

Abstract
Cancer stem cells (CSC) and genes have been linked to cancer development and therapeutic resistance, but the signaling mechanisms regulating CSC genes and phenotype are incompletely understood. CK2 has emerged as a key signal serine/threonine kinase that modulates diverse signal cascades regulating cell fate and growth. We previously showed that CK2 is often aberrantly expressed and activated in head and neck squamous cell carcinomas (HNSCC), concomitantly with mutant (mt) tumor suppressor TP53, and inactivation of its family member, TAp73. Unexpectedly, we observed that classical stem cell genes Nanog, Sox2, and Oct4, are overexpressed in HNSCC with inactivated TAp73 and mtTP53. However, the potential relationship between CK2, TAp73 inactivation, and CSC phenotype is unknown. We reveal that inhibition of CK2 by pharmacologic inhibitors or siRNA inhibits the expression of CSC genes and side population (SP), while enhancing TAp73 mRNA and protein expression. Conversely, CK2 inhibitor attenuation of CSC protein expression and the SP by was abrogated by TAp73 siRNA. Bioinformatic analysis uncovered a single predicted CK2 threonine phosphorylation site (T27) within the N-terminal transactivation domain of TAp73. Nuclear CK2 and TAp73 interaction, confirmed by co-immunoprecipitation, was attenuated by CK2 inhibitor, or a T27A point-mutation of this predicted CK2 threonine phospho-acceptor site of TAp73. Further, T27A mutation attenuated phosphorylation, while enhancing TAp73 function in repressing CSC gene expression and SP cells. A new CK2 inhibitor, CX-4945, inhibited CSC related SP cells, clonogenic survival, and spheroid formation. Our study unveils a novel regulatory mechanism whereby aberrant CK2 signaling inhibits TAp73 to promote the expression of CSC genes and phenotype.
Abbreviations: CK2, Casein Kinase 2; CSC, Cancer Stem Cells; DMAT, 2-Dimethylamino-4,5,6,7-tetrabromo-1H-benzimidazole; HNSCC, Head and neck squamous cell carcinoma; HEKA, Human epidermal keratinocytes; HOK, Human oral keratinocytes; mt, Mutant; SP, Side population; TAp73, Transactivating p73; TP53, Transforming Protein p53; UM-SCC, University of Michigan Squamous Cell Carcinoma; wt, Wild-type
Introduction
The development of cancers has recently been linked to a small subset of cells capable of reproducing the cancer cell population and forming tumors, designated as tumor-initiating or cancer stem cells (CSC) [1], [2]. In head and neck squamous cell carcinomas (HNSCC), cells with CSC-like phenotype and tumor forming properties have been identified in tumors and cell lines [2], [3], [4], [5], [6]. Recently, HNSCC CSC were found to be enriched within the “side population” (SP) of cells excluding Hoechst dye 33342 by fluorescence activated cell sorter analysis [6], a phenotype also associated with export and resistance to chemotherapy. Such isolated SP cells, when compared to non-SP cells, differentially expressed stem cell gene markers BMI-1 and ABCG2 transporter, formed self-replicating spheroids in vitro, and initiated tumors, characteristic of CSCs. Genes encoding key stem cell factors that promote the developmental stem cell phenotype, including Sox2, Oct4 and Nanog, are also increased within tumors and CSC in HNSCC [7]. Sox2, Oct4, and Nanog activation, target gene regulation, and the CSC phenotype are inducible, supporting their functional importance in HNSCC CSCs. However, the signal and transcription factors orchestrating expression of these genes and the CSC phenotype in HNSCC are incompletely understood.
Among possible candidates, CK2 (formerly casein kinase II) has emerged as a key signal serine/threonine kinase that modulates diverse proteins and target cascades to regulate cell fate and growth [8]. CK2 is dysregulated in most cancers examined, including HNSCC, where it is aberrantly expressed and activated [8], [9], [10]. CK2 is detected as a tetrameric complex comprised of catalytic α and/or α′ and regulatory β subunits in the cytoplasm that mediate cell signaling. Additionally, catalytic CK2α subunits have also been found to be localized to the nucleus and complexed with chromatin, suggesting a potential role for CK2α in regulating gene transcription and expression [10]. Supporting this possibility, we demonstrated that CK2α is a key mediator repressing expression and function of the critical transcription factor and tumor suppressor TP53, in a subset of HNSCC with wild type TP53 genotype [11]. Knockdown of CK2 by siRNA, particularly CK2α, increased TP53 mRNA and protein expression, inducing TP53-mediated growth arrest and apoptosis in vitro, and inhibiting tumorigenesis of wtTP53 HNSCC xenografts in vivo [11]. Intriguingly, TP53 activated by ultraviolet light-induced DNA damage has also been previously implicated in terminating embryonic stem cell renewal, by suppressing Nanog transcription and expression [12]. Unfortunately, TP53 is directly mutated in the majority of epithelial malignancies, and > 70% of HNSCC [13], compromising its potential to suppress CSC gene expression and tumorigenesis. However, the TP53 family also includes p63 and p73, which are implicated in regulation of self-renewal and programmed cell death and differentiation of squamous epithelia [14], [15]. These observations raise the question whether these TP53 homologues that control physiological epithelial self-renewal and differentiation may also be dysregulated by CK2 to unleash the expression of stem cell genes and phenotype in cancer.
We recently showed that HNSCC with mtTP53 often retain and overexpress related family member, TAp73, which has the potential to replace TP53 function [16]. TAp73 has an N-terminal transactivation (TA) domain which shares homology, transactivating, and tumor suppressor function with TP53. In HNSCC with mtTP53, our studies revealed that TAp73 is capable of repressing expression of key TP53 target growth arrest and apoptotic genes including p21, NOXA and PUMA. However, although overexpressed, TAp73 is inactivated by a reversible mechanism involving inflammatory signaling and displacement from p53 promoter response elements by ΔNp63α, a p63 isoform lacking the full N-terminal TA domain. Whether and how CK2 signaling may contribute to TAp73 inactivation, and CSC gene expression and phenotype, is unknown, but could provide a potential mechanism to target for prevention of malignant progression in cells after mutation of TP53.
In the present study, we noted from gene expression profiling that Sox2, Oct4 and Nanog gene expression is increased in HNSCC lines in which TAp73 was increased but inactivated, and in the side population previously demonstrated to contain CSC [6]. Thus, we hypothesized that CK2 signaling may inactivate TAp73 to promote CSC gene expression and phenotype in HNSCC with mtTP53. Here, we examined whether CK2 mediates inactivation of TAp73, to orchestrate expression of key CSC-related transcription factor genes Nanog, Sox2 and Oct4, the side population, clonogenic survival, and sphere forming CSC phenotypes in HNSCC expressing TAp73 with mtTP53.
Materials and Methods
Cell Lines
The UM-SCC cell lines were obtained from Dr. Thomas E. Carey, University of Michigan, and re-genotyped and origin confirmed in 2010 [17]. Genotyped stocks were frozen and used within 3 months of thawing. Expression of TP53, p63, and p73 isoforms and TP53 sequence for exons 4 to 9 was confirmed in our laboratory as previously reported [16], [18]. Primary human epidermal keratinocytes (HEKA) or Oral Keratinocytes (HOK) were cultured in accordance with the supplier's protocol (Invitrogen) and used within 5 passages.
Reagents, siRNA and Plasmid Transfection
CK2 inhibitor 2-dimethylamino-4,5,6,7-tetrabromo-1H-benzimidazole (DMAT) was from Calbiochem and used as described previously [11]. CX-4945 is a novel selective CK2 inhibitor [19] obtained from Cylene Pharmaceuticals under a Materials Transfer Agreement with NIDCD. The oligonucleotide sequences for TAp73 specific siRNA inhibition were: 5′r(CGGAUUCCAGCAUGGACGU)d(TT)3′and 5′r(ACGUCCAUGCUGGAAUCCG).
d(TT)3′ (Integrated DNA Technologies, IDT). The CK2 specific siRNAs were from Dharmacon/Thermo Scientific, CK2A1, siGENOME SMARTpool (Cat# M-003475-03); CK2A2 ON-TARGET plus SMARTpool (Cat# L-004752-00); CK2B, ON-TARGETplus SMARTpool (Cat# L-007679-00); Control siRNA, ON-TARGETplus Non-targeting Pool (Cat# D-001810-10-05). The p53/p73 specific response element pG13-luc, PUMA-luc, and p21/WAF1-luc luciferase reporter genes were kindly provided by Dr. Alex Zaika, Vanderbilt University [20]. The expression vector containing a human Flag-pcDNA3-TAp73 was kindly provided by Dr. Zhi-Min Yuan, Harvard University [21]. The TAp73-T27A mutant, in which Thr-27 was substituted to Ala (T27A), was synthesized by GENEWIZ, Inc, and sequence verified. All transfections were performed using Lipofectamine 2000 according to the manufacturer's instructions (Invitrogen/Life Technology). Each sample was assayed in triplicate and data were presented as mean ± SD.
Western Blot and Coimmunopreciptiations
Western blot analysis and co-immunoprecipitation was performed as previously [16] with antibodies indicated, CK2α (Santa Cruz, sc-6479), CK2α′ (Santa Cruz, sc-6481), Nanog (Cell Signaling, 4903), Oct4 (Cell Signaling, 4286), Sox2 (Cell Signaling, 2748), beta-actin (Cell Signaling, 4967), TAp73 (IMGENEX,IMG-246), p73 (IMGENEX,IMG-259A), Oct-1 (Santa Cruz, sc-53830), Flag antibody(Sigma, M2), PUMA (Cell Signaling, 4976).
Real time RT-PCR
RNA isolation and cDNA synthesis were performed as previously [16]. PCR primers for TAp73(GGCTGCGACGGCTGCAGAGC; GCTCAGCAGATTGAACTGGGCCAT)were synthesized by Invitrogen, and other primers used were purchased (Applied Biosystems). Amplification conditions were: 2 minutes at 50°C and 10 minutes at 95°C, followed by 40 cycles of 15 seconds at 95°C and 1 minute at 60°C, carried out using an ABI Prism 7700 Sequence Detection System (Applied Biosystems). Relative gene expression values were calculated after normalization to 18S rRNA.
Flow cytometric analysis
Flow cytometric assay for SP cells in HNSCC was adapted from Tabor et al. [6]. We cultured the lines with control diluent culture medium, DMAT, CX-4945 and/or transfected them with different siRNAs where indicated. Both floating and adherent cells detached using trypsin-EDTA (In vitrogen) were collected, centrifuged, washed and resuspended in DMEM containing 2% FCS (staining medium) and preincubated in a 1.5-ml Eppendorf tube at 37°C for 10 minutes. Cells were labeled in the same medium at 37°C for 90 minutes with 2.5 μg/ml Hoechst 33342 dye (Sigma-Aldrich, St. Louis, MO), either alone or in combination with 50 mM verapamil (Sigma-Aldrich), as negative control. Cells were centrifuged and resuspended in cold DMEM and filtered through 40 μm mesh. Propidium iodine (BD Biosciences, San Diego, CA) was added at 2 μg/ml for detection of dead cells. Cells were washed twice with cold PBS, then fixed with cold 70% ethanol and kept overnight at 4°C. Then, 3 to 5 × 104 cells were analyzed by a FACSVantage fluorescence-activated cell sorter (BD Biosciences) using dual-wavelength analysis (blue, 424 nm; red, 630 nm) after excitation with 350-nm UV light. Propidium iodide-positive dead cells (< 15%) were excluded from the analysis.
In vitro kinase assays
The Flag-cDNA3-TAp73 and Flag-cDNA3-TAp73-T27A fusion proteins were expressed in UM-SCC-46 cells and immobilized on anti-Flag-agarose beads. Anti-Flag-agarose beads were incubated with 10 mg of protein lysates of UM-SCC-46 cell 48°C for 2 hours, then washed three times with buffer (20 mM Tris–HCl at pH 7.5, 200 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 1% Triton X-100, 0.1 mM dithiothreitol, 1 mM PMSF protease inhibitor). The samples were then incubated in 400 ml buffer (100 mM Tris, pH 8.0 20 mM MgCl2, 100 mM NaCl, 50 mM KCl and 100 mM ATP containing 5 μCi of γ-[32P]-ATP) with 300 U recombinant CK2 (α2β2, New England Biolabs, P6010S) at 30°C for 30 minutes. The kinase reactions were terminated by washing the samples twice and re-suspending the samples in SDS sample buffer. The samples were boiled for 5 minutes and the proteins resolved by SDS-PAGE. Phosphorylation of Flag-TAp73 was assessed by SDS-PAGE and autoradiography of the dried gels. Loading of the recombination TAp73 protein was compared by Coomassie BlueTM-stained SDS-polyacrylamide gels.
Identification of CK2 motif in TAp73
CK2 phosphorylation sites on TAp73 were predicted using Scansite. T27 was identified as a CK2 phorphorylation site on the human TAp73 gene. Coincidence with human T27, a similar CK2 phosphorylation site T31 was predicted on mouse TAp73 gene using the Scansite program.
Clonogenic Assay
Human UM-SCC-1 and UM-SCC-46 were plated as 200cell/well in 6-well plates. Each cell line was plated in triplicate and incubated for 4 hours in CO2 incubator at 37°C to allow cells attach to the dish. Then cells were immediately treated with 0.5, 1 and 5 μM CK2 inhibitor CX4945 with DMSO as negative control. The culture medium is same as for sphere formation assay, below. After 14 days the cells were washed, fixed and stained with 0.5% crystal violet. The colonies with ≥ 50 cells were counted.
Sphere formation assay
Human UM-SCC-1 and UM-SCC-46 cells were plated as 500cell/well in 6-well ultra-low adherent dish and treated with 0.5, 1 and 5 μM CK2 inhibitor CX4945 with DMSO as negative control. The culture medium is modified as serum free Keratinocyte-SFM medium (GIBCO) containing EGF (10 ng/ml) (StemCell) and FGF (5 ng/ml) (StemCell). Spheres ≥ 50 μm were counted under the microscopy after 14 days. SCC13 cells form monolayer colonies instead of sphere and only colonies with ≥ 100 cells were counted.
Results
Expression of CSC-related markers is increased in a HNSCC subset overexpressing inactivated TAp73 and mtTP53, and their CSC-like side population
Nanog, Oct4 and Sox2 are established stem cell markers, for which expression has been studied in only a limited number of HNSCC lines, and the mechanism(s) regulating their overexpression has not been fully determined [7]. We surveyed expression of these CSC markers in a panel of 9 UM-SCC lines. We observed increased mRNA and/or protein levels of these CSC markers in a subset of cell lines compared with human epidermal keratinocytes (HEKA) or oral keratinocytes (HOK) as controls (Figure 1A and B). Interestingly, Nanog, Oct4 and Sox2 protein expression was increased in four cell lines (UM-SCC-22A, -B, -38, and -46), we previously found to exhibit increased expression but attenuated function of tumor suppressor TAp73 and mtTP53 [16]. Higher relative mRNA expression of Sox2 detected by qRT-PCR in UM-SCC-22A, -22B, -38, -46 cells was due to low signal in control HEKA. The results were verified in independent experiments, and consistent with low Sox2 protein in control cells in Figure 1B by Western blot.
Figure 1.
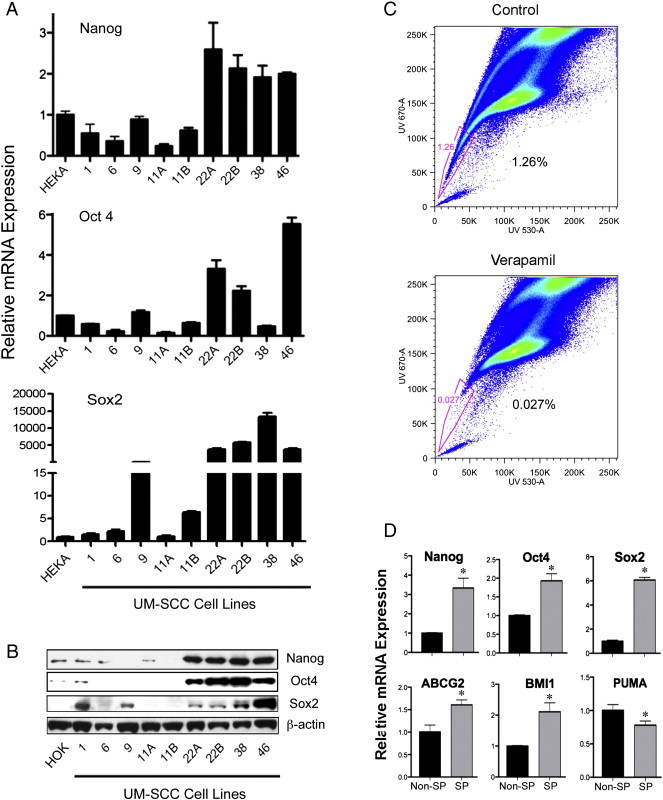
A subset of HNSCC cell lines and side population (SP) cells exhibit increased expression of CSC-related markers. A. Basal mRNA levels for Nanog, Oct4, and Sox2 were quantified by qRT-PCR in HEKA and nine UM-SCC cell lines, including 3 lines with deficient expression of wt TP 53 (UM-SCC-1, -6, -9), 2 lines lacking mtTP53 (UM-SCC-11A, -11B) and 4 lines overexpressing TAp73 with mt TP53 (UM-SCC-22A, -22B, -38 and -46)(16). Higher relative expression of Sox2 in UM-SCC was due to low signal in control HEKA, verified in an independent experiment and consistent with low Sox2 protein in control in panel B by Western blot. B. Basal expression of Nanog, Oct4, and Sox2 proteins was analyzed by Western blot in whole cell lysates of control HOK and 9 UM-SCC cell lines, with β-actin used as control for loading. C. UM-SCC-46 cell lines were labeled with Hoechst 33342 dye and analyzed by flow cytometry without and with treatment with verapamil. HoechstLow SP cells highlighted within the window indicated are quantified. D. The mRNA expression of CSC related gene markers Nanog, Oct4, Sox2, BMI1, ABCG2, and pro-apoptotic gene PUMA were assessed in non-SP and SP sub-populations sorted by FACS, using QRT-PCR. The expression level in non-SP cells is normalized to 1.0. Columns, mean between triplicate samples; bars, SD. *, Student t test, P < .05.
The side population (SP), which exhibits low Hoechst dye 33342 uptake by UV fluorescence activated cell sorter (FACS) analysis, has previously been shown to be enriched for CSC markers, anchorage independent sphere formation, and tumorigenic capacity in a variety of cancers, including HNSCC [6]. We identified the SP in UM-SCC-46 from the subset expressing the CSC markers with TAp73 and mtTP53 (Figure 1C). The SP gated in UM-SCC-46 was significantly reduced by treatment with verapamil, a blocker of calcium-dependent Hoechst dye exclusion, previously shown to be a feature of the SP containing CSC in HNSCC and other cancers that express ABC transporters [6]. The SP displayed significantly increased expression of Nanog, Oct4, and Sox2 (Figure 1D), which are key transcription factors implicated in epithelial SP and CSC [1], [2], [7]. Further, we confirmed that the SP isolated by FACS exhibited significantly increased expression of mRNA for independent CSC markers BMI-1, and transporter ABCG2, compared with non-SP cells (Figure 1D, P < .05), as previously reported for HNSCC [6]. By contrast, expression of pro-apoptotic gene PUMA, a TP53/TAp73 target which exhibits reduced expression in HNSCC [16], was also slightly reduced in SP cells (Figure 1D, P < .05). Thus, increased expression of multiple established CSC transcription factors and markers is detected in a subset of HNSCC lines, and enriched in the SP of UM-SCC-46, one of the lines with increased expression of inactivated TAp73 with mtTP53.
CK2 inhibitor or siRNA reduce CSC gene and protein expression and the side population
As we previously observed that inhibition of protein kinase CK2 inhibits HNSCC tumorigenesis [11], we examined if established CK2 inhibitor (DMAT) or silencing RNAs (siRNA) could inhibit the CSC markers, utilizing two independent cell lines from the subset overexpressing these CSC markers and TAp73. CK2 inhibitor DMAT significantly reduced Nanog, Oct4, and Sox2 mRNA expression in both UM-SCC-22A and -46 (Figure 2A). DMAT had corresponding inhibitory effects on expression of these proteins in UM-SCC-46 (Figure 2B), and -22A (Suppl. Figure 1A). Combining siRNAs targeting both CK2α/α′ catalytic subunits inhibited most of the CSC marker mRNAs in the 2 cell lines except Oct4 in UM-SCC-22A (Figure 2C), but did inhibit all three CSC markers at the protein level in UM-SCC-46 (Figure 2D) and -22A (Suppl. Figure1B). The individual CK2α and α′ subunit siRNAs had a variable effect on expression of the different CSC marker mRNA and proteins (Figure 2, C and D, Suppl. Figure 1, A and B), similar to that observed previously for multiple genes in other cell lines [11]. Corresponding to the effects of DMAT on the CSC markers, is a marked reduction in SP cells (Figure 2E). Normalized to control (0.97 = 100%), DMAT decreased SP by 65% and 96% at 10 and 20 μM, respectively (Figure 2E). Similarly, siRNA targeting both CK2α/α′ catalytic subunits potently reduced the SP, when compared with transfection with scrambled control siRNA (Figure 2F). These inhibitory effects of CK2 inhibitor DMAT or siRNA suggest that CK2 may be important in regulation of CSC genes and the side population phenotype in HNSCC.
Figure 2.
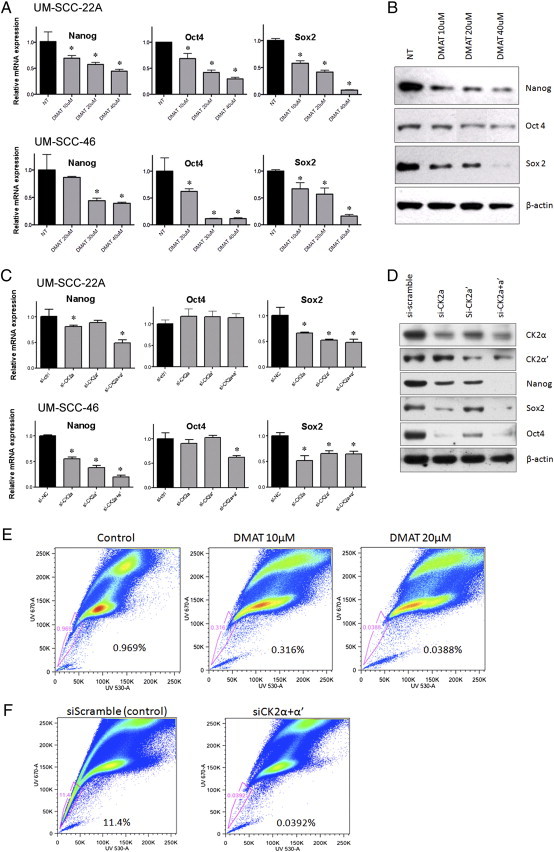
CK2 inhibition by DMAT or CK2α/α′ siRNA suppresses CSC-related SP cells and markers in two independent HNSCC lines. A. CK2 inhibitor DMAT attenuates CSC marker gene Nanog, Oct4, and Sox2 mRNA expression in UM-SCC-22A (Upper panel) and UM-SCC-46 (Lower panel). Quantitation of target genes was assessed using QRT-PCR with the treatment of increasing concentration of DMAT. Bars, SD. *Reduced versus non-treated, student t test, P < .05. B. Western blot showing Nanog, Oct4, and Sox2 protein expression are decreased in whole cell extracts from UM-SCC-46 48 hours after treatment with increasing concentrations of DMAT. β-Actin was used as loading control. Similar results for UM-SCC-22A are shown in Suppl Figure 1A. C. Effect of individual and combined CK2α and α′ siRNA knockdown on CSC marker Nanog, Oct4, and Sox2 mRNA expression in UM-SCC-22A (Upper panel) and UM-SCC-46 (Lower panel). The expression level in control siRNA-treated sample is normalized to 1.0. Columns, mean between triplicate samples; bars, SD. *Student t test, P < 0.05. D. Effect of individual and combined CK2α and α′ siRNA knockdown on CSC marker protein expression in UM-SCC-46, 48 hours after transfection with siRNA of CK2α, CK2α′, CK2α + α′, and scramble siRNA control. Nanog, Sox2, Oct4, CK2α, CK2α′ were detected, with β-actin used as loading control. Similar results for UM-SCC-22A are shown in Suppl Figure 1B. E. CK2 inhibitor DMAT and F. CK2α/α′ siRNAs inhibit SP cell detection in HNSCC. UM-SCC-46 cells pre-treated with 10 or 20 μM DMAT or DMSO diluent containing medium alone, or 100 nM scrambled control or CK2α/α′ siRNAs were labeled with Hoechst 33342 dye, and analyzed by flow cytometry. The percent HoechstLow SP cells highlighted within the window indicated were reduced by CK2 inhibitors.
CK2 inhibitor and siRNA increase expression and function of TP53 family member TAp73 which acts as a suppressor of CSC genes and the side population
We previously observed that CK2α suppresses TP53 and TAp63 family member gene expression [11]. Thus, we explored if CK2α modulates the homologous TAp73 tumor suppressor isoform, and whether CSC gene signatures and the SP cell phenotype are regulated by a mechanism involving TAp73. CK2 inhibition by DMAT or CK2α siRNA similarly enhanced expression of TAp73 mRNA in UM-SCC-46 (Figure 3A), supporting a role for CK2α in repression of TAp73 gene expression. DMAT also increased TAp73 but not faster migrating ΔNp73 isoforms as detected by TAp73 or p73 antibodies when compared to constitutive Oct1 as a loading control in nuclear extracts (Figure 3B), a requisite for possible tumor suppressor function in gene regulation. Conversely, TAp73 siRNA knockdown resulted in dose dependent enhancement of Oct4 and Nanog mRNA expression (Figure 3C). TAp73 siRNA knockdown in UM-SCC-22A cells enhanced Sox2 mRNA expression (Suppl. Figure 2A), but not in UM-SCC-46 cells (Suppl. Figure 2B). Furthermore, a decrease of Sox2, Oct4 and Nanog protein expression with CK2 inhibitor DMAT treatment was attenuated by TAp73 siRNA knockdown (Figure 3D). Together, these findings suggest CK2 inhibitor modulation of TAp73 expression and/or function may contribute to repression of these proteins. To further confirm the potential of CK2 inhibition to enhance TAp73 function as a tumor suppressor, we examined the effects of DMAT and CK2α siRNA on classical TP53/TAp73 inducible genes. CK2 inhibition by DMAT or CK2α siRNA had a reciprocal effect, enhancing TAp73 inducible TP53/TAp73 response element specific reporter pG13, as well as growth arrest and apoptotic genes CDKN2A(p21), and PUMA (Suppl Figure 3A, B) [16], [20]. This effect of CK2 inhibition was also confirmed to be TAp73 dependent, requiring co-expression of TAp73-Flag (Suppl Figure 3C-E), in cell line UM-SCC-1, which is deficient for TAp73 [16]. Furthermore, DMAT inhibited SP cells, while TAp73 knockdown by siRNA strongly increased the number of SP cells, in the absence or presence of DMAT (Figure 3E). Together, these results support the hypothesis that CK2-mediated inactivation of TAp73 promotes CSC gene expression and the SP phenotype, while inhibiting growth arrest and apoptotic genes, and that this is reversible by CK2 inhibition.
Figure 3.
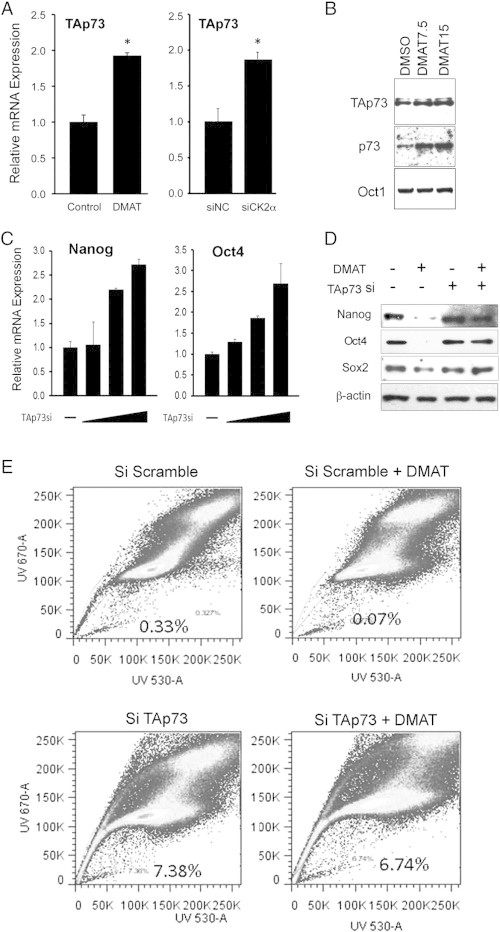
CK2 inhibition enhanced TAp73 expression and TAp73 dependent suppression of CSC-related markers and SP cells. A. TAp73 mRNA expression was significantly increased 48 hours after treatment with CK2 inhibitor DMAT (left) or transfection with CK2α siRNA (right) in UM-SCC46 cells. B. TAp73 and total p73 protein expression was increased in nuclear extracts 48 hours after UM-SCC-46 cells were treated with increasing concentrations of CK2 inhibitor DMAT, as detected by Western blot. Nuclear Oct1 is shown as a constitutive loading control. C. CSC-related Oct4 and Nanog mRNA expression was increased in UM-SCC-46 48 hours after transfection with increasing concentration of 50, 100 and 200 nM TAp73 siRNA. D. CSC-related Sox2, Oct4, and Nanog proteins were decreased 48 hours after DMAT treatment of UM-SCC-46, while TAp73 siRNA knockdown attenuated this effect. E. UM-SCC-46 cells were labeled with Hoechst 33342 dye and analyzed by flow cytometry 48 hours after transfection with control scrambled siRNA or TAp73 siRNA −/+ DMAT. SP cell number decreased after DMAT treatment, while DMAT showed no significant effect on SP cells after TAp73 knockdown.
CK2α interaction with TAp73 is inhibited by DMAT and T27A mutation of a predicted CK2 phospho-acceptor site in the transactivation domain of TAp73
Bioinformatic analysis of TAp73 as a potential substrate for CK2 serine/threonine kinase uncovered a single high probability motif containing threonine at amino acid position 27 (T27) within the transactivation (TA) domain of human TAp73 (Figure 4A; Suppl Figure 4). Supporting the importance of the predicted site, the highly conserved homologous site is found in the TA domain of human (T27) and mouse (T31) in a region of predicted surface accessibility of TAp73 protein (Suppl Figure 4). Reciprocal co-immunoprecipitations established that interaction occurs between CK2α and TAp73, and this interaction is attenuated by CK2 inhibitor DMAT in a dose dependent manner (Figure 4B). Immunoprecipitation of TAp73 with anti-TAp73, or cells transfected with TAp73-Flag with anti-Flag antibodies, showed enhanced CK2 interaction, whereas this interaction was markedly reduced upon equivalent expression of a sequence-verified T27A point-mutant of TAp73-Flag (Figure 4, C and E, top panel). Increased expression of TAp73-Flag was accompanied by increased phosphorylation, while equivalent overexpression of TAp73 with T27A point mutation of the predicted CK2 phosphoacceptor site showed markedly reduced phosphorylation when cell lysates were incubated with recombinant CK2α2β2 in an in vitro kinase assay (Figure 4, D and E, top panel). These results reveal that CK2α-TAp73 interaction and phosphorylation of TAp73 is inhibited by DMAT or T27A mutation of a predicted CK2 phosphoacceptor site in the transactivation domain of TAp73.
Figure 4.
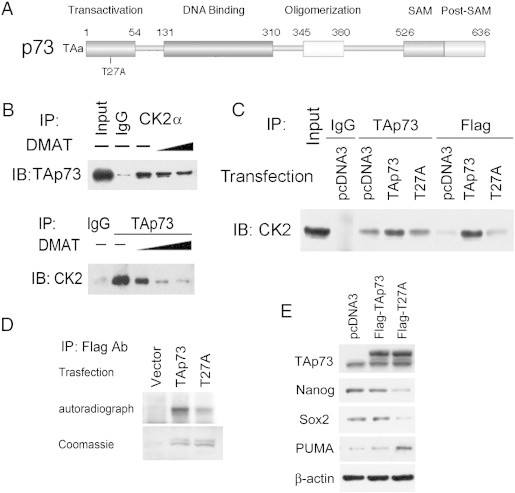
CK2 and TAp73 interaction is inhibited by CK2 inhibitor DMAT or T27A point mutation within a predicted CK2 phospho-acceptor motif in TAp73. A. CK2 interaction with TAp73 is predicted by presence of a high probability CK2 phosphorylation site at Threonine 27 (T27) within the TA domain of human TAp73 using Motifscan (Suppl Figure 4). B. Immunoprecipitation (IP) with anti-CK2α or TAp73 antibodies demonstrates reciprocal interaction between CK2α and TAp73 on immunoblotting (IB) in whole cell lysates of UM-SCC-46 cells. The interaction is decreased after treatment with increasing amount of CK2 specific inhibitor DMAT (10, 20 μM). C. Interaction between CK2α and TAp73 is decreased 48 hours after transfection with Flag-T27A-TAp73 mutant when compared with Flag-TAp73 control. Whole cell lysates from UM-SCC-46 cells were immunoprecipitated (IP) with TAp73 or Flag antibodies, and then immunoblotted (IB) with CK2α antibody. Physical interaction between CK2α and TAp73 was increased after over-expression of wild type TAp73, but decreased between Flag-T27A and CK2α. D. In vitro kinase assay shows decreased phosphorylation of TAp73 after T27A mutation. Lysates from cells transfected with empty vector, Flag-TAp73, or Flag-T27A were incubated with the recombinant CK2α2β2 protein in the presence of [γ-32P]ATP. The reaction mixtures were separated by SDS-PAGE and subjected to autoradiography (top panel). Bottom panel shows the Coomassie Brilliant Blue staining of the Flag-TAp73 fusion proteins as the loading control. E. Top panel, equivalent expression of Flag-TAp73 and Flag-T27A TAp73 in lysates used for C, D, E, obtained 48 hours after UM-SCC-46 cells were transfected with empty vector, wild type TAp73, or T27A mutant form of TAp73; lower panels, T27A TAp73 enhances inhibition of CSC markers and expression of proapoptotic protein PUMA. The protein expression of TAp73, Nanog, Sox2 and PUMA in whole cell lysates were assessed by Western blot, with β-actin as the loading control.
Mutation of the predicted CK2 T27A phosphoacceptor site enhances TAp73 inhibition of CSC marker expression and SP cells
Overexpression of TAp73, which exhibited increased CK2 interaction and phosphorylation (Figure 4, C and D), resulted in only slight inhibition of CSC markers Nanog and Sox2, or reciprocal enhancement of TAp73-inducible proapototic protein PUMA (Figure 4E). However, similar expression of the CK2 phospho-acceptor mutant T27A-TAp73 that exhibited reduced interaction and phosphorylation (Figure 4C and D), strongly repressed the CSC markers, and reciprocally enhanced TAp73 inducible proapoptotic protein PUMA (Figure 4E). Consistent with these effects, overexpression of TAp73 only partially reduced SP cells, while T27A-TAp73 strongly reduced SP cells detected (Figure 5A). Treatment with CK2 inhibitor DMAT resulted in inhibition of SP cells detected in empty vector control and TAp73 transfected cells, but had little additional effect after near complete SP inhibition observed with overexpression of T27A-TAp73 (Figure 5B). These data support that pharmacologic CK2 inhibition or prevention of T27 phosphorylation by T27A mutation enhances the repressive effect of TAp73 on these CSC markers and SP cells.
Figure 5.
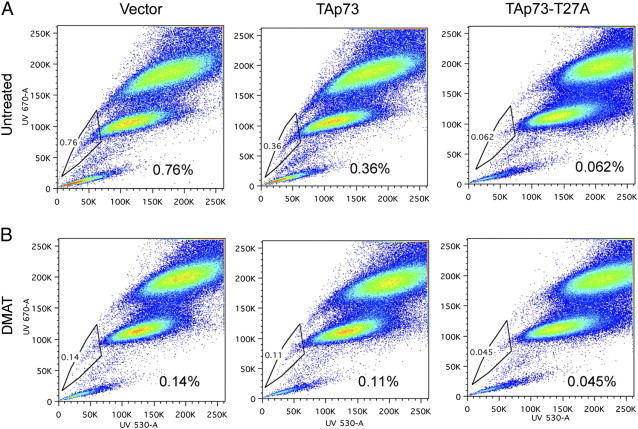
TAp73-T27A mutation of CK2 phosphoacceptor site and CK2 inhibitor DMAT suppress CSC SP cells. 48 hours after transfection with empty vector control, Flag-TAp73, or Flag-T27A TAp73, UM-SCC-46 cells were labeled with Hoechst 33342 dye and analyzed by flow cytometry. A. Without treatment. B. Treatment with DMAT 20 μM. UM-SCC-46 cells were examined for Hoechst dye uptake. HoechstLow SP cells are highlighted, and % total cells are shown.
CK2 promotes clonogenic survival and CSC spheres
To further examine if CK2 regulates CSC phenotypes, we investigated its role in clonogenic colony and tumor spheroid formation, two CSC features previously shown to correspond to SP and enhanced tumorigenicity in HNSCC (6). We used a novel selective CK2 inhibitor, CX4945, currently under investigation in clinical trials [19]. CK2 inhibitor CX-4945 significantly reduced clonogenic survival (Figure 6A) and sphere formation (Figure 6B) in UM-SCC-1 and -46, in a dose dependent manner (*P < .05). The morphologic effects of CK2 inhibitor on colony (Figure 6C) and sphere formation (Figure 6D) are linked with the SP CSC phenotype [5]. Together, these data indicate the potential of CK2 inhibition to inhibit SP, clonogenic and sphere phenotypic features, that are characteristic of CSC in HNSCC [5].
Figure 6.
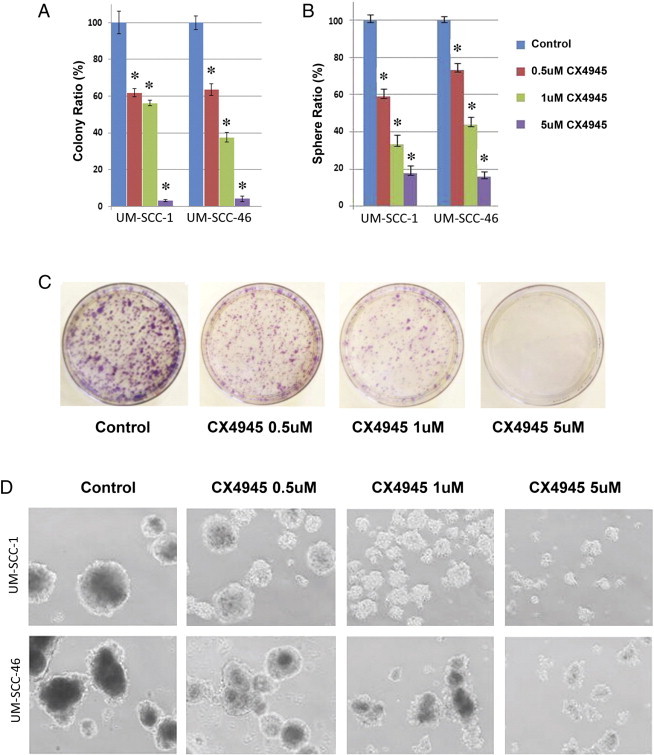
Pharmacological CK2 inhibitor CX-4945 inhibits clonogenic survival and sphere formation by HNSCC. CK2 inhibitor CX4945 inhibits clonogenic survival (A) and sphere formation (B) of UM-SCC-1 and UM-SCC-46 lines in a dose dependent manner between 0.5 to 5 μM in 3 replicate experiments. *Students t test, P < .05. C. Photomicrographs showing CX-4945 inhibition of colony formation in UM-SCC-46. D. Photomicrographs showing CX-4945 inhibition of tumor sphere formation in UM-SCC-1 and -46.
Discussion
Here, we unveil a new mechanism by which kinase CK2-mediated TAp73 phosphorylation and inactivation, enables enhanced expression of stem cell genes Nanog, Sox2, Oct4 and phenotypes that are implicated in CSC of HNSCC and other cancers (Figure 7) [1], [7]. Recent data available from The Cancer Genome Atlas provides evidence for amplification of Sox2, Oct4 and/or Nanog genes in over 20% of tumors, indicating that these genes may also be deregulated by direct genomic alterations in HNSCC [13], [22]. Unexpectedly, we observed that expression of these CSC transcription factors is increased in a subset of HNSCC lines with increased expression of inactivated TAp73 and mtTP53 [16]. Pharmacologic or siRNA inhibition of CK2 further enhanced TAp73 expression. CK2 inhibition or mutational inactivation of a predicted CK2 phospho-acceptor motif in the transactivation domain of TAp73 then restored p73 function, repressing mRNA and protein expression of these CSC transcription factor genes (Figure 7). Conversely, we found CK2 inhibition reciprocally induced TAp73-inducible growth arrest and proapoptotic genes CDKN2A(p21) and PUMA in HNSCC [16; Suppl Figure 1], indicating a pivotal role as a switch regulating genes that determine cell fate. Concordantly, CK2 inhibition attenuated the SP subset, clonogenicity, and sphere formation, linked to CSC phenotype and tumorgenicity in HNSCC and other cancers [6].
Figure 7.
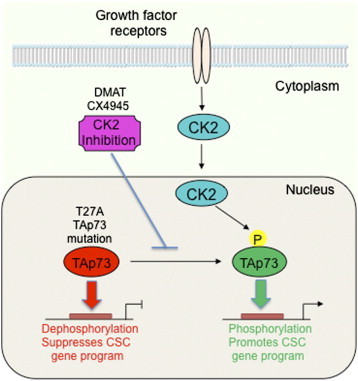
Model of the role of CK2 phosphorylation and inhibition of TAp73 in CSC gene program and phenotype. CK2 theonine/serine kinase mediates phosphorylation and inactivation of tumor suppressor TAp73, promoting expression of CSC genes and phenotype. CK2 inhibition by small molecule inhibitors DMAT or CX-4945, or T27A mutation of the CK2 phosphoacceptor motif of TAp73 uncovers TAp73 as a suppressor of CSC genes and phenotype.
We found that CK2 inhibition enhanced TAp73 expression and dependent repression of several known CSC transcription factor genes in two independent lines from an HNSCC subset overexpressing TAp73 with mtTP53. We recently demonstrated that CK2 similarly contributes to repression of TP53 and TAp63 mRNA and protein in a subset of HNSCC with wtTP53 genotype, an effect also reversible by CK2 inhibition [11]. Together, our current and prior studies indicate an important role of CK2 in pan-repression of expression and function of tumor suppressor TP53 and isoforms TAp63, and TAp73 in HNSCC subsets with wt or mtTP53. With the prevalence of TP53 mutation or inactivation in nearly all HNSCC [13], identification of key kinases reversibly regulating TAp73 inactivation is of potential biologic and therapeutic importance. Because TAp73 is rarely mutated and may function as a tumor suppressor in place of TP53 [15], [16], druggable kinase targets such as CK2 capable of reactivating TAp73 have potential for prevention or therapy.
CK2α/α′ kinase subunits are reported to be overexpressed and form nuclear complexes associated with chromatin in HNSCC and other cancers [8], [10], but their nuclear function was heretofore obscure. Previously, we observed predominantly nuclear localization of both CK2α/α′ catalytic subunits and TAp73 in HNSCC [11], [16]. We establish here that nuclear CK2 and TAp73 interact by co-IP, and this interaction is blocked by CK2 inhibitor DMAT, supporting their role in a common mechanism reversible by CK2 inhibitors. The effect of CK2 inhibitor in suppressing CSC target protein expression and SP phenotype was abrogated by TAp73 siRNA. Conversely, CK2 inhibition enhanced expression of growth arrest and pro-apoptotic genes CDKN2A(p21) and PUMA required TAp73, together supporting their dependence on the tumor suppressor function of TAp73. Further defining the nature of this interaction, we identified a threonine site (T27) within a single high probability and conserved CK2 motif within the N-terminal domain critical for CK2 interaction and transactivation function of TAp73. T27A substitution of this site attenuated CK2-TAp73 interaction, TAp73 phosphorylation, reciprocal expression of CSC marker and PUMA proteins, and the SP phenotype. CK2 inhibitor CX-4945, which is currently in clinical trials, also inhibited SCC cell clonogenicity and sphere formation in vitro. Examination of effects of CK2α/α′ inhibition delivered by nanoparticles in specimens from our previous study further indicated that CK2 inhibition can inhibit tumor growth, and enhance p73 expression in tumor in vivo [11, Y. Bian and C. Van Waes, unpublished observations]. Together, these studies implicate CK2-TAp73 interaction and T27 phosphorylation as a molecular switch that promotes cancer stem cell genes, side population, and phenotype.
A novel finding of this study is the identification of a conserved CK2 T27 phospho-acceptor site in the transactivation domain of human and murine TAp73, which is important in regulating the functional activity of TAp73. T27 was also reported as a functional inhibitory phosphorylation site for Polo-like Kinase-1 (Plk1) [[23], [24]], another key regulator promoting cell cycle progression and survival [25]. Interestingly, the TA domain of TP53 lacks this motif, but contains a different T18 motif phosphorylated by kinases ATM/ATR, that stabilize its expression and function, to promote growth arrest and apoptosis in DNA damage responses [26]. Thus, while TP53 and TAp73 can both function to suppress growth, the contrasting functions of ATR/ATM and CK2 and respective phosphorylation sites in TP53 and TAp73 proteins could underlie the important functional differences in their evolutionary roles in regulating growth arrest or promotion in damaged and replicating epithelia.
Here we reveal that CK2 inhibition and TAp73 activation mediates repression of Nanog, Oct4 and Sox2 mRNA and protein expression, and CSC SP phenotype, while promoting expression of known TAp73 inducible growth arrest and proapoptotic genes such as p21WAF1 and PUMA [14], [15], [16]. TP53 was previously implicated in binding the promoter and repressing Nanog mRNA and protein expression, and promoting differentiation of embryonic stem cells in response to DNA damage [12]. Recently, overexpression of the ΔNp73 isoform, lacking the N-terminal region and full transactivating domain, was also shown to enhance Sox2, Oct4 expression and generation of human induced pluripotent stem cells [27], supporting a broader overlap in function of TP53 and TAp73 in repressing these stem cell genes. Preliminary analysis of the promoters of Oct4, Nanog, and Sox2 reveal that they contain predicted sites for TP53/TAp73 and other transcription factors, but it is also possible that TAp73 regulated intermediary transcription factors or microRNAs co-modulate CSC genes.
Prior studies suggest that CK2 may modulate the CSC phenotype through a variety of transcriptional mechanisms. A recent study demonstrates that CK2α is associated with Hedgehog (Hh)-Gli1 and Notch1 pathway transcription factors in lung cancers, where knockdown of CK2α reduced Hh/Gli1 and Notch1 signaling and the stem-like side population [28], [29]. Knockout of the regulatory CK2β subunit in mice inhibited transcription factor Olig2, embryonic neural stem cell proliferation, and oligodendrocyte development [30]. Increased CK2α relative to CK2β is linked to upregulation of SNAIL1, TWIST1, ZEB1/2, and epithelial mesenchymal transition of breast cancer cells [31]. These observations suggest CK2 can enhance multiple pathways important in CSC. Although it is not yet known if TAp73 directly or indirectly regulates CSC genes, preliminary chromatin immunoprecipitation sequence analysis of the promoters of Oct4, Nanog, and Sox2 reveal that they contain sites for TAp73 binding (H Cheng and C Van Waes, unpublished observations). Future chromatin immunoprecipitation sequencing studies for CK2α, TAp73 and other transcription factors in promoter regulation of these genes may enhance understanding of the mechanisms and potential targets involved in orchestrating this stem cell gene program.
Here, we find that small molecule CK2 inhibitors DMAT or CX-4945 enhance TAp73 expression and function, and inhibit SP and CSC phenotypes in HNSCC with mtTP53 in vitro. Interestingly, CK2 inhibition was shown to inhibit PI3K/Akt axis signaling via Akt serine 129 [21] and induce TAp73 (this study), similar to that observed with a selective PI3K-mTOR inhibitor in our recent study [32]. Studies by others show that mTOR inhibitor rapamycin also potentiates TAp73 expression, modulates TAp73 dependent p21 expression and growth inhibition in vitro, and stem cell related gene signatures linked to prognosis in rhabdomysarcoma [33], [34]. These observations suggest CK2 and PI3K/mTOR/Akt axis signaling as well as CK2 phosphorylation of TAp73 may contribute to repression of TAp73 expression and function. We have demonstrated that potent inhibition of CK2 by antisense nanoparticles [11], or PI3K-mTOR inhibitor [32] can inhibit tumorigenesis of HNSCC in vivo. However preliminary preclinical murine xenograft studies with CK2 inhibitor CX-4945, demonstrate relatively limited inhibitory effects on established tumors in vivo, when compared with CX-4945 in vitro, where higher concentrations may be achievable. Ongoing studies suggest that inhibition of CK2 or PI3K-mTOR together with other prosurvival signal pathways involved in resistance may enhance the effects on tumor growth and survival. Thus, CK2 and other inhibitors that enhance reactivation of wtTP53 and/or TAp73 function merit further investigation for prevention and therapy of SCC.
The following are the supplementary data related to this article.
CK2 inhibition by DMAT or CK2α/α′ siRNA suppresses CSC-related SP cells and markers in UM-SCC-22A. A. Western blot showing Nanog, Oct4, and Sox2 protein expression are decreased in whole cell extracts from UM-SCC-46 48h after treatment with increasing concentrations of DMAT. β-actin was used as loading control. B. Effect of individual and combined CK2α and α′ siRNA knockdown on CSC marker protein expression in UM-SCC-22A, 48h after transfection with siRNA of CK2α, CK2α′, CK2α+α′, and scramble siRNA control. Nanog, Sox2, Oct4, CK2α, CK2α′ were detected, with β-actin used as loading control.
TAp73 and CSC-related Nanog, and Sox2 mRNA expression 48 hours after transfection and knockdown with TAp73 siRNA in. A.UM-SCC-22A and B. UM-SCC-46 cells. Cells were transfected with lipofection reagent alone, or with 100 nM control or TAp73 siRNA as described in methods. Left panels, TAp73 siRNA efficiently repressed TAp73 mRNA in both lines. Middle panels, Nanog mRNA expression was increased in both UM-SCC-22A and -46 cells. Right panels, SOX2 mRNA was increased in UM-SCC-22A but not -46 cells. Ctrl vs TAp73, * p<0.05.
CK2 inhibitor DMAT and CK2α siRNA enhancement of TP53/TAp73 response element, p21 and PUMA luciferase reporter activity is TAp73 dependent. A. CK2 inhibitor DMAT or control containing DMSO diluent or B. CK2α or control siRNA were co-transfected with pG13-Luc containing TP53/TAp73 responsive elements, or p21-Luc or PUMA-luc promoters containing TP53/TAp73 sites in UM-SCC-46,, and luciferase reporter activity was assayed after 48 hours. C. pG13-, p21-, and PUMA-luc were co-transfected without or with Flag-TAp73 and CK2α siRNA in UM-SCC-1 cell which lacks TAp73 and TP53 expression (16), and luciferase activity was assayed after 48 hours. Fold luciferase activity normalized to co-transfected beta-galactosidase reporter is shown.
Motif Scan identifies conserved CK2 Threonine motif at T27 in human and T31 in murine TAp73. The protein query for transcription factor TAp73 is represented schematically as a line, with phosphorylation site predictions for Ser/Thr residues of CK2 (including splice variations) by the Scansite program. A conserved homologous CK2 phosphorylation site was predicted in a region of surface accessibility in human (Upper panel, T27) and mouse (Lower panel, T31) TAp73 protein.
Acknowledgments
Reading and helpful comments of Drs. James Battey and James Mitchell are appreciated.
Footnotes
Grant support: HL, XXQ, XY, JZ, YB, ZC and CVW, supported by NIDCD Intramural Research Projects Z01-DC-000016 and DC-000073. CY, supported by the HHMI-NIH Scholars Program.
Conflicts of Interest: CX-4945 was obtained by Materials Transfer Agreement from Cylene Pharmaceuticals.
Contributor Information
Zhong Chen, Email: chenz@nidcd.nih.gov.
Carter Van Waes, Email: vanwaesc@nidcd.nih.gov.
References
- 1.D'Angelo R.C., Wicha M.S. Stem cells in normal development and cancer. Prog Mol Biol Transl Sci. 2010;95:113–158. doi: 10.1016/B978-0-12-385071-3.00006-X. [DOI] [PubMed] [Google Scholar]
- 2.Prince M.E., Ailes L.E. Cancer stem cells in head and neck cancer. J Clin Oncol. 2008;26:2871–2875. doi: 10.1200/JCO.2007.15.1613. [DOI] [PubMed] [Google Scholar]
- 3.Prince M.E., Sivanandan R., Kaczorowski A., Wolf G.T., Kaplan M.J., Dalerba P., Weissman I.L., Clarke M.F., Ailles L.E. Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proc Natl Acad Sci U S A. 2007;104:973–978. doi: 10.1073/pnas.0610117104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Clay M.R., Tabor M., Owen J.H., Carey T.E., Bradford C.R., Wolf G.T., Wicha M.S., Prince M.E. Single-marker identification of head and neck squamous cell carcinoma cancer stem cells with aldehyde dehydrogenase. Head Neck. 2010;32:1195–1201. doi: 10.1002/hed.21315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Janisiewicz A.M., Shin J.H., Murillo-Sauca O., Kwok S., Le Q.T., Kong C., Kaplan M.J., Sunwoo J.B. CD44(+) cells have cancer stem cell-like properties in nasopharyngeal carcinoma. Int Forum Allergy Rhinol. 2012;2:465–470. doi: 10.1002/alr.21068. [DOI] [PubMed] [Google Scholar]
- 6.Tabor M.H., Clay M.R., Owen J.H., Bradford C.R., Carey T.E., Wolf G.T., Prince M.E. Head and neck cancer stem cells: the side population. Laryngoscope. 2011;121:527–533. doi: 10.1002/lary.21032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bourguignon L.Y., Wong G., Earle C., Chen L. Hyaluronan-CD44v3 interaction with Oct4-Sox2-Nanog promotes miR-302 expression leading to self-renewal, clonal formation, and cisplatin resistance in cancer stem cells from head and neck squamous cell carcinoma. J Biol Chem. 2012;287:32800–32824. doi: 10.1074/jbc.M111.308528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Trembley J.H., Wang G., Unger G., Slaton J., Ahmed K. CK2: a key player in cancer biology. Cell Mol Life Sci. 2009;66:1858–1867. doi: 10.1007/s00018-009-9154-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gapany M., Faust R.A., Tawfic S., Davis A., Adams G.L., Ahmed K. Association of elevated protein kinase CK2 activity with aggressive behavior of squamous cell carcinoma of the head and neck. Mol Med. 1995;1:659–666. [PMC free article] [PubMed] [Google Scholar]
- 10.Faust R.A., Gapany M., Tristani P., Davis A., Adams G.L., Ahmed K. Elevated protein kinase CK2 activity in chromatin of head and neck tumors: association with malignant transformation. Cancer Lett. 1996;101:31–35. doi: 10.1016/0304-3835(96)04110-9. [DOI] [PubMed] [Google Scholar]
- 11.Brown M.S., Diallo O.T., Hu M., Ehsanian R., Yang X., Arun P., Lu H., Korman V., Unger G., Ahmed K. CK2 modulation of NF-kappaB, TP53, and the malignant phenotype in head and neck cancer by anti-CK2 oligonucleotides in vitro or in vivo via sub-50-nm nanocapsules. Clin Cancer Res. 2010;16:2295–2307. doi: 10.1158/1078-0432.CCR-09-3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lin T., Chao C., Saito S., Mazur S.J., Murphy M.E., Appella E., Xu Y. p53 induces differentiation of mouse embryonic stem cells by suppressing Nanog expression. Nat Cell Biol. 2005;7:165–171. doi: 10.1038/ncb1211. [DOI] [PubMed] [Google Scholar]
- 13.Grandis J.R., Hayes D.N., El-Naggar A.K. Proceedings of The Cancer Genome Atlas' 2nd Annual Scientific Symposium, Crystal City, VA. 2012. Comprehensive genomic characterization of squamous cell carcinoma of the head and neck; p. 22. [Google Scholar]
- 14.King K.E., Weinberg W.C. P63: Defining roles in the morphogenesis, homeostasis and neoplasia of the epidermis. Cancer Res. 2008;68:5122–5131. doi: 10.1002/mc.20337. [DOI] [PubMed] [Google Scholar]
- 15.Young De, Ellisen L.W. P63 and p73 in human cancer: defining the network. Oncogene. 2007;26:5169–5183. doi: 10.1038/sj.onc.1210337. [DOI] [PubMed] [Google Scholar]
- 16.Lu H., Yang X., Duggal P., Allen C.T., Yan B., Cohen J., Nottingham L., Romano R.A., Sinha S., King K.E. TNF-α promotes c-REL/ΔNp63α interaction and TAp73 dissociation from key genes that mediate growth arrest and apoptosis in head and neck cancer. Cancer Res. 2011;71:6867–6877. doi: 10.1158/0008-5472.CAN-11-2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brenner J.C., Graham M.P., Kumar B., Saunders L.M., Kupfer R., Lyons R.H., Bradford C.R., Carey T.E. Genotyping of 73 UM-SCC head and neck squamous cell carcinoma cell lines. Head Neck. 2010;32:417–426. doi: 10.1002/hed.21198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yan B., Yang X., Lee T.L., Friedman J., Tang J., Van Waes C., Chen Z. Genome-wide identification of novel expression signatures reveal distinct patterns and prevalence of binding motifs for p53, nuclear factor-kappaB and other signal transcription factors in head and neck squamous cell carcinoma. Genome Biol. 2007;8:R78. doi: 10.1186/gb-2007-8-5-r78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Siddiqui-Jain A., Drygin D., Streiner N., Chua P., Pierre F., O'Brien S.E., Bliesath J., Omori M., Huser N., Ho C. CX-4945, an orally bioavailable selective inhibitor of protein kinase CK2, inhibits prosurvival and angiogenic signaling and exhibits antitumor efficacy. Cancer Res. 2010;70:10288–10298. doi: 10.1158/0008-5472.CAN-10-1893. [DOI] [PubMed] [Google Scholar]
- 20.Wei J., O'Brien D., Vilgelm A., Piazuelo M.B., Correa P., Washington M.K., El-Rifai W., Peek R.M., Zaika A. Interaction of Helicobacter pylori with gastric epithelial cells is mediated by the p53 protein family. Gastroenterology. 2008;134:1412–1423. doi: 10.1053/j.gastro.2008.01.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tsai K.K., Yuan Z.M. c-Abl stabilizes p73 by a phosphorylation-augmented interaction. Cancer Res. 2003;63:3418–3424. [PubMed] [Google Scholar]
- 22.http://www.cbioportal.org/public-portal/
- 23.Soond S.M., Barry S.P., Melino G., Knight R.A., Latchman D.S., Stephanou A. p73-mediated transcriptional activity is negatively regulated by polo-like kinase 1. Cell Cycle. 2008;7:1214–1223. doi: 10.4161/cc.7.9.5777. [DOI] [PubMed] [Google Scholar]
- 24.Koida N., Ozaki T., Yamamoto H., Ono S., Koda T., Ando K., Okoshi R., Kamijo T., Omura K., Nakagawara A. Inhibitory role of Plk1 in the regulation of p73-dependent apoptosis through physical interaction and phosphorylation. J Biol Chem. 2008;283:8555–8563. doi: 10.1074/jbc.M710608200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cholewa B.D., Liu X., Ahmad N. The role of polo-like kinase 1 in carcinogenesis: cause or consequence. Cancer Res. 2013;73:6848–6855. doi: 10.1158/0008-5472.CAN-13-2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jenkins L.M., Durell S.R., Mazur S.J., Appella E. p53 N-terminal phosphorylation: a defining layer of complex regulation. Carcinogenesis. 2012;33:1441–1449. doi: 10.1093/carcin/bgs145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lin Y., Cheng Z., Yang Z., Zheng J., Lin T. DNp73 improves generation efficiency of human induced pluripotent stem cells. BMC Cell Biol. 2012;13:9. doi: 10.1186/1471-2121-13-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang S., Wang Y., Mao J.H., Hsieh D., Kim I.J., Hu L.M., Xu Z., Long H., Jablons D.M., You L. Inhibition of CK2α down-regulates Hedgehog/Gli signaling leading to a reduction of a stem-like side population in human lung cancer cells. PLoS One. 2012;7:e38996. doi: 10.1371/journal.pone.0038996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang S., Long H., Yang Y.L., Wang Y., Hsieh D., Li W., Au A., Stoppler H.J., Xu Z., Jablons D.M. Inhibition of CK2α down-regulates Notch1 signalling in lung cancer cells. J Cell Mol Med. 2013;17:854–862. doi: 10.1111/jcmm.12068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huillard E., Ziercher L., Blond O., Wong M., Deloulme J.C., Souchelnytskyi S., Baudier J., Cochet C., Buchou T. Disruption of CK2beta in embryonic neural stem cells compromises proliferation and oligodendrogenesis in the mouse telencephalon. Mol Cell Biol. 2010;30:2737–2749. doi: 10.1128/MCB.01566-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Deshiere A., Duchemin-Pelletier E., Spreux E., Ciais D., Combes F., Vandenbrouck Y., Couté Y., Mikaelian I., Giusiano S., Charpin C. Unbalanced expression of CK2 kinase subunits is sufficient to drive epithelial-to-mesenchymal transition by Snail1 induction. Oncogene. 2013;32:1373–1383. doi: 10.1038/onc.2012.165. [DOI] [PubMed] [Google Scholar]
- 32.Herzog A., Bian Y., Vander Broek R., Hall B., Coupar J., Cheng H., Sowers A.L., Cook J.D., Mitchell J.B., Chen Z. PI3K/mTOR inhibitor PF-04691502 antitumor activity is enhanced with induction of wild-type TP53 in human xenograft and murine knockout models of head and neck cancer. Clin Cancer Res. 2013;19:3808–3819. doi: 10.1158/1078-0432.CCR-12-2716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rosenbluth J.M., Mays D.J., Pino M.F., Tang L.J., Pietenpol J.A. A gene signature-based approach identifies mTOR as a regulator of p73. Mol Cell Biol. 2008;28:5951–5964. doi: 10.1128/MCB.00305-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rosenbluth J.M., Mays D.J., Jiang A., Shyr Y., Pietenpol J.A. Differential regulation of the p73 cistrome by mammalian target of rapamycin reveals transcriptional programs of mesenchymal differentiation and tumorigenesis. Proc Natl Acad Sci U S A. 2011;108:2076–2081. doi: 10.1073/pnas.1011936108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
CK2 inhibition by DMAT or CK2α/α′ siRNA suppresses CSC-related SP cells and markers in UM-SCC-22A. A. Western blot showing Nanog, Oct4, and Sox2 protein expression are decreased in whole cell extracts from UM-SCC-46 48h after treatment with increasing concentrations of DMAT. β-actin was used as loading control. B. Effect of individual and combined CK2α and α′ siRNA knockdown on CSC marker protein expression in UM-SCC-22A, 48h after transfection with siRNA of CK2α, CK2α′, CK2α+α′, and scramble siRNA control. Nanog, Sox2, Oct4, CK2α, CK2α′ were detected, with β-actin used as loading control.
TAp73 and CSC-related Nanog, and Sox2 mRNA expression 48 hours after transfection and knockdown with TAp73 siRNA in. A.UM-SCC-22A and B. UM-SCC-46 cells. Cells were transfected with lipofection reagent alone, or with 100 nM control or TAp73 siRNA as described in methods. Left panels, TAp73 siRNA efficiently repressed TAp73 mRNA in both lines. Middle panels, Nanog mRNA expression was increased in both UM-SCC-22A and -46 cells. Right panels, SOX2 mRNA was increased in UM-SCC-22A but not -46 cells. Ctrl vs TAp73, * p<0.05.
CK2 inhibitor DMAT and CK2α siRNA enhancement of TP53/TAp73 response element, p21 and PUMA luciferase reporter activity is TAp73 dependent. A. CK2 inhibitor DMAT or control containing DMSO diluent or B. CK2α or control siRNA were co-transfected with pG13-Luc containing TP53/TAp73 responsive elements, or p21-Luc or PUMA-luc promoters containing TP53/TAp73 sites in UM-SCC-46,, and luciferase reporter activity was assayed after 48 hours. C. pG13-, p21-, and PUMA-luc were co-transfected without or with Flag-TAp73 and CK2α siRNA in UM-SCC-1 cell which lacks TAp73 and TP53 expression (16), and luciferase activity was assayed after 48 hours. Fold luciferase activity normalized to co-transfected beta-galactosidase reporter is shown.
Motif Scan identifies conserved CK2 Threonine motif at T27 in human and T31 in murine TAp73. The protein query for transcription factor TAp73 is represented schematically as a line, with phosphorylation site predictions for Ser/Thr residues of CK2 (including splice variations) by the Scansite program. A conserved homologous CK2 phosphorylation site was predicted in a region of surface accessibility in human (Upper panel, T27) and mouse (Lower panel, T31) TAp73 protein.