The IC50 values of leflunomide and A77 1726 to inhibit S6 phosphorylation in cell culture were approximately between 50 and 75 μM, consistent with the results obtained from in vitro kinase assay revealing the IC50 values of leflunomide and A77 1726 to inhibit S6K1 approximately at 55 and 80 μM, respectively. A77 1726 or leflunomide may also inhibit S6K2 but is unlikely able to inhibit other serine/threonine protein kinases A, G, and C such as AKT and mTOR. The IC50 values of leflunomide and A77 1726 required to inhibit S6K1 are physiologically relevant. The pharmacokinetics of leflunomide favorably fit its potential use in oncology. Plasma concentrations of A77 1726 in rheumatoid arthritis patients treated with leflunomide (20 mg/day) can reach 200 μM [39]. The serum concentrations of A77 1726 in mice treated with leflunomide at a dose of 35 mg/kg had a remarkably long half-life of 15 hours. A77 1726 peaks at 500 μM within 4 hours and remains at 250 μM at 24 hours after a single dose of 35 mg/kg leflunomide [40]. Both these concentrations are sufficient to inhibit S6K1 activity. We anticipate that the dose of leflunomide used in preclinical studies and in patients will allow the concentration of A77 1726 in tumor tissue to exceed its IC50 values and to inhibit S6K1 activity.
In summary, our study demonstrated the ability of leflunomide and its active metabolite to inhibit S6K1 activity and to induce the feedback activation of the PI3K and MAP kinase pathways (Figure 7). We further showed that the feedback activation of the MAP kinase pathway by A77 1726 led to the accelerated S cell cycle entry and DNA replication; however, depletion of pyrimidine nucleotide synthesis through direct inhibition of DHO-DHase activity as well as indirect inhibition of CAD activity stalled the cell cycle in the S phase (Figure 7). A77 1726 inhibited cell proliferation in part by inhibiting S6K1 activity.
Footnotes
Conflict of interest: All authors declare no competing interest.
Grant support: This project was supported in part by a melanoma research grant (X.X.) from CinKate Corporation (Oak Park, IL), by Diabetes Pilot Research (X.X.), by the Piccolo Family Foundation (X.X.), National Institutes of Health (AR057404) (J.Z), and by the American Cancer Society, Illinois Division (L.U.).
References
- 1.Zhang S, Yu D. PI(3)king apart PTEN's role in cancer. Clin Cancer Res. 2010;16:4325–4330. doi: 10.1158/1078-0432.CCR-09-2990. [DOI] [PubMed] [Google Scholar]
- 2.Wander SA, Hennessy BT, Slingerland JM. Next-generation mTOR inhibitors in clinical oncology: how pathway complexity informs therapeutic strategy. J Clin Invest. 2011;121:1231–1241. doi: 10.1172/JCI44145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Emerling BM, Akcakanat A. Targeting PI3K/mTOR signaling in cancer. Cancer Res. 2011;71:7351–7359. doi: 10.1158/0008-5472.CAN-11-1699. [DOI] [PubMed] [Google Scholar]
- 4.Xu X, Blinder L, Shen J, Gong H, Finnegan A, Williams JW, Chong AS. In vivo mechanism by which leflunomide controls lymphoproliferative and autoimmune disease in MRL/MpJ-lpr/lpr mice. J Immunol. 1997;159:167–174. [PubMed] [Google Scholar]
- 5.Xu X, Shen J, Mall JW, Myers JA, Huang W, Blinder L, Saclarides TJ, Williams JW, Chong AS. In vitro and in vivo antitumor activity of a novel immunomodulatory drug, leflunomide: mechanisms of action. Biochem Pharmacol. 1999;58:1405–1413. doi: 10.1016/s0006-2952(99)00228-2. [DOI] [PubMed] [Google Scholar]
- 6.Xu X, Williams JW, Gong H, Finnegan A, Chong AS. Two activities of the immunosuppressive metabolite of leflunomide, A77 1726. Inhibition of pyrimidine nucleotide synthesis and protein tyrosine phosphorylation. Biochem Pharmacol. 1996;52:527–534. doi: 10.1016/0006-2952(96)00303-6. [DOI] [PubMed] [Google Scholar]
- 7.Xu X, Williams JW, Bremer EG, Finnegan A, Chong AS. Inhibition of protein tyrosine phosphorylation in T cells by a novel immunosuppressive agent, leflunomide. J Biol Chem. 1995;270:12398–12403. doi: 10.1074/jbc.270.21.12398. [DOI] [PubMed] [Google Scholar]
- 8.Ruckemann K, Fairbanks LD, Carrey EA, Hawrylowicz CM, Richards DF, Kirschbaum B, Simmonds HA. Leflunomide inhibits pyrimidine de novo synthesis in mitogen-stimulated T-lymphocytes from healthy humans. J Biol Chem. 1998;273:21682–21691. doi: 10.1074/jbc.273.34.21682. [DOI] [PubMed] [Google Scholar]
- 9.Elder RT, Xu X, Williams JW, Gong H, Finnegan A, Finnegan A, Chong AS. The immunosuppressive metabolite of leflunomide, A77 1726, affects murine T cells through two biochemical mechanisms. J Immunol. 1997;159:22–27. [PubMed] [Google Scholar]
- 10.Siemasko K, Chong AS, Jäck HM, Gong H, Williams JW, Finnegan A. Inhibition of JAK3 and STAT6 tyrosine phosphorylation by the immunosuppressive drug leflunomide leads to a block in IgG1 production. J Immunol. 1998;160:1581–1588. [PubMed] [Google Scholar]
- 11.Siemasko KF, Chong AS, Williams JW, Bremer EG, Finnegan A. Regulation of B cell function by the immunosuppressive agent leflunomide. Transplantation. 1996;61:635–642. doi: 10.1097/00007890-199602270-00020. [DOI] [PubMed] [Google Scholar]
- 12.Williamson RA, Yea CM, Robson PA, Curnock AP, Gadher S, Hambleton AB, Woodward K, Bruneau JM, Hambleton P, Spinella-Jaegle S. Dihydroorotate dehydrogenase is a target for the biological effects of leflunomide. Transplant Proc. 1996;28:3088–3091. [PubMed] [Google Scholar]
- 13.Bruneau JM, Yea CM, Spinella-Jaegle S, Fudali C, Woodward K, Robson PA, Sautes C, Westwood R, Kuo EA, Williamson RA. Purification of human dihydro-orotate dehydrogenase and its inhibition by A77 1726, the active metabolite of leflunomide. Biochem J. 1998;336(Pt 2):299–303. doi: 10.1042/bj3360299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.White RM, Cech J, Ratanasirintrawoot S, Lin CY, Rahl PB, Burke CJ, Langdon E, Tomlinson ML, Mosher J, Kaufman C. DHODH modulates transcriptional elongation in the neural crest and melanoma. Nature. 2011;471:518–522. doi: 10.1038/nature09882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fenton TR and Gout IT. Functions and regulation of the 70 kDa ribosomal S6 kinases. Int J Biochem Cell Biol43, 47–59. [DOI] [PubMed]
- 16.Sahin F, Kannangai R, Adegbola O, Wang J, Su G, Torbenson M. mTOR and P70 S6 kinase expression in primary liver neoplasms. Clin Cancer Res. 2004;10:8421–8425. doi: 10.1158/1078-0432.CCR-04-0941. [DOI] [PubMed] [Google Scholar]
- 17.Perez-Tenorio G, Karlsson E, Waltersson MA, Olsson B, Holmlund B, Nordenskjold B, Fornander T, Skoog L, and Stal O. Clinical potential of the mTOR targets S6K1 and S6K2 in breast cancer. Breast Cancer Res Treat128, 713–723. [DOI] [PubMed]
- 18.Noh WC, Mondesire WH, Peng J, Jian W, Zhang H, Dong J, Mills GB, Hung MC, Meric-Bernstam F. Determinants of rapamycin sensitivity in breast cancer cells. Clin Cancer Res. 2004;10:1013–1023. doi: 10.1158/1078-0432.ccr-03-0043. [DOI] [PubMed] [Google Scholar]
- 19.Ben-Sahra I, Howell JJ, Asara JM, and Manning BD. Stimulation of de novo pyrimidine synthesis by growth signaling through mTOR and S6K1. Science339, 1323–1328. [DOI] [PMC free article] [PubMed]
- 20.Robitaille AM, Christen S, Shimobayashi M, Cornu M, Fava LL, et al. Quantitative phosphoproteomics reveal mTORC1 activates de novo pyrimidine synthesis. Science339, 1320–1323. [DOI] [PubMed]
- 21.Dibble CC, Asara JM, Manning BD. Characterization of Rictor phosphorylation sites reveals direct regulation of mTOR complex 2 by S6K1. Mol Cell Biol. 2009;29:5657–5670. doi: 10.1128/MCB.00735-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rosner M, Schipany K, and Hengstschlager M. p70 S6K1 nuclear localization depends on its mTOR-mediated phosphorylation at T389, but not on its kinase activity towards S6. Amino Acids42, 2251–2256. [DOI] [PubMed]
- 23.Tremblay F, Brûlé S, Hee Um S, Li Y, Masuda K, Roden M, Sun XJ, Krebs M, Polakiewicz RD, Thomas G. Identification of IRS-1 Ser-1101 as a target of S6K1 in nutrient- and obesity-induced insulin resistance. Proc Natl Acad Sci U S A. 2007;104:14056–14061. doi: 10.1073/pnas.0706517104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carracedo A, Ma L, Teruya-Feldstein J, Rojo F, Salmena L, Alimonti A, Egia A, Sasaki AT, Thomas G, Kozma SC. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J Clin Invest. 2008;118:3065–3074. doi: 10.1172/JCI34739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baumann P, Mandl-Weber S, Volkl A, Adam C, Bumeder I, Oduncu F, Schmidmaier R. Dihydroorotate dehydrogenase inhibitor A771726 (leflunomide) induces apoptosis and diminishes proliferation of multiple myeloma cells. Mol Cancer Ther. 2009;8:366–375. doi: 10.1158/1535-7163.MCT-08-0664. [DOI] [PubMed] [Google Scholar]
- 26.Liacini A, Seamone ME, Muruve DA, and Tibbles LA. Anti-BK virus mechanisms of sirolimus and leflunomide alone and in combination: toward a new therapy for BK virus infection. Transplantation90, 1450–1457. [DOI] [PubMed]
- 27.Sawamukai N, Saito K, Yamaoka K, Nakayamada S, Ra C, Tanaka Y. Leflunomide inhibits PDK1/Akt pathway and induces apoptosis of human mast cells. J Immunol. 2007;179:6479–6484. doi: 10.4049/jimmunol.179.10.6479. [DOI] [PubMed] [Google Scholar]
- 28.Copps KD, White MF. Regulation of insulin sensitivity by serine/threonine phosphorylation of insulin receptor substrate proteins IRS1 and IRS2. Diabetologia. 2012;55:2565–2582. doi: 10.1007/s00125-012-2644-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wan X, Harkavy B, Shen N, Grohar P, Helman LJ. Rapamycin induces feedback activation of Akt signaling through an IGF-1R-dependent mechanism. Oncogene. 2007;26:1932–1940. doi: 10.1038/sj.onc.1209990. [DOI] [PubMed] [Google Scholar]
- 30.O'Reilly KE, Rojo F, She QB, Solit D, Mills GB, Smith D, Lane H, Hofmann F, Hicklin DJ, Ludwig DL. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006;66:1500–1508. doi: 10.1158/0008-5472.CAN-05-2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tamburini J, Chapuis N, Bardet V, Park S, Sujobert P, Willems L, Ifrah N, Dreyfus F, Mayeux P, Lacombe C. Mammalian target of rapamycin (mTOR) inhibition activates phosphatidylinositol 3-kinase/Akt by up-regulating insulin-like growth factor-1 receptor signaling in acute myeloid leukemia: rationale for therapeutic inhibition of both pathways. Blood. 2008;111:379–382. doi: 10.1182/blood-2007-03-080796. [DOI] [PubMed] [Google Scholar]
- 32.Pearce LR, Alton GR, Richter DT, Kath JC, Lingardo L, Chapman J, Hwang C, Alessi DR. Characterization of PF-4708671, a novel and highly specific inhibitor of p70 ribosomal S6 kinase (S6K1) Biochem J. 2010;431:245–255. doi: 10.1042/BJ20101024. [DOI] [PubMed] [Google Scholar]
- 33.Bae EJ, Yang YM, Kim SG. Abrogation of hyperosmotic impairment of insulin signaling by a novel class of 1,2-dithiole-3-thiones through the inhibition of S6K1 activation. Mol Pharmacol. 2008;73:1502–1512. doi: 10.1124/mol.107.044347. [DOI] [PubMed] [Google Scholar]
- 34.Cook MR, Pinchot SN, Jaskula-Sztul R, Luo J, Kunnimalaiyaan M, and Chen H. Identification of a novel Raf-1 pathway activator that inhibits gastrointestinal carcinoid cell growth. Mol Cancer Ther9, 429–437. [DOI] [PMC free article] [PubMed]
- 35.Basu G, Mohapatra A, Manipadam MT, Mani SE, John GT. Leflunomide with low-dose everolimus for treatment of Kaposi's sarcoma in a renal allograft recipient. Nephrol Dial Transplant. 2011;26:3412–3415. doi: 10.1093/ndt/gfr416. [DOI] [PubMed] [Google Scholar]
- 36.Hail N, Jr., Chen P, and Bushman LR. Teriflunomide (leflunomide) promotes cytostatic, antioxidant, and apoptotic effects in transformed prostate epithelial cells: evidence supporting a role for teriflunomide in prostate cancer chemoprevention. Neoplasia12, 464–475. [DOI] [PMC free article] [PubMed]
- 37.Huang M, Wang Y, Collins M, Mitchell BS, Graves LM. A77 1726 induces differentiation of human myeloid leukemia K562 cells by depletion of intracellular CTP pools. Mol Pharmacol. 2002;62:463–472. doi: 10.1124/mol.62.3.463. [DOI] [PubMed] [Google Scholar]
- 38.O'Donnell EF, Kopparapu PR, Koch DC, Jang HS, Phillips JL, Tanguay RL, Kerkvliet NI, and Kolluri SK. The aryl hydrocarbon receptor mediates leflunomide-induced growth inhibition of melanoma cells. PLoS One7, e40926. [DOI] [PMC free article] [PubMed]
- 39.Chan V, Charles BG, Tett SE. Population pharmacokinetics and association between A77 1726 plasma concentrations and disease activity measures following administration of leflunomide to people with rheumatoid arthritis. Br J Clin Pharmacol. 2005;60:257–264. doi: 10.1111/j.1365-2125.2005.02415.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chong AS, Huang W, Liu W, Luo J, Shen J, Xu W, Ma L, Blinder L, Xiao F, Xu X. In vivo activity of leflunomide: pharmacokinetic analyses and mechanism of immunosuppression. Transplantation. 1999;68:100–109. doi: 10.1097/00007890-199907150-00020. [DOI] [PubMed] [Google Scholar]