

Abstract
Melanomas are highly radioresistant tumors, mainly due to efficient DNA double-strand break (DSB) repair. Dbait (which stands for DNA strand break bait) molecules mimic DSBs and trap DNA repair proteins, thereby inhibiting repair of DNA damage induced by radiation therapy (RT). First, the cytotoxic efficacy of Dbait in combination with RT was evaluated in vitro in SK28 and 501mel human melanoma cell lines. Though the extent of RT-induced damage was not increased by Dbait, it persisted for longer revealing a repair defect. Dbait enhanced RT efficacy independently of RT doses. We further assayed the capacity of DT01 (clinical form of Dbait) to enhance efficacy of “palliative” RT (10 × 3 Gy) or “radical” RT (20 × 3 Gy), in an SK28 xenografted model. Inhibition of repair of RT-induced DSB by DT01 was revealed by the significant increase of micronuclei in tumors treated with combined treatment. Mice treated with DT01 and RT combination had significantly better tumor growth control and longer survival compared to RT alone with the “palliative” protocol [tumor growth delay (TGD) by 5.7-fold; median survival: 119 vs 67 days] or the “radical” protocol (TGD by 3.2-fold; median survival: 221 vs 109 days). Only animals that received the combined treatment showed complete responses. No additional toxicity was observed in any DT01-treated groups. This preclinical study provides encouraging results for a combination of a new DNA repair inhibitor, DT01, with RT, in the absence of toxicity. A first-in-human phase I study is currently under way in the palliative management of melanoma in-transit metastases (DRIIM trial).
Abbreviations: DNA-PK, DNA-dependent kinase; DSB, double-strand break; PARP, poly-adenyl-ribose polymerase; RT, radiation therapy; SSB, single-strand break; TGD, tumor growth delay
Introduction
Melanoma is the fifth and sixth most common cancer in men and women, respectively, and has the fastest growing incidence of any cancer [1]. First-line treatment for non-metastatic melanoma is, when possible, surgical excision of the primary tumor and affected lymph nodes, with eventual adjuvant radiation therapy (RT) [2], [3]. Melanoma is characterized by symptomatic locoregional recurrence, distant metastases, and despite recent advances, poor response to systemic drugs [4]. Although RT is important for locoregional control and palliation in oncology, this approach is not always incorporated into the management of melanoma. Reluctance to use RT can be traced to findings of early experimental and clinical reports suggesting that melanoma was resistant to radiation [5], [6]. RT for melanoma was described as “futile” [7] and “should not be relied on for the cure of these lesions” [8]. In the early 1970s, in vitro studies revealed that melanoma cells are less sensitive to RT only at lower doses and show increased cell death with higher doses per fraction [9]. In the modern era, many reports have demonstrated that RT could have a potential place in the management of melanoma (e.g., post-lymphadenectomy, brain metastases, and so on) although controversy remains [10]. Most studies use a hypofractionated (dose per fraction > 2.5 Gy) schedule of RT. Greater use of RT, especially in association with molecular therapy that could enhance its efficacy, may allow substantial improvements to multidisciplinary melanoma management [10], [11].
RT cytotoxicity is mainly due to DNA damage. DNA double-strand breaks (DSBs) are the most severe RT-induced DNA damage and are lethal to the cell if not repaired [12]. DSBs are produced directly and indirectly by RT. Indirect DSB most often occur during replication if initial damage is unrepaired. For example, when a replication fork encounters an unrepaired single-strand break (SSB), the fork is blocked and leads to the conversion of this SSB in DSB [13]. The ability of cancer cells to recognize DNA damage and initiate repair is an important mechanism of radioresistance [14], [15]. Inhibition of DNA repair could make cancer cells more vulnerable to the DNA damaging therapies like RT [16]. Therefore, to inhibit DNA repair, we designed an innovative family of molecules named Dbait (which stands for DNA strand break bait). Dbait consists of 32 bp deoxyribonucleotides forming an intramolecular DNA double helix that mimics DNA lesions. They act as a bait for DNA damage signaling enzymes, the poly-adenyl-ribose polymerase (PARP), and the DNA-dependent kinase (DNA-PK), inducing a “false” DNA damage signal and ultimately inhibiting recruitment at the damage site of many proteins involved in DSB and SSB repair (SSBR) pathways (Figure 1) [17], [18].
Figure 1.
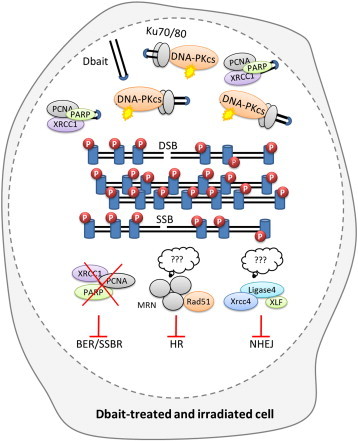
Schematic overview of the mechanism of action of Dbait. In irradiated and Dbait-treated cells, DNA repair signaling is perturbed leading to global inhibition of DNA repair pathways. On the one hand, Dbait recognition by the DNA-PK complex induces DNA-PK activation ( ) and initiates the uncoordinated phosphorylation of its nuclear targets visualized by pan-nuclear γ-H2AX (
) and initiates the uncoordinated phosphorylation of its nuclear targets visualized by pan-nuclear γ-H2AX ( ). Upon creation of a DSB in the DNA, the DNA damage signaling apparatus induced by Dbait is dispersed across all the modified chromatin and inhibits the recruitment of the factors required for DSB repair at the site of the damage. This leads to both non-homologous end joining (NHEJ) and homologous recombination (HR) inhibition. On the other hand, Dbait can also be bound by PARP (major protein involved in BER and SSBR) leading to its autoPARylation and the further recruitment of different BER and SSBR proteins on Dbait molecules. These proteins are thus hijacked far from the damage leading to BER/SSBR inhibition.
). Upon creation of a DSB in the DNA, the DNA damage signaling apparatus induced by Dbait is dispersed across all the modified chromatin and inhibits the recruitment of the factors required for DSB repair at the site of the damage. This leads to both non-homologous end joining (NHEJ) and homologous recombination (HR) inhibition. On the other hand, Dbait can also be bound by PARP (major protein involved in BER and SSBR) leading to its autoPARylation and the further recruitment of different BER and SSBR proteins on Dbait molecules. These proteins are thus hijacked far from the damage leading to BER/SSBR inhibition.
We report an analysis of the potential of Dbait for clinical application to sensitize skin melanoma to RT. First, we demonstrated the radiosensitizing properties of Dbait in vitro. Then, we performed animal studies to determine the clinical potential and applicability of combined Dbait + RT treatment for human melanoma. The data presented here were used to design the clinical trial DRIIM (clinicaltrials.gov/ct2/show/NCT01469455).
Material and Methods
Cell Culture and Dbait Molecules
The human skin melanoma cell lines SK28 and 501mel were obtained from American Type Culture Collection (ATCC) (Manassas, VA). Cells were grown in complete RPMI (Gibco, Cergy Pontoise, France) supplemented with 10% FBS (ATGC, Orléans, France), 1% sodium pyruvate, streptomycin (100 mg/ml), and penicillin (100 mg/ml; Invitrogen, Carlsbad, CA). Cells were maintained at 37°C under a 5% CO2 atmosphere, at 100% humidity. Dbait molecules are 32 bp oligonucleotides that are made by automated solid-phase oligonucleotide synthesis methods (Eurogentec, Seraing, Belgium). The sequence of the Dbait molecule used is 5′-GCTGTGCCCACAACCCAGCAAACAAGCCTAGA-(H)-TCTAGGCTTGTTTGCTGG GTTGTGGGCACAGC-3′, where H is a hexaethylene glycol linker and the underlined letters are phosphorodiamidate nucleosides. For animal studies, a 5′-cholesterol linked form of Dbait (called DT01), which avoids the use of toxic transfection agents, diluted in 5% glucose was used [19].
In Vitro Dbait and Irradiation Treatments and Clonogenic Survival Assay
DT01 used in vivo does not efficiently transfect cells in vitro. Therefore, we used 1.25 mg/l Dbait, or transfection control, complexed with 11 kDa polyethylenimine (PEI; Polypus Transfection, Illkirch, France) to treat the cells. Cells were seeded at 300,000 cells per 60-mm dishes the day before transfection. Then, cells were transfected with Dbait or control in 1200 μl of serum-free RPMI for 5 hours. At the end of transfection, the medium was removed and replaced with complete RPMI (Gibco) already heated to 37°C. The irradiation was then performed as single exposures at increasing doses (1- to 8-Gy irradiation), delivered by a 137Cs unit (0.5 Gy/min). Twenty-four hours after irradiation, cells were seeded at 100 to 10,000 cells per 25-cm3 flask in complete medium. After 12 days of incubation, the cell clones were stained with crystal violet. Colonies with more than 50 cells were counted by microscopic inspection and the radiation-surviving fractions were determined. The intrinsic radiosensitivity was evaluated by using two parameters: the surviving fraction at 2 Gy (SF2) and the area under the survival curve. Survival curves were obtained in accordance with the linear quadratic model (Kaleida Graph software 4.0).
Antibodies and Immunologic Techniques for H2AX Labeling
SK28 and 501mel cells were seeded at 300,000 cells per 60-mm dishes, 24 hours before treatment. Cells were transfected by incubation with 1.25 mg/l Dbait or transfection control for 5 hours. When irradiated, the cells were then subjected to 2.5-Gy irradiation. Immunocytochemistry of the phosphorylated form of the H2AX histone (γ-H2AX) was done as previously described [18] immediately after irradiation and/or Dbait treatment. Monoclonal antibodies against γ-H2AX (Millipore, Billerica, MA) were used. Microscopy was performed at room temperature with the Leica SP5 confocal system, attached to a DMI6000 stand, with a 40 or 63 ×/1.4 oil immersion objective. Images were processed with the freely available ImageJ software (http://rsb.info.nih.gov.gate1.inist.fr/ij/) and the LOCI bioformat plug-in (http://www.loci.wisc.edu/ome/formats.html) to access images generated by the Leica SP5 confocal system.
Assessment of DNA Breaks by Single-Cell Electrophoresis
SK28 and 501mel cells were seeded at 100,000 cells per 35-mm dishes. Twenty-four hours later, cells were transfected with 1.25 mg/l Dbait or control for 5 hours and were left untreated or were treated with 8-Gy irradiation. DNA breaks were assessed by single-cell electrophoresis (alkaline Comet assay) immediately, 5, 30, 60, and 180 minutes and 24 hours after irradiation, as previously described [18]. DNA damage was quantified as the Comet tail moment, which represents the extent of DNA damage (both SSB and DSB) in individual cells.
Assessment of Dbait Activity in DNA-PK Inhibited or Depleted Cells
SK28 cells were seeded in six-well plates at a concentration of 1.5 × 105 cells on the day before shRNA transduction to obtain 50% of confluence for the viral infection. Subconfluent SK28 cells were transduced with lentiviruses that expressed either the control, non-targeting shRNA (shCTL; sc-108080; Santa Cruz Biotechnology, (Dallas, Texas, USA)), or shRNA targeting DNA-PKcs (shDNA-PK; sc-35200-V; Santa Cruz Biotechnology) at a multiplicity of infection of 3 using polybrene (5 μg/ml). The most efficiently depleted cells (80% of knockdown of DNA-PKcs gene) were selected; 300,00 cells were seeded in 60-mm dishes. Twenty-four hours later, cells were transfected by incubation with 1.25 mg/l Dbait or transfection control for 5 hours. Immediately after treatment, cells were immunostained with mouse monoclonal anti–DNA-PKcs (Ab-4, Cocktail; Thermo Scientific, (Waltham, MA, USA)) or mouse monoclonal anti–γ-H2AX antibodies (Millipore) and counterstained with 4',6-diamidino-2-phenylindole (DAPI). For DNA-PK inhibition, cells were incubated for 1 hour in serum-free medium containing 25 μM NU7026 and then incubated in serum-free medium containing 25 μM NU7026 and 1.25 mg/l control or Dbait for 5 hours. Immunocytochemistry of the phosphorylated form of γ-H2AX was conducted immediately after control or Dbait treatment.
Dbait and Irradiation Treatment in Mice
SK28 human melanoma xenograft tumors were obtained by injecting 4 × 106 tumor cells into the flank of adult female nude mice (Janvier, Le Genest Saint Isle, France). The animals were housed in our animal facility for at least 1 week before starting the experiments. There were six animals per cage under controlled conditions of light and dark cycles (12 hours:12 hours), relative humidity (55%), and temperature (21°C). Food and tap water were available ad libitum. When subcutaneous tumors reached approximately 125 mm3, mice were separated into homogeneous groups to receive different treatment protocols: no treatment (NT), RT alone for 2 weeks (RT2w: 10 × 3 Gy), RT alone for 4 weeks (RT4w: 20 × 3 Gy), DT01 alone for 2 weeks (DT012w: 6 × 4 mg), RT + DT01 for 2 weeks (RT2w + DT012w: 10 × 3 Gy + 6 × 4 mg) or 4 weeks (RT4w + DT014w: 20 × 3 Gy + 12 × 4 mg), and RT + DT01 followed by a second cycle of DT01 alone 6 weeks later (RT2w + DT012w + DT012w: 10 × 3 Gy + 6 × 4 mg + 6 × 4 mg). Initially, we checked mock-treated animals to ensure the absence of any changes in tumor growth or survival compared to animals treated with or without RT (Figure S1). A 137Cs unit (0.5 Gy/min) with a shield designed to protect approximately two thirds of the animal’s body was used to administer RT. Doses were controlled by thermoluminescence dosimetry. Four milligrams of DT01 was given 5 hours before RT sessions (2 mg intratumoral and 2 mg peritumoral subcutaneous injections), once every 2 days. All animals were treated daily with chloroquine per os (40 mg/kg), for 1 week before DT01 treatment and then during the whole treatment period. Administration of chloroquine favors the release of oligonucleotides, like DT01, from endosomes into the cytoplasm improving its cellular uptake [19], [20]. In all experiments, tumors were measured with a digital caliper every 2 to 3 days and any local skin toxicity was noted. Tumor volumes were calculated using the following formula: length × width × width/2. Mice were weighed every week and followed up for 280 days. For ethical reasons, the animals were sacrificed when tumors reached 1500 mm3. The Local Committee on Ethics of Animal Experimentation approved all experiments.
Micronuclei Assessment
Micronuclei contain fragments of chromosomes generally generated during defective DNA repair [21]. One month after initial treatment, tumors were fixed in formalin and then embedded in paraffin. Sections were cut and stained with hematoxylin, eosin, and saffron. Percent of micronuclei was estimated in the non-necrotic and proliferative area by quantifying the cells presenting micronuclei in the cytoplasm, according to their described characteristics [22] in at least 1000 cells.
Statistical Analysis
Random-effects model was used for comparison between clonogenic cell survival assays [23]. The type of interaction was described as additive when corresponding to the effect being equal to that of the theoretically calculated effects of RT or Dbait alone and supra-additive when the effect of concurrent use of both RT and Dbait is considered to be more efficient than the calculated effect of single use. The term synergy is considered to correspond to supra-additivity [24]. Two-sided unpaired t tests were used for comparison between micronuclei formation and tumor growth delay (TGD). Comet tail moment data were analyzed as previously described [25], by two-way analysis of variance of the medians, followed by Bonferroni post hoc tests. TGD was calculated by subtracting the mean tumor volume quadrupling time of the control group from tumor volume quadrupling times of individual mice in each treatment group. The mean TGD was calculated for each treatment group from the individual measurements. Survival curves were assessed by Kaplan-Meier estimates and compared using the nonparametric log-rank test since the data do not follow a normal distribution. S-Plus 6.2 version software (MathSoft, (Cambridge, MA, USA)) and statEL (Ad Science, (Paris, France)) were used for statistical analyses.
Results
Dbait Disorganizes and Inhibits Repair of Radio-Induced DNA Damage in Melanoma Cell Lines Leading to Inhibition of Proliferation
We have previously shown that Dbait treatment led to the activation of DNA-PK [17]. This hyperactivation triggers phosphorylation of the histone H2AX all along the chromatin, however not specifically at radio-induced DSB sites. This disorganized H2AX phosphorylation prevents subsequent detection of the radio-induced DSBs and therefore their repair (Figure 1). Here, we checked the ability of Dbait to induce DNA-PK activation in the melanoma cell lines SK28 and 501mel by monitoring the H2AX phosphorylation. Dbait treatment, with or without RT, resulted in pan-nuclear phosphorylation of H2AX in at least 60% of the cells, evidencing DNA-PK activation by Dbait in these cells (Figure 2A). The Dbait-induced H2AX phosphorylation displays a characteristic pan-nuclear distribution different from the γ-H2AX foci formed at the radio-induced DNA DSB (Figure 2B). There were no such signals in the shDNA-PK mutants or in cells with inhibited DNA-PK (Figure 2, A and C), demonstrating that H2AX pan-nuclear phosphorylation was strictly dependent on DNA-PK activation by Dbait.
Figure 2.
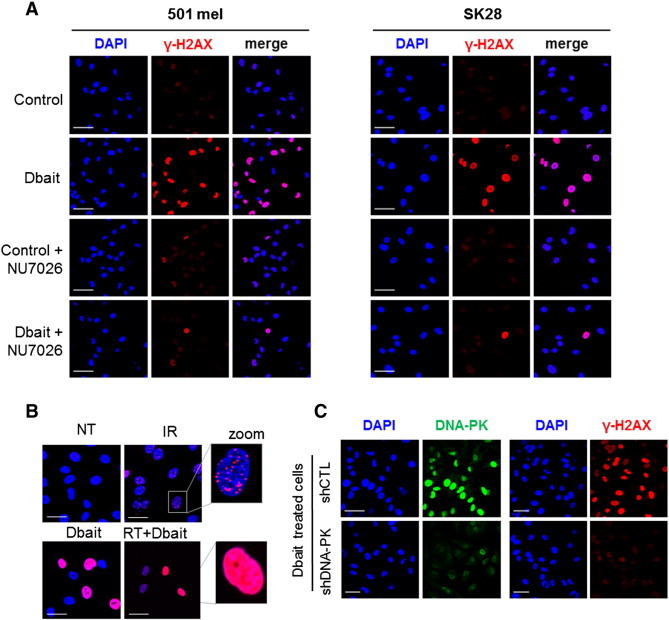
Effect of Dbait on H2AX phosphorylation. (A) SK28 and 501mel melanoma cells were transfected with an inactive control oligonucleotide or Dbait ± NU7026 (DNA-PK inhibitor). Immunofluorescence of γ-H2AX (red) and chromatin (DAPI; blue) was visualized. Dbait treatment led to non-localized pan-nuclear H2AX phosphorylation evidencing Dbait activity. This activity was dependent on DNA-PK activation. Bar, 50 μm. (B) SK28 melanoma cells were transfected with an inactive control oligonucleotide or Dbait. Immunofluorescence of γ-H2AX (red) and chromatin (DAPI; blue) was visualized immediately after irradiation and/or Dbait treatment. Irradiation alone resulted in localized γ-H2AX foci representing radio-induced DNA DSBs; Dbait treatment with or without irradiation led to non-localized pan-nuclear H2AX phosphorylation evidencing Dbait activity. Bar, 30 μm. (C) SK28 cells were transduced with lentiviruses that express either control, non-targeting shRNA, or shRNA targeting DNA-PKcs. After Dbait transfection, cells were immunostained with mouse monoclonal anti–DNA-PKcs or anti–γ-H2AX. Dbait activity was not detected in cells transduced with shRNA targeting DNA-PKcs. Bar, 50 μm.
The consequences on clonogenic cell survival of Dbait treatment associated to RT were investigated in 501mel and SK28 cells. Survival analyzed 12 days after treatment revealed a high sensitivity of 501mel to stand-alone treatment of Dbait with only 35% of cells forming colonies after Dbait treatment (Figure 3, A and B). In contrast, SK28 cells were less affected by Dbait treatment alone with 85% of cells forming colonies. Similarly, melanoma cells showed different sensitivities to RT. SK28 cells were more radioresistant than 501mel (surviving fraction at 2 Gy, 0.89 vs 0.61; area under the survival curve, 5.2 vs 3.6). However, both cell lines demonstrated an increased lethality after combined treatment compared to irradiation alone (P < .001; Figure 3, A and B). In 501mel cells, association of Dbait + RT led mainly to an additive increase of cell toxicity, while highly RT-resistant SK28 cells were significantly radiosensitized with combined treatment for all RT doses (P < .001). Interestingly, a loss of the initial shoulder region of the curve was observed in the SK28-radiosensitized cell line (Figure 3A).
Figure 3.
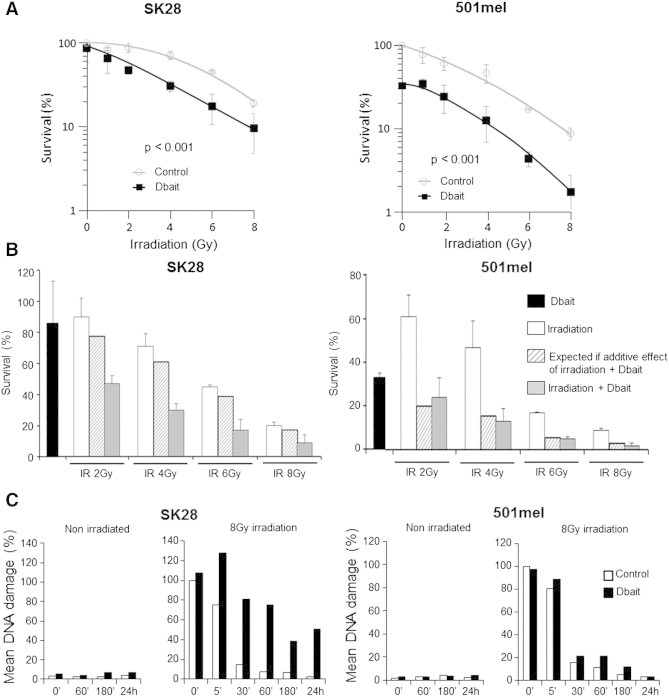
Effect of Dbait on cell survival and repair of DNA breaks induced by RT. (A) Cell survival assay. Transfected SK28 and 501mel cells were irradiated with 1 to 8 Gy, and clonogenic survival was estimated 12 days later. (B) Simulation of expected additive effect of Dbait and irradiation on cell survival. Data are represented as median values ± standard error. (C) The extent of DNA damage in transfected cells was analyzed by alkaline Comet assay at various times (0, 60, and 180 minutes and 24 hours) after Dbait or control treatment and at various times (0, 5, 30, 60, and 180 minutes and 24 hours) after combination with irradiation (8 Gy) in SK28 and 501mel cell lines.
To confirm that radiosensitization by Dbait was due to the inhibition of repair of radio-induced DNA SSB and DSB damage, we carried out alkaline Comet assays (Figure 3C). Dbait treatment alone did not induced DNA damage in both cell lines. In SK28 control–transfected cells, RT induced DNA damage, which gradually decreased over time due to repair activity. In contrast, in Dbait-treated cells, RT-induced DNA damage was not initially greater, but persisted for longer (P < .05) as expected if break repair was inhibited. While displaying greater radiosensitivity compared to SK28, 501mel shows rapid DNA repair, which was only slightly reduced by Dbait treatment. The damage repair revealed by the Comet assay could be less accurate in 501mel cells and responsible for the late death revealed by the low clonal survival.
Radiosensitizing Effect of Dbait In Vivo with a “Palliative” RT Protocol
We used athymic nude mice bearing SK28 melanoma tumor xenografts to investigate whether the radiosensitizing effect observed in vitro was also reproducible in vivo. SK28 xenografts were chosen since they were more radioresistant in vitro. The experimental design was aimed at confirming the clinical relevance and applicability of the approach (Figure 4A). Consistent with one of the current palliative RT protocols used for patients, 10 fractions of 3 Gy were given locally over a 2-week period. DT01 was administered locally 5 hours before RT every 2 days (six sessions in total; Figure 4A). Tumor growth was slightly lower following RT2w or DT012w treatments alone than in untreated animals. However, the combination of the two treatments (RT2w + Dbait2w) significantly decreased tumor growth (P < .004 vs RT2w or DT012w; Figure 4B and Table 1). Moreover, the RT2w + DT012w combined therapy resulted in a significant enhancement of survival (P < .01 vs RT2w or DT012w; Figure 4C and Table 1). To maintain treatment efficacy, we added a second cycle of DT01 administration at the time of tumor escape, 6 weeks after the initial RT2w + DT012w treatment. The second cycle of DT01 alone significantly improved the control of tumor growth leading to an enhanced survival (P < .0001 vs RT2w + DT012w; Figure 4C and Table 1). Moreover, this additional treatment triggered a complete response in 4 of the 18 animals (22%; Table 1). Importantly, no significant increase in skin toxicity was observed in irradiated and Dbait-treated healthy tissues surrounding the tumor during follow-up.
Figure 4.
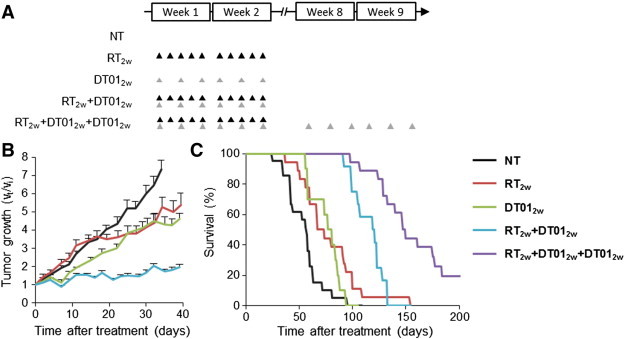
Radiosensitization of SK28 tumors by DT01 with a “palliative” RT protocol. Mice bearing subcutaneous SK28 melanoma tumor xenografts were untreated (NT), treated for 2 weeks with 10 fractions of 3 Gy (RT2w), treated for 2 weeks with six administrations of 4 mg of DT01 (DT012w), both treatments (RT2w + DT012w), or both treatments followed with a second cycle of DT01 6 weeks later (RT2w + DT012w + DT012w). (A) Treatment schedule. Each RT 3-Gy fraction is represented by a black triangle. Each DT01 4-mg administration is represented by a gray triangle. (B) Tumor growth. DT01 with RT significantly increased tumor growth control. (C) Survival. Mice were monitored until tumors reached 1500 mm3 (death time). Survival was significantly improved in mice treated with RT2w + DT012w and RT2w + DT012w + DT012w. Vt, tumor volume at a given time point; Vi, initial volume before treatment.
Table 1.
Comparison of the Response to Treatment of SK28 Melanoma Xenografted Animals Treated with Various Protocols.
| Treatment | Number of Mice | Median Survival in Days | RR (P Value) | Mean TGD in Days (%) | SD TGD in Days | Complete Response (%) |
|---|---|---|---|---|---|---|
| NT | 21 | 57 | 0 (100) | 11 | 0 (0) | |
| DT012w | 10 | 81 | 0.48 (3 × 10− 2)⁎ | 11 (149) | 10 | 0 (0) |
| RT2w | 18 | 67 | 0.41 (2 × 10− 3)† | 10 (147) | 16 | 0 (0) |
| RT2w + DT012w | 12 | 119 | 0.38 (4 × 10− 3)‡ | 57 (362) | 18 | 0 (0) |
| RT2w + DT012w + DT012w | 18 | 150 | 0.21(5)§ | 75 (449) | 76 | 4 (22) |
| RT4w | 12 | 109 | 0.24 (5)¶ | 39 (278) | 44 | 1 (8) |
| RT4w + DT014w | 11 | 221 | 0.19 (5)# | 126 (681) | 52 | 7 (64) |
RR, relative risk. Statistics: ⁎DT012wversus NT, †RT2wversus NT, ‡RT2w + DT012wversus RT2w, §RT2w + DT012w + DT012wversus RT2w + DT012w, ¶RT4wversus NT, #RT4w + DT014wversus RT4w.
To confirm that DT01 treatment inhibits DNA repair in vivo, we monitored micronuclei formation in the proliferative area of the treated tumors. Micronuclei are small additional nuclei, located in the cytoplasm, that contain fragments of chromosomes largely generated during defective DNA repair [21]. As expected after DNA repair inhibitor treatment, there were twice as many micronuclei formed in cells following RT2w + DT012w treatment than RT2w or DT012w alone (P < .03; Figure 5).
Figure 5.
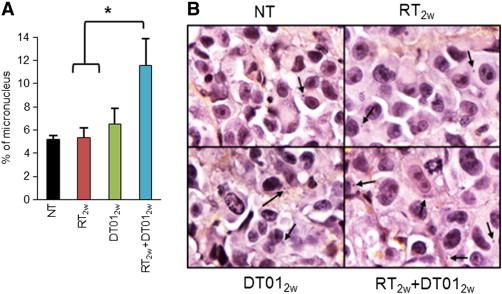
Micronuclei detection in SK28 melanoma tumor xenografts treated with a “palliative” RT protocol. One month after initial treatment by RT2w, DT012w, and RT2w + DT012w according to the “palliative” protocol (Figure 4), tumors were removed and the numbers of micronuclei were quantified. Percentage of micronucleus was estimated in the non-necrotic and proliferative area by quantifying the cells presenting micronucleus in the cytoplasm within at least 1000 cells. (A) Results are reported as mean (± standard error) percentages. There were twice as many micronuclei formed in cells following RT2w + DT012w treatment than RT2w or DT012w treatments (P < .03). (B) Representative images of hematoxylin, eosin, and safran labeled tumor sections with micronuclei detection (arrows).
Radiosensitizing Effect of Dbait In Vivo with a “Radical” RT Protocol
The RT protocol (10 × 3 Gy) used in the experiment reported above is a palliative protocol proposed to patients suffering from non-operable cutaneous melanoma. We further investigated whether Dbait could increase the efficacy of a more “radical” RT protocol. We performed the treatment cycle over 4 weeks, such that the total doses were 20 × 3 Gy of irradiation (biologic equivalent dose of 37 × 2 Gy using a linear quadratic model and α/β = 2.5 [26]) and 12 × 4 mg of Dbait (Figure 6A). RT4w + Dbait4w significantly extended TGD to 39 days versus 10 days for RT2w + Dbait2w (Figure 6B; P < .01), resulting in doubling the median survival (221 vs 119 days; P < .0001; Figure 6C and Table 1). In addition, this treatment led to seven complete responses among the 11 animals treated (64%; Table 1). As previously observed, doubling the RT and Dbait doses was not associated with additional local adverse effects.
Figure 6.
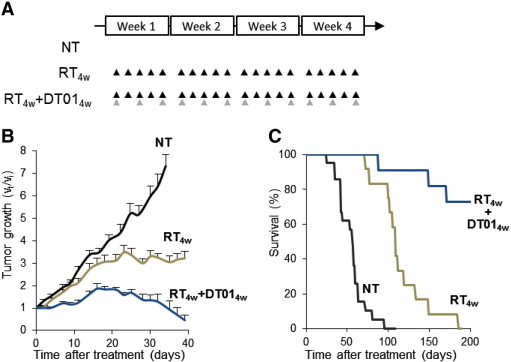
Radiosensitization of SK28 tumors by DT01 with a “radical” RT protocol. Mice bearing subcutaneous SK28 melanoma tumor xenografts were untreated (NT), treated for 4 weeks with 10 fractions of 3 Gy (RT4w), or treated with combined RT and DT01 treatments (RT4w + DT014w). (A) Treatment schedule. Each RT 3-Gy fraction is represented by a black triangle. Each DT01 4-mg local administration is represented by a gray triangle. (B) Tumor growth. DT01 with RT significantly increased tumor growth control. (C) Survival. Mice were monitored until tumors reached 1500 mm3 (death time). Survival was significantly improved in mice treated with RT4w + DT014w. Vt, tumor volume at a given time point; Vi, initial volume before treatment.
Discussion
Efficient DNA repair activity in cancer cells increases the resistance of cancer to RT [27], [28]. Over the last decade, many molecules inhibiting various DNA repair pathways by targeting their key enzymes have been developed [29], [30], [31], [32], [33]. These strategies, based on specific target inhibition, may be thwarted by target mutation or activation of another repair pathway. For example, PARP inhibitors require an additional defect in homologous recombination to be effective [29]. In contrast, Dbait is not a specific enzyme inhibitor but represents a new drug strategy targeting the entire DNA DSB repair system through perturbation of DNA repair signaling [17], [18]. On the one hand, the DNA DSB signaling apparatus induced by Dbait is dispersed across all the modified chromatin and inhibits the recruitment of the factors required for DSB repair at the site of damage. However, Dbait can also be bound by PARP [major protein involved in base excision repair (BER) and SSBR] leading to its autoPARylation and further recruitment of different BER and SSBR proteins on Dbait molecules. These proteins are thus hijacked far from the site of damage leading to BER/SSBR inhibition [34].
In this preclinical study, Dbait was used to radiosensitize human melanoma both in vitro and in vivo. In vitro, Dbait led to an increase of cell toxicity in both cell lines. This increase was additive in one cell line and synergic in the other. Although a synergic effect is preferred for radiosensitization, both additive and synergic effects can be of clinical interest, particularly if the molecule is not toxic for healthy tissues, which is the case for Dbait in this preclinical study. Interestingly, the synergic effect was observed in the cell line that was found to be the more radioresistant. In this cell line, the shoulder region of the survival curve was no longer observed with the combined treatment. This could be consistent with Dbait mechanism of action as this part of the curve is often described as representative of the capacity of DNA repair of the cells [35], [36], [37]. For the in vivo portion of the work, the experimental design was intended to test the clinical relevance and applicability of these findings, both for the palliative RT protocol and local administration of DT01. A recent review reported the potential applications of intralesional agents in the management of cutaneous malignancy [38]. There have also been reports of intralesional injection of accessible melanoma tumors with standard chemotherapeutic agents (cisplatin and bleomycin) and with drugs generating a local immune response [39], [40], [41], [42]. Similarly, the association of intralesional injections and subsequent RT has been assessed in the management of accessible metastatic or recurrent melanoma [43], [44]. Here, we found that for some tumors with necrosis or high interstitial pressure, it was preferable to administer DT01 locally at the periphery of the tumor to favor delivery to the proliferative area. Therefore, we combined both intratumoral and peritumoral injections for DT01 administration. This design had the advantage of healthy tissue at the periphery of the tumor receiving both DT01 and RT and therefore a good indicator of healthy skin tolerance of combined treatment (Supplementary Figure S2).
The antitumor activity of Dbait in association with RT has already been demonstrated [18], [19]. However, it has been observed that Dbait without transfectants or modifications had no antitumor activity as it did not enter into the tumor cells. Although Dbait complexed with PEI formulations displayed a strong antitumor activity, they revealed a weak effective dose/toxicity ratio of 0.8 with healthy skin necrosis due to the transfection agent (PEI). The necessity of using toxic transfection agents prevented progress toward clinical applications. In this study, we used an innovative formulation of Dbait, linked to cholesterol (DT01), which is efficiently taken up by tumor cells in the absence of adjuvant vectors [19], [45] and, therefore, could be proposed to patients. In vivo, DT01 seems as effective as Dbait linked with PEI but much less toxic. DT01 is a novel chemical family, belonging to the oligonucleotide pharmacological class. DT01 toxicology studies in Wistar rats and cynomolgus monkeys show that the only undesirable adverse effects observed are inflammatory responses at the injection sites that were slight to moderate, dose-dependent, and reversible after a 2-week recovery period [46]. DT01 administration in mice does not increase the sensitivity of healthy tissue surrounding the tumor to irradiation. As Dbait does not induce cell cycle arrest in melanoma cell lines (Supplementary Figure S3)[17], the specificity of action of DT01 in tumor cells may be due to the impaired cell cycle controls (checkpoints) frequent in tumor cells that allow cells to divide despite DT01 consequent unrepaired breaks and therefore enter mitotic catastrophe. This impaired cell cycle controls are often associated with p53 mutations [47]. In contrast, non-tumor cells with proficient cell cycle control stop dividing until repair completion, which can take place after DT01 disappearance [48]. Therefore, DT01 simply prevents DNA repair of RT-induced damage without inducing new lesions on chromosomes, leading to toxicity of dividing tumor cells and not of healthy tissues.
Here, we demonstrate that DT01 exhibited a moderate stand-alone antitumor activity, which could be explained by an accumulation of unrepairable spontaneous DNA breaks during cell proliferation as suggested by the presence of micronuclei in tumors. Conversion of this basal damage and replication stress into severe lesions (unrepaired DSB) by DT01-induced DNA repair inhibition might trigger cell death and thus the moderate stand-alone activity of the molecule. When the treatments were combined, DT01 improved RT efficacy leading to a pronounced increase in animal survival. In patients, in case of recurrence in an already irradiated zone, a second full cycle of treatment combining RT + Dbait will not be feasible. We therefore tested additional DT01 treatment alone: this led to a longer control of tumor growth and, in some cases, even complete response. The tumor growth control combined to micronuclei assessment, 50 days after the first treatment cycle, showed that the first RT + DT01 treatment triggered long-term tumor genetic instability conferring dependency of the tumor to constitutive repair activities [49], [50], [51]. This dependency could sensitize the tumor cells to a second cycle with a stand-alone DT01 treatment. These findings suggest that it may be valuable to repeat Dbait administration after the initial irradiation.
Our results provide the preclinical proof of concept that combining RT with DT01 inhibition of DNA repair could be of benefit to patients with cutaneous melanoma. A first-in-human phase I trial (DRIIM: clinicaltrials.gov/ct2/show/NCT01469455) is currently under way to evaluate the tolerance and efficacy of local DT01 administration in association with RT in patients suffering from in-transit metastases of melanoma. DT01 and RT are delivered according to the palliative protocol described in this manuscript. Patients who show treatment-associated benefits may be proposed an additional course of DT01 treatment after the end of follow-up. If DT01 safety and efficacy are confirmed, the preclinical results we report suggest that a clinical trial with more radical doses of RT and DT01 could be considered. This approach may provide an innovative option for neoadjuvant or radical treatment strategies for inoperable, locoregional, newly diagnosed, or recurrent melanoma.
Acknowledgments
We thank Nathalie Berthault (Institut Curie) for statistical analyses, Marie-Christine Lienafa (DNA Therapeutics), Christophe Roulin (Institut Curie), and Aurélie Herbette (DNA Therapeutics) for their technical assistance, and Nirmitha Herath for her advice and review of this paper. This work benefited from the technical support of the Institut Curie animal facility, microscopy, and experimental RT platforms. This study was supported by the Institut Curie (translational program), the Centre National de la Recherche Scientifique, the Institut National de la Santé et de la Recherche Médicale, and the Agence Nationale de la Recherche (ANR OSEMPB03003). Conflict of interest statement: F.D., M.S., and M.Q. are employees of DNA Therapeutics. M.D. and J-S.S. are cofounders of DNA Therapeutics. All other authors have no conflicts of interest.
Footnotes
This article refers to supplementary materials, which are designated by Figures S1 to S3 and are available online at www.neoplasia.com.
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.neo.2014.08.008.
Appendix A. Supplementary Materials
Figure S1. Injections with vehicle alone (5% glucose) do not affect RT efficacy. Animals with SK28 human melanoma xenograft received six subcutaneous and intratumoral injections of 5% glucose solution (vehicle of Dbait; gray) or no injections (black) and full protocol of radiotherapy for 2 weeks. Survival was monitored as described in the Material and Methods section. The data from nine independent experiments are presented. Statistical analysis: P < .3; RR = 0.86.
Figure S2. Detection of DT01 activity (Dbait linked with cholesterol) in tumors. γ-H2AX (representing DT01 activity) was monitored in LU1205 tumors 3 hours after subcutaneous and intratumoral injections of 2 mg of Dbait. Bar, 500 μm.
Figure S3. Flow cytometric cell cycle analysis. Quantification of the 501mel and SK28 cell distribution in cell cycle 6 and 24 hours after Dbait treatment. DNA was labeled with propidium iodide.
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62:10–29. doi: 10.3322/caac.20138. [DOI] [PubMed] [Google Scholar]
- 2.Jost L. Cutaneous malignant melanoma: ESMO clinical recommendations for diagnosis, treatment and follow-up. Ann Oncol. 2007;18(Suppl. 2):ii71–ii73. doi: 10.1093/annonc/mdm045. [DOI] [PubMed] [Google Scholar]
- 3.Burmeister BH, Henderson MA, Ainslie J, Fisher R, Di Iulio J, Smithers BM, Hong A, Shannon K, Scolyer RA, Carruthers S. Adjuvant radiotherapy versus observation alone for patients at risk of lymph-node field relapse after therapeutic lymphadenectomy for melanoma: a randomised trial. Lancet Oncol. 2012;13:589–597. doi: 10.1016/S1470-2045(12)70138-9. [DOI] [PubMed] [Google Scholar]
- 4.Lutzky J. New therapeutic options in the medical management of advanced melanoma. Semin Cutan Med Surg. 2010;29:249–257. doi: 10.1016/j.sder.2010.10.006. [DOI] [PubMed] [Google Scholar]
- 5.Harwood AR, Cummings BJ. Radiotherapy for malignant melanoma: a re-appraisal. Cancer Treat Rev. 1981;8:271–282. doi: 10.1016/s0305-7372(81)80011-4. [DOI] [PubMed] [Google Scholar]
- 6.Rofstad EK. Radiation sensitivity in vitro of primary tumors and metastatic lesions of malignant melanoma. Cancer Res. 1992;52:4453–4457. [PubMed] [Google Scholar]
- 7.Paterson R. The radical X-ray treatment of carcinomata. Br J Radiol. 1936;9:671–679. [Google Scholar]
- 8.MacKee GM, Cipollaro AC, Montgomery H. Lea and Febiger; Philadelphia: 1946. X-rays and radium in the treatment of diseases of the skin. [Google Scholar]
- 9.Barranco SC, Romsdahl MM, Humphrey RM. The radiation response of human malignant melanoma cells grown in vitro. Cancer Res. 1971;31:830–833. [PubMed] [Google Scholar]
- 10.Khan N, Khan MK, Almasan A, Singh AD, Macklis R. The evolving role of radiation therapy in the management of malignant melanoma. Int J Radiat Oncol Biol Phys. 2011;80:645–654. doi: 10.1016/j.ijrobp.2010.12.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stevens G, McKay MJ. Dispelling the myths surrounding radiotherapy for treatment of cutaneous melanoma. Lancet Oncol. 2006;7:575–583. doi: 10.1016/S1470-2045(06)70758-6. [DOI] [PubMed] [Google Scholar]
- 12.Radford IR. The level of induced DNA double-strand breakage correlates with cell killing after X-irradiation. Int J Radiat Biol Relat Stud Phys Chem Med. 1985;48:45–54. doi: 10.1080/09553008514551051. [DOI] [PubMed] [Google Scholar]
- 13.Kuzminov A. Single-strand interruptions in replicating chromosomes cause double-strand breaks. Proc Natl Acad Sci U S A. 2001;98:8241–8246. doi: 10.1073/pnas.131009198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bradbury PA, Middleton MR. DNA repair pathways in drug resistance in melanoma. Anticancer Drugs. 2004;15:421–426. doi: 10.1097/01.cad.0000127665.74096.93. [DOI] [PubMed] [Google Scholar]
- 15.Wang C, Lees-Miller SP. Detection and repair of ionizing radiation-induced DNA double strand breaks: new developments in nonhomologous end joining. Int J Radiat Oncol Biol Phys. 2013;86:440–449. doi: 10.1016/j.ijrobp.2013.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Helleday T, Petermann E, Lundin C, Hodgson B, Sharma RA. DNA repair pathways as targets for cancer therapy. Nat Rev Cancer. 2008;8:193–204. doi: 10.1038/nrc2342. [DOI] [PubMed] [Google Scholar]
- 17.Quanz M, Chassoux D, Berthault N, Agrario C, Sun J-S, Dutreix M. Hyperactivation of DNA-PK by double-strand break mimicking molecules disorganizes DNA damage response. PLoS One. 2009;4:e6298. doi: 10.1371/journal.pone.0006298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Quanz M, Berthault N, Roulin C, Roy M, Herbette A, Agrario C, Alberti C, Josserand V, Coll J-L, Sastre-Garau X. Small-molecule drugs mimicking DNA damage: a new strategy for sensitizing tumors to radiotherapy. Clin Cancer Res. 2009;15:1308–1316. doi: 10.1158/1078-0432.CCR-08-2108. [DOI] [PubMed] [Google Scholar]
- 19.Berthault N, Maury B, Agrario C, Herbette A, Sun JS, Peyrieras N, Dutreix M. Comparison of distribution and activity of nanoparticles with short interfering DNA (Dbait) in various living systems. Cancer Gene Ther. 2011;18:695–706. doi: 10.1038/cgt.2011.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shiraishi T, Nielsen PE. Enhanced delivery of cell-penetrating peptide-peptide nucleic acid conjugates by endosomal disruption. Nat Protoc. 2006;1:633–636. doi: 10.1038/nprot.2006.92. [DOI] [PubMed] [Google Scholar]
- 21.Fenech M, Kirsch-Volders M, Natarajan AT, Surralles J, Crott JW, Parry J, Norppa H, Eastmond DA, Tucker JD, Thomas P. Molecular mechanisms of micronucleus, nucleoplasmic bridge and nuclear bud formation in mammalian and human cells. Mutagenesis. 2011;26:125–132. doi: 10.1093/mutage/geq052. [DOI] [PubMed] [Google Scholar]
- 22.Titenko-Holland N., Moore L.E., Smith M.T. Measurement and characterization of micronuclei in exfoliated human cells by fluorescence in situ hybridization with a centromeric probe. Mutat Res. 1994;312:39–50. doi: 10.1016/0165-1161(94)90007-8. [DOI] [PubMed] [Google Scholar]
- 23.Verbeke G, Molenberghs G. Springer; New York: 2009. Linear mixed models for longitudinal data. [Google Scholar]
- 24.Raitanen M, Rantanen V, Kulmala J, Helenius H, Grénman R, Grénman S. Supra-additive effect with concurrent paclitaxel and cisplatin in vulvar squamous cell carcinoma in vitro. Int J Cancer. 2002;100:238–243. doi: 10.1002/ijc.10472. [DOI] [PubMed] [Google Scholar]
- 25.Duez P, Dehon G, Kumps A, Dubois J. Statistics of the Comet assay: a key to discriminate between genotoxic effects. Mutagenesis. 2003;18:159–166. doi: 10.1093/mutage/18.2.159. [DOI] [PubMed] [Google Scholar]
- 26.Overgaard J, Overgaard M, Hansen PV, von der Maase H. Some factors of importance in the radiation treatment of malignant melanoma. Radiother Oncol. 1986;5:183–192. doi: 10.1016/s0167-8140(86)80048-2. [DOI] [PubMed] [Google Scholar]
- 27.Zhu Y, Hu J, Hu Y, Liu W. Targeting DNA repair pathways: a novel approach to reduce cancer therapeutic resistance. Cancer Treat Rev. 2009;35:590–596. doi: 10.1016/j.ctrv.2009.06.005. [DOI] [PubMed] [Google Scholar]
- 28.Lord CJ, Ashworth A. The DNA damage response and cancer therapy. Nature. 2012;481:287–294. doi: 10.1038/nature10760. [DOI] [PubMed] [Google Scholar]
- 29.Chalmers AJ, Lakshman M, Chan N, Bristow RG. Poly(ADP-ribose) polymerase inhibition as a model for synthetic lethality in developing radiation oncology targets. Semin Radiat Oncol. 2010;20:274–281. doi: 10.1016/j.semradonc.2010.06.001. [DOI] [PubMed] [Google Scholar]
- 30.Sarkaria JN. Identifying inhibitors of ATM and ATR kinase activities. Methods Mol Med. 2003;85:49–56. doi: 10.1385/1-59259-380-1:49. [DOI] [PubMed] [Google Scholar]
- 31.Shinohara ET, Geng L, Tan J, Chen H, Shir Y, Edwards E, Halbrook J, Kesicki EA, Kashishian A, Hallahan DE. DNA-dependent protein kinase is a molecular target for the development of noncytotoxic radiation-sensitizing drugs. Cancer Res. 2005;65:4987–4992. doi: 10.1158/0008-5472.CAN-04-4250. [DOI] [PubMed] [Google Scholar]
- 32.Kashishian A, Douangpanya H, Clark D, Schlachter ST, Eary CT, Schiro JG, Huang H, Burgess LE, Kesicki EA, Halbrook J. DNA-dependent protein kinase inhibitors as drug candidates for the treatment of cancer. Mol Cancer Ther. 2003;2:1257–1264. [PubMed] [Google Scholar]
- 33.Raju U, Riesterer O, Wang Z-Q, Molkentine DP, Molkentine JM, Johnson FM, Glisson B, Milas L, Ang KK. Dasatinib, a multi-kinase inhibitor increased radiation sensitivity by interfering with nuclear localization of epidermal growth factor receptor and by blocking DNA repair pathways. Radiother Oncol. 2012;105:241–249. doi: 10.1016/j.radonc.2012.08.010. [DOI] [PubMed] [Google Scholar]
- 34.Croset A, Cordelières FP, Berthault N, Buhler C, Sun J-S, Quanz M, Dutreix M. Inhibition of DNA damage repair by artificial activation of PARP with siDNA. Nucleic Acids Res. 2013;41:7344–7355. doi: 10.1093/nar/gkt522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Curtis SB. Lethal and potentially lethal lesions induced by radiation—a unified repair model. Radiat Res. 1986;106:252–270. [PubMed] [Google Scholar]
- 36.Seymour CB, Mothersill C. Lethal mutations, the survival curve shoulder and split-dose recovery. Int J Radiat Biol. 1989;56:999–1010. doi: 10.1080/09553008914552451. [DOI] [PubMed] [Google Scholar]
- 37.Goodhead DT. Saturable repair models of radiation action in mammalian cells. Radiat Res Suppl. 1985;8:S58–S67. [PubMed] [Google Scholar]
- 38.Good LM, Miller MD, High WA. Intralesional agents in the management of cutaneous malignancy: a review. J Am Acad Dermatol. 2011;64:413–422. doi: 10.1016/j.jaad.2009.12.013. [DOI] [PubMed] [Google Scholar]
- 39.Thompson JF, Hersey P, Wachter E. Chemoablation of metastatic melanoma using intralesional Rose Bengal. Melanoma Res. 2008;18:405–411. doi: 10.1097/CMR.0b013e32831328c7. [DOI] [PubMed] [Google Scholar]
- 40.Oratz R, Hauschild A, Sebastian G, Schadendorf D, Castro D, Bröcker E-B, Orenberg EK. Intratumoral cisplatin/adrenaline injectable gel for the treatment of patients with cutaneous and soft tissue metastases of malignant melanoma. Melanoma Res. 2003;13:59–66. doi: 10.1097/00008390-200302000-00010. [DOI] [PubMed] [Google Scholar]
- 41.Green DS, Bodman-Smith MD, Dalgleish AG, Fischer MD. Phase I/II study of topical imiquimod and intralesional interleukin-2 in the treatment of accessible metastases in malignant melanoma. Br J Dermatol. 2007;156:337–345. doi: 10.1111/j.1365-2133.2006.07664.x. [DOI] [PubMed] [Google Scholar]
- 42.Byrne CM, Thompson JF, Johnston H, Hersey P, Quinn MJ, Michael Hughes T, McCarthy WH. Treatment of metastatic melanoma using electroporation therapy with bleomycin (electrochemotherapy) Melanoma Res. 2005;15:45–51. doi: 10.1097/00008390-200502000-00008. [DOI] [PubMed] [Google Scholar]
- 43.Ogawa Y, Kubota K, Ue H, Tadokoro M, Matsui R, Yamanishi T, Hamada N, Kariya S, Nishioka A, Nakajima H. Safety and effectiveness of a new enzyme-targeting radiosensitization treatment (KORTUC II) for intratumoral injection for low-LET radioresistant tumors. Int J Oncol. 2011;39:553–560. doi: 10.3892/ijo.2011.1069. [DOI] [PubMed] [Google Scholar]
- 44.Foote MC, Burmeister BH, Thomas J, Mark Smithers B. A novel treatment for metastatic melanoma with intralesional rose bengal and radiotherapy: a case series. Melanoma Res. 2010;20:48–51. doi: 10.1097/CMR.0b013e328331caa2. [DOI] [PubMed] [Google Scholar]
- 45.Manoharan M. Oligonucleotide conjugates as potential antisense drugs with improved uptake, biodistribution, targeted delivery, and mechanism of action. Antisense Nucleic Acid Drug Dev. 2002;12:103–128. doi: 10.1089/108729002760070849. [DOI] [PubMed] [Google Scholar]
- 46.Schlegel A, Buhler C, Devun F, Agrario C, Urien S, Lokiec F, Sun J-S, Dutreix M. Pharmacokinetics and toxicity in rats and monkeys of coDbait: a therapeutic double-stranded DNA oligonucleotide conjugated to cholesterol. Mol Ther Nucleic Acids. 2012;1:e33. doi: 10.1038/mtna.2012.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Roemer K. Mutant p53: gain-of-function oncoproteins and wild-type p53 inactivators. Biol Chem. 1999;380:879–887. doi: 10.1515/BC.1999.108. [DOI] [PubMed] [Google Scholar]
- 48.Vermeulen K, Van Bockstaele DR, Berneman ZN. The cell cycle: a review of regulation, deregulation and therapeutic targets in cancer. Cell Prolif. 2003;36:131–149. doi: 10.1046/j.1365-2184.2003.00266.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shaheen M, Allen C, Nickoloff JA, Hromas R. Synthetic lethality: exploiting the addiction of cancer to DNA repair. Blood. 2011;117:6074–6082. doi: 10.1182/blood-2011-01-313734. [DOI] [PubMed] [Google Scholar]
- 50.Jiang H, Reinhardt HC, Bartkova J, Tommiska J, Blomqvist C, Nevanlinna H, Bartek J, Yaffe MB, Hemann MT. The combined status of ATM and p53 link tumor development with therapeutic response. Genes Dev. 2009;23:1895–1909. doi: 10.1101/gad.1815309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Williamson EA, Hromas R. Repressing DNA repair to enhance chemotherapy: targeting MyD88 in colon cancer. J Natl Cancer Inst. 2013;105:926–927. doi: 10.1093/jnci/djt148. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Injections with vehicle alone (5% glucose) do not affect RT efficacy. Animals with SK28 human melanoma xenograft received six subcutaneous and intratumoral injections of 5% glucose solution (vehicle of Dbait; gray) or no injections (black) and full protocol of radiotherapy for 2 weeks. Survival was monitored as described in the Material and Methods section. The data from nine independent experiments are presented. Statistical analysis: P < .3; RR = 0.86.
Figure S2. Detection of DT01 activity (Dbait linked with cholesterol) in tumors. γ-H2AX (representing DT01 activity) was monitored in LU1205 tumors 3 hours after subcutaneous and intratumoral injections of 2 mg of Dbait. Bar, 500 μm.
Figure S3. Flow cytometric cell cycle analysis. Quantification of the 501mel and SK28 cell distribution in cell cycle 6 and 24 hours after Dbait treatment. DNA was labeled with propidium iodide.