

Abstract
Systems biology has gained a tremendous amount of interest in the last few years. This is partly due to the realization that traditional approaches focusing only on a few molecules at a time cannot describe the impact of aberrant or modulated molecular environments across a whole system. Furthermore, a hypothesis-driven study aims to prove or disprove its postulations, whereas a hypothesis-free systems approach can yield an unbiased and novel testable hypothesis as an end-result. This latter approach foregoes assumptions which predict how a biological system should react to an altered microenvironment within a cellular context, across a tissue or impacting on distant organs. Additionally, re-use of existing data by systematic data mining and re-stratification, one of the cornerstones of integrative systems biology, is also gaining attention. While tremendous efforts using a systems methodology have already yielded excellent results, it is apparent that a lack of suitable analytic tools and purpose-built databases poses a major bottleneck in applying a systematic workflow. This review addresses the current approaches used in systems analysis and obstacles often encountered in large-scale data analysis and integration which tend to go unnoticed, but have a direct impact on the final outcome of a systems approach. Its wide applicability, ranging from basic research, disease descriptors, pharmacological studies, to personalized medicine, makes this emerging approach well suited to address biological and medical questions where conventional methods are not ideal.
Keywords: Systems biology, Data integration, Pathway mapping, Computational biology
1. Introduction
In 1958 Francis Crick first discussed the central dogma of molecular biology: that information is transferred sequentially in one direction from nucleic acid to protein and cannot move in the opposite direction, which is often summarized by the phrase “DNA makes RNA makes protein” [1]. While the central dogma is still a core part of our understanding of the molecular machinery that facilitates life, the picture is of course now much more complex (Fig. 1), as has been previously discussed [2,3].
Fig. 1.
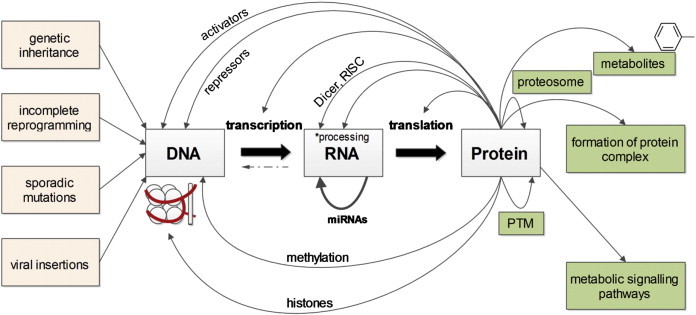
A summary of the molecular processes occurring between the common biomolecules of the cell. Arrows represent the transfer of information and/or the regulation of these transfer processes. *‘Processing’ of RNA indicates both common events such as capping or splicing and less common RNA editing events. DNA, deoxyribonucleic acid; RNA, ribonucleic acid; PTM, posttranslational modification; RISC, RNA-induced silencing complex.
We now know that in addition to the genetic information stored as the code in the form of the four bases guanine, thymine, adenine and cytosine, there is also information carried in by the modification of these bases, e.g. methylation or hydroxymethylation of cytosine. Another type of epigenetic (above genetic) modification is to the histones, the proteins which bind to DNA to form chromatin. These epigenetic changes can act in concert [4] and they contribute to changes in levels of gene expression [5] and can direct which parts of the genetic code form a resulting mature transcript via alternative promoter selection and alternative splicing [6,7]. While epigenetic changes are mostly erased during gametogenesis, they have been shown in some cases to persist through generations [8].
Similarly RNA transcripts may undergo base modifications although these are much less extensively studied [9]. While most transcripts are protein-coding as suggested by the summarization of the central dogma quoted above, many are non-coding. One class of transcripts called microRNAs serve to downregulate gene expression by cleaving their specific target mRNA sequences. Some of these miRNAs seem to target thousands of specific RNAs and are extremely highly conserved across eukaryotes [10]. As transcripts can undergo alternative splicing one gene may encode a large number of proteins by the removal of exons from pre-mRNA [11]. The protein products of these transcripts also undergo post-translational modifications before forming a mature protein product [12], so that along with splice variants and alternative start sites, one multi-exon gene has the potential to form of a vast array of proteins. Proteins of course interact with each other and with metabolites, but also assist in various nucleic acid related processes such as transcription [13] and miRNA-directed downregulation [14].
Molecular biology has undoubtedly been transformed by large-scale sequencing initiatives such as the human genome project (HGP), as evidenced by the techniques and tools which have arisen from it. ‘Completed’ genome sequences allowed the development of genome-wide DNA microarrays which soon showed how different tissues in a complex organism may have very diverse patterns of gene expression [15]. The ‘next generation’ and ‘third generation’ sequencing machines, whose production was no doubt encouraged by the HGP have allowed for the categorization of the microbiome — the collective genomes of the community of microorganisms of a particular environment. In humans this microbiotic community (or microbiota) is vast, diverse between individuals, can provide metabolic function and is implicated in various disease states — it is sometimes discussed as an organ itself [16].
Pleiotropy is the term used to describe how one gene (or rather its products) can affect several traits. This often occurs through one molecular cause having a physiological consequence with related physiological consequences or due to the gene being involved in multiple pathways with different physiological outcomes. In some cases the product of the gene may have multiple molecular functions [17] and it has been suggested that alternative splicing may contribute to pleiotropy in some cases [18]. Genes may even act as an activator and a repressor of the same process due to alternative splicing [19].
An insight from one gene expression study which investigated several individual tissues of Drosophila melanogaster showed that many genes which had known functions in one tissue were also highly expressed in unexpected tissues [20]. This suggests that many genes whose function is thought to be well understood may have alternative functions in different environments and/or are helping contribute to completely distinct physiological states.
The biophysical microenvironment in the form of changes of the micro-topographic cell characteristics can induce modifications at an epigenetic level, promoting changes on the patterns of histone acetylation and methylation therefore modifying cell reprogramming efficiency [21]. Other biophysical constraints such as cell-to-cell contact, stiffness, surface tension, and magnetic field are also involved in the interaction with molecular networks with the last being governed by non-linear dynamics [22].
The field of bioinformatics is broad, but is perhaps best described by the large biological datasets which must be manipulated via computation and tested with robust statistics. The key challenges of bioinformatics are those of data storage and algorithm development and the goal is usually a description of that individual dataset, possibly by statistics alone or by also integrating annotations in an automatic manner and interpreting the enrichment of these.
Systems biology is sometimes misunderstood as a new word for bioinformatics. While it is true that there is a certain amount of intersection between the fields, a systems biologist seeks to use bioinformatics tools, data, and databases in conjunction with clinical data and information from the literature, in order to describe as much of a system as possible or as is appropriate to the task. The addition of clinical and laboratory measurements means that systems biology need not be entirely reductionist, or at least that some additional higher scale measurements may act as a surrogate for missing low-level data. A systems approach may also make use of complex mathematical models to simulate the system or determine putative associations.
Due to the diverse range of data types and analysis methods, a systems biologist should work in collaboration with clinical scientists, lab scientists and possibly (bio)informaticians and (bio)statisticians depending on the requirements of the task and the skill set of the individual. A systems biologist may work in a team including a bioinformatician, in which case it is not necessary to do the omics data pre-processing, but should at least have a conceptual understanding of where the data has come from, including the power, limitations and possible biases involved.
Another source of confusion may be that systems biology is necessarily about kinetic mathematical models, irrespective of size. While this may be a useful methodology to a systems biologist, this is only one possible aspect and such a modeling effort should relate to a large system or should be used to validate a crucial part of that system; systems biology should aim to describe the complexity of biological systems rather than extremely small abstract sets of reactions.
The approach is commonly applied to studying normal functions, diseases, prediction of drug effects (on-target and off-target) and comparison of different species. The desired outcome of many of these applications is to establish a mode of action informed from diverse methods describing events on different biological scales, and often to use this model to make informative and useful predictions such as the identification of biomarkers or possible off-target drug effects. There is no one exact procedure to follow to reach this conclusion, as each result could suggest a different approach required, or the iteration of that analysis with a higher or lower stringency. It is, in this sense, directed partly by experience; knowing which analysis is most appropriate for data with particular features. Each step should however be well-founded in evidence and potential sources of bias sought out and accounted for where possible.
This review seeks to provide an overview of systems biology, introducing important concepts from molecular biology, statistics and bioinformatics. A good grounding in key components of these subjects is important as these fields provide the majority of the data, analysis methods and the biological framework under which systems biology is practiced. Examples of the various applications of systems biology will be described and some of the challenges for future work will be identified.
2. Data
2.1. Platforms & preprocessing
Systems biology requires large datasets, preferably with a wide genomic scope and unbiased toward particular pathways or processes. Most often the information required to form the basis of an analysis is the transcriptome, proteome or metabolome, as the data itself or fold changes derived from it may be mapped to biological pathways and used to infer systemic differences between groups.
Microarrays emerged relatively early with respect to other genome-wide technologies and may be used to gather information on a range of molecules and molecular events, but most notably they are used to measure gene expression. They are relatively cheap compared to some other omics methods, and the procedures for processing the data are now mostly well-devised. Manufacturing differs from company to company but the general concept remains the same: a probe or a set of genomically proximate probes is designed to hybridize to a transcript and fluoresce upon hybridization, and the level of fluorescence will inform on the abundance of the target molecule. The probes have genome-wide coverage and are bound to the microarray (or ‘chip’) in a 2D array of ‘spots’ or first bound to microscopic beads which are deposited in a 2D array of wells. Comparisons are only robust between samples within the same probe or probe set, not between probes or probe sets as different binding affinities, genetic variation etc. may exist between the sequences compared and act to obscure true biological variation.
When using this type of data one should study both the manufacturer's annotation materials and the literature to identify any improvements that might be made to the analysis. For example the Illumina Human Methylation 450 Beadchip has many probes which may truly report SNP differences rather than differential methylation and like many microarrays suffers from cross-hybridization [23]. It is also advantageous to either check for the latest version of annotation or create novel annotation using public resources [24]. For creating new microarray annotation, protein databases etc. one may require a large scale sequence alignment and clustering software which can handle large amounts of data and run on relatively low-powered machines such as LSCluster [25].
Just as some probes can annotate several genes, some genes are detected by several probes; it is a many-to-many (m–n) relationship that exists between probe sets and genes. Often it is the case that different probe sets for the same genes are highly correlated and where they are not, it suggests that either an un-annotated cross-hybridization or alternative splicing is occurring.
RNA-seq is an emerging alternative method for estimating gene expression by sequencing cDNA and aligning it to a reference genome. RNA-seq is a more powerful technology in that it can detect genetic variation and levels of different splice variants. Microarrays are however cheaper and simpler to analyze and it has been suggested that the two technologies should be used together to gather the broadest transcriptome coverage [26].
While any large dataset may be useful, those of most interest are the final functional products, i.e. proteins and metabolites. Mass spectrometry (MS) is most often the tool to collect this data on a large scale. It is used in combination with a method to separate the components of a sample prior to MS such as liquid chromatography, gas chromatography or capillary electrophoresis. MS itself involves ionizing these molecules, fragmenting them and measuring their mass/charge ratios. These peaks then allow the identification of the molecule relating to each profile of mass/charge peaks. Different types of separation and different MS equipment have different strengths and limitations. For example different separation techniques will be more/less suitable for differently sized or charged molecules and offer a different degree of chromatographic resolution [27,28].
Deviations in quantified MS peak intensities may arise from technical variations and result in unreliable run-to-run reproducibility. The normalization of peak intensity across all samples of a proteomics or metabolomics dataset is therefore an important step in preprocessing [29]. As the normalization affects downstream analysis it is crucial to select an appropriate method and crucial that normalization factors are independent from biological factors [30]. Out of the many possible normalization methods proposed for MS data Total Ion Count (TIC) is emerging as a gold standard. Total Ion Count simply acts to scale each spectrum such that the total area under the curve is equal [31]. After preprocessing the peaks must then be associated to proteins/metabolites.
2.2. Databases and identifiers
One of the initial challenges of working with large datasets – especially proteomics and metabolomics datasets – is the association between useful identifiers and the data itself. Many different sets of IDs exist for both, and some have become defunct over time as support for a database is dropped. While some issues such as this may be unavoidable, both database curators and users should adhere to good general practices to minimize other problems. For example, when an ID is assigned to a molecule, it should be permanently associated to it. If the molecule is deemed to be a contamination, unusual fusion protein or spurious reading then, rather than the entry being removed entirely, it should remain in the database, but marked as spurious and possibly set as being unsearchable. This way a molecule may not be removed then later returned to a database under a different ID. Conversely one ID will never be attributed to two different molecules.
Using gene symbols as IDs is not recommended as they can be duplicated and are prone to change. One recent example is CDH1, which is the Human Genome Organisation (HUGO)-designated gene name for Cadherin-1, however CDH1 is also used throughout the literature as a gene name for CDC20-like protein 1 or Fizzy-related protein homolog FZR1. Any semi- or fully-automated data extraction from the published literature may therefore inadvertently attribute molecular properties to the wrong entry. Furthermore, in some species gene symbols may contain Greek characters which are often not correctly read by various tools (e.g. “αTry” in D. melanogaster). Despite this many tools only accept these IDs (sometimes with Greek letters converted to roman words, i.e. “alphaTry”), and therefore it is often necessary to convert between types of IDs. While some conversion tools are available these issues cannot properly be addressed without a concerted effort between researchers, organizations and publishers.
If possible IDs should be constrained to the 36 (English) alphanumerical possibilities, which even with a modest ID length would allow for an adequate number of possibilities. Even with only 4 characters of this set there would be 1,679,616 resulting IDs. Whatever length of ID selected length should be retained, e.g. ‘0001’ rather than ‘1’, to avoid errors of truncation. Finally if other characters (e.g. Greek) are used in order to represent common IDs in one part of a database, the rest of the database and its associated interfaces and the user workflow proceeding from it should be made fully unicode-compatible. Care should also be taken not to use IDs from restricted-access commercial databases in publications.
The main resource for sequence data including RNAseq is the Sequence Read Archive — a public database of NGS data [32]. The International Sequence Database Collaboration (INSDC) is a collaboration between the European Bioinformatics Institute, National Center for Biotechnology Information and the DNA Data Bank of Japan who provide access to SRA, both through their own graphical user interfaces and via programmatic routes.
The Universal Protein Resource (UniProt) is a comprehensive database of protein structures and annotations. It is divided into four main components: UniProt Knowledgebase (UniProtKB), UniProt Reference Clusters (UniRef), UniProt Archive (UniParc), and UniProt Metagenomic and Environmental Sequences (UniMES) [33]. UniProtKB is divided into two sections. UniProt/TrEMBL stores translations of the sequences found in EMBL-Bank/GenBank/DDBJ, many of which are derived from SRA. While the sharing of information between large bioinformatics databases adds to the power of these databases, it can also lead to the reproduction of errors if sufficient curation is not implemented. Many UniProt/TrEMBL entries share identical sequences, and naming errors (e.g. “HLQ-DQB1” as opposed to “HLA-DQB1”) may occur throughout several resources. UniProt/Swiss-Prot is intended to contain one entry per gene and is the gold standard in protein sequence referencing and the assembly of pertinent molecular functions.
The Human Metabolome Database describes small molecule metabolites in humans. It contains over 40,000 metabolites and links three types of data: chemical, clinical, and molecular biology/biochemistry [34]. Several similar databases exist for other species. KEGG Pathway is a large, well-curated database with zoomable maps which show large sections of the metabolisms of various species [35]. MetaCyc is a similar database containing fewer compounds but a greater number of pathways and reactions, and with more extensive pathway descriptions [36]. Pathway Commons uses data from open-access databases to link out to a software called Cytoscape [37,38].
While the large datasets used for systems biology are often generated from samples gathered specifically for the study, public datasets may also be used either in conjunction with a new dataset or as the main dataset(s) for the analyses themselves. NCBI's GEO [39] and EBI's ArrayExpress [40] are the main databases for array-based data, however other data types are also now included in GEO such as mass spectrometry data. Array-based and sequencing-based data in GEO and ArrayExpress must adhere to "minimum information about a microarray experiment" (MIAME) or “minimum information about sequencing experiments” (MINSEQE) respectively [41].
2.3. Public datasets
There is currently much less support for sharing of proteomics and metabolomics datasets despite the growing need, and relatively few public datasets available. The Proteomics Identification Database (PRIDE) contains over 25,000 proteomics experiments [35]. Metabolights is a repository hosted by EBI and launched in 2012, which currently houses 39 experiments [42]. Standards for reporting proteomics and metabolomics experiments are coordinated by Human Proteome Organisation's Proteomics Standards Initiative (HUPO-PSI), and Metabolomics Standards Initiative (MSI) respectively.
While standards such as MIAME exist to ensure a certain amount of information is uploaded along with the data in public databases, some useful sample info e.g. on biological confounders or potential batch effects may be missing, or only initially gathered for some datasets. Additionally if one makes use of several different datasets the batch effects of using different cohorts should be taken into account, even if all potential biological confounders are accounted for and the same technology was used to gather the information.
Another issue with public databases of this type which are storing human data is that there is no consensus on whether consent for the uploading of the data is implicit, or whether additional consent should be sought. A survey showed that patients felt that it was very (69%) or somewhat (21%) important to be asked for additional consent [43]. This is clearly a cause for some ethical concern and in future foresight should be applied in writing ethical consent statements regarding omics data open access.
In some cases rather than searching for experimental data relating to a specific disease one may simply wish to know about the normal expression across different tissues of an organism. This can help identify possible sources of cross-talk, or help to study normal function or to compare systems in multiple species. BioGPS and EBI's Gene Expression Atlas offer similar services in this area, including graphical output for each gene, and useful related links [44,45].
Meta-analysis of multiple public datasets can be extremely powerful. For example public gene expression data from GEO was used to determine the minimum number of genes required to impute the remainder of the human genome. Genometry found that only 1000 ‘landmark’ genes were required to estimate the remaining genome expression, expanding on previous work in this area [46], and have generated a microarray to assay only these genes so that costs are reduced and an increased number of samples may be studied. The source of the data must be scrutinized however, as it can cause a bias in downstream analysis — i.e. if 50% of the data in GEO were to correspond to cancer experiments then a bias would exist in the selection of the 1000 genes.
2.4. Pathways and annotation
In addition to databases containing experimental data, annotation databases may also be extremely useful. Broadly speaking this data comes from two original sources — experimental data itself held in a public repository or reported in scientific literature, and predictions based on sequence alignments, structural similarities or expression profiles. One of the most common annotations in molecular biology is gene ontology (GO) [47]. There are three broad categories of these: biological process, cellular component, and molecular function — each term is part of a greater hierarchy with these divisions at the top.
Online Mendelian Inheritance in Man (OMIM) is a database of human genes and genetic phenotypes. The NCBI Taxonomy Database was developed in consultation with the INSDC [48]. Almost 280,000 species have been described by Taxonomy representing an estimated 10% of the described species of life on the planet. GeneCards is a very useful source of summary information collecting data from many different popular resources [49].
Reactome supports pathway enrichment analyses on that basis. WikiPathways is a pathway database with an open and collaborative ethos [50]. PathVisio is a tool which is used to view and edit pathways from WikiPathways and its website provides helpful user tutorials [51]. To search for additional pathway tools one can use Pathguide, which contains information on over 500 resources (http://pathguide.org).
3. Algorithms and implementations
3.1. Preprocessing & quality control
A wide variety of methods may be used for both preprocessing and downstream analysis, found as stand-alone software or on shared platforms. The following is far from a comprehensive list of methods or the tools that implement them but serves as an overview (Fig. 2 shows these methods ordered into a workflow). ‘R’ is a scripting language and environment primarily developed for statistical computing. It is particularly useful in bioinformatics and systems biology because of the number of relevant packages available, largely through the open source Bioconductor project, which contains hundreds of packages alone [52].
Fig. 2.
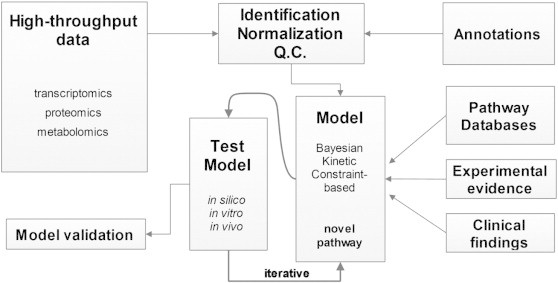
A general workflow of systems biology. This figure summarizes the overall approach, at each step highlighting some of the options available. These options often depend on the type of the data and the techniques used to gather it. QC, quality control.
R has a diverse range of uses, from preprocessing, to statistical testing and on to downstream analysis, and in particular it has great utility in microarray processing, although many of the packages initially developed for microarray analysis have since been applied to proteomics and metabolomics. Many graphical user interface (GUI) alternatives exist, however often what is gained in speed and simplicity is lost in flexibility and power, and many of these GUI applications such as Partek, SPSS and IPA are commercial.
Affymetrix microarray probe sets are dispersed randomly across their chips so that if a spatial effect does occur it is unlikely to greatly affect the final probe set value. Illumina seeks to further reduce spatial effects by randomly assigning the well used for each probe on each individual chip. While this could be of utility, if the file describing the coordinates of the probes is not extracted at the time of scanning then the locations of the beads are lost and spatial examination cannot later be investigated. Due to these measures taken by the manufacturers spatial correction is often foregone and rather packages are used to identify extremely dubious chips and simply remove them from analysis, however packages do exist that aim to correct for smaller spatial effects [53].
One popular pre-processing procedure is called Robust Multi-array Average (RMA), which background adjusts, quantile normalizes, log-transforms and summarizes from individual probe values down to probe set values [54]. A log2 transformation is performed in order to acquire a more normal distribution to allow the use of parametric tests. The log2 scale is also beneficial to the interpretation of fold changes as upregulations and downregulations are scaled equally around zero, as opposed to raw downregulations being found between zero and one and upregulations being found between one and infinity.
There are now a diverse range of mass spectrometers used to generate MS data in proteomics, with various advantages and limitations. Similarly there are also a considerable number of algorithms developed to query and cross compare the tandem MS data [55]. The most popular programs/packages used to identify proteins from raw MS data are MASCOT, SeQuest, OMSSA and X!Tandem [56]. The emergence of new tools, e.g. Morpheus [57], and development of specialized tools such as MaxQuant [58], specifically aimed at high resolution MS data, will accelerate protein identification.
IDEOM is an Excel interface used for the analysis of LC/MS and GC/MS metabolomics data [59]. It alleviates the requirement for either scripting skills or in-depth understanding of preprocessing procedures in obtaining a filtered, interpretable list of metabolites from a raw input file and makes use of mzMatch [60] and XCMS [61] to preprocess and identify metabolite IDs and then populate worksheets with metabolite data and provide graphs of statistical output.
After pre-processing the data should be in the format of a large matrix with rows by biomolecule and columns by patient (or possibly the transpose). Distance matrices may be useful at this stage, showing either distances between samples or between molecules and often displayed as a heatmap. The ‘distance’ is most commonly Euclidean distance, Manhattan distance or some type of correlation (Pearson, Spearman etc.).
Dimensionality reduction methods reduce the number of rows describing each patient so that the data may be plotted on a 1D, 2D or 3D graph. The points on the graph are often colored according to various features — plotting these points and coloring by certain variables may prove informative as to quality control by identifying samples which are extremely different from others, or displaying the association between clusters and various variables.
Sammon mapping seeks to compress a highly dimensional dataset down to a plottable number of dimensions [62]. Sammon mapping seeks to do this while minimizing what is described as stress, which is a representation of the error in the distances between points in the new data space as compared to the original. Principal Components Analysis (PCA) on the other hand seeks to essentially tilt the axis through the data space, such that the ‘first’ principle component (denoted “PC1”) captures the maximum amount of variance possible and all components remain orthogonal to each other [63]. The order of the naming of the components is determined by ranking the list based on the amount of variance described by each, i.e. PC2 captures the second most variance. While all the data is maintained (unlike in Sammon mapping) scree plots may be used to show the amount of variance for each component and to decide how many principle components are worthy of examination.
Each PC can be described as correlating to a certain extent with each original input variable, such that if a particular PC separates the samples into two clusters, those variables mostly responsible may be identified. A technique which can be used in combination with PCA is varimax rotation, in which the top PCs are selected and further rotated such that for each varimax-rotated component the variance across the correlations with the input variables is maximized — therefore each PC may be said to largely correlate to a small number of input variables [64,65]. In some cases, where data is highly dimensional and highly correlated, it can be useful to input these data into statistical tests — this way each varimax-rotated PC would be representative of a group of biomolecules and each group can be tested alongside the other in the same model, rather than doing iterative tests in which different molecules may be accounting for the same difference in the dependant variable.
3.2. Statistical analysis
Several possibilities exist for statistical analysis on a list of biomolecules or groupings of biomolecules, each with its own set of assumptions. Standard statistical tests to compare two groups such as the nonparametric Wilcoxon rank sum and parametric Student's t-test may be used depending on the assumptions appropriate to the data. Alternatively the independent variable of interest may be continuous in which case a different technique such as simple linear regression is used. Linearity of relationships should be considered and non-linear regression and mutual information employed where appropriate [66]. Non-linear relationships are poorly accounted for by linear approaches and can be responsible for the apparent noise of a system [67].
These may be iterated over every probe set and the results should be multi-test adjusted. These tests seek to identify those results with a low (e.g. < 0.05) probability of occurring by chance, and so if 100 tests were done then 5 results would be expected to show positive without multitest correction. Bonferroni correction is direct and intuitive and simply involves multiplying each p-value by the number of tests done. It is very stringent however, especially with a very large number of tests, so other less stringent methods have been developed (such as the Benjamini–Hochberg correction), which often take into account the rank of each test as ordered by p value [68].
New methods have been developed with the advent of genome-wide technologies. Limma is an R package which was developed to facilitate gene expression microarray analysis [69]. With Limma one may model batch effects, technical replicates, time series and complex multifactor experiments. It also provides the option to do a moderated t-test, essentially borrowing information from other genes.
Rank products is a heuristic method which calculates statistical significance based on ranks of fold changes, makes few assumptions about the data and is especially useful when few biological replicates are available. It is non-parametric and can be used with various high throughput data types, including a mixed data type meta-analysis and has been implemented as a package in R.
3.3. Pathway analysis
Cytoscape is a tool primarily designed for network visualization and analysis; it makes use of a wide variety of plug-ins to extend its functionality which are designed by the scientific community. However, many useful plug-ins such as MiMI [50] have not been updated to be compatible with current versions.
ClueGO [70] is a popular Cytoscape plug-in used for term enrichment analysis — i.e. determining if any terms are associated more frequently with the top of a ranked list of genes, or with a sub-set of a larger gene list. As the name suggests this is done with GOs as the terms, and allows the user to subcategorize based on the three main categories or by evidence codes. It also provides the capability to analyze with KEGG, WikiPathways and Reactome terms. Term enrichment is a good way of making a preliminary analysis on the data and identifying areas to focus on. For additional functionality, CluePedia [71] can be added to ClueGO to produce networks with custom correlation scores and other data plotted as edges between genes and nodes.
Cytoscape has a “pathway database” ‘app category’ containing plug-ins which derive data from a variety of information sources and provide some appropriate tools for pathway editing and enrichment analysis: CyKEGGParser manipulates KEGG files [72]; ReactomeFIPlugIn facilitates pathway enrichment analysis based on the Reactome database [73]; an alternative interface to WikiPathways is provided; and Metscape allows users to build and analyze networks of genes and compounds, use gene expression and metabolomics data to identify enriched pathways and their metabolic consequences and rely on data from several different sources [74]. These are just a few examples of the tools available in this category. There are many similar tools under several other related categories and many of them are found repeatedly across categories. Large datasets may also be used to infer novel relationships by Bayesian inference models. These are used to calculate the probability that a relationship exists between two molecules based upon the observation, or generally-speaking they test a hypothesis based on priors. The key challenges to this approach are having enough power (data) to infer the number of possible interactions and another is the lack of standards for accepting or rejecting relationships, however the ability to recreate a well-accepted interaction can at least be used to benchmark different methods [75].
3.4. Mathematical modeling
Kinetic models are useful for mathematically modeling/simulating the dynamics of a system in silico, and may be constructed using the SimBiology package for MatLab. Broadly-speaking there are two classes of these types of models: deterministic and stochastic. Deterministic models predict an average outcome; they do not take stochastic fluctuations into account. Deterministic approaches are therefore more appropriate for systems with large numbers of molecules, where small fluctuations have negligible impact. Ordinary differential equations (ODEs) involve functions of one independent variable and derivatives of the functions with respect to it. These are used to model dynamics over time only. Partial differential equations (PDEs) can be used instead to model dynamics over several independent variables i.e. multiple spatial axes as well as time.
Exact stochastic approaches such as the Gillespie algorithm can be useful, especially for modeling reactions with a small amount of molecules where small fluctuations can have a large impact on the system [76]. With this approach distributions are used rather than averages, and each reaction is explicitly modeled instead of the average approach used in deterministic modeling. In order to create accurate models of large networks it may be best to utilize both approaches — deterministic for large populations and stochastic for lowly-populated important reactions. One of the biggest challenges for all kinetic modeling is to accurately obtain or estimate the reaction constants and other parameters as required. Another stochastic modeling approach outside of kinetic modeling is agent-based modeling (ABM). ABM was initially developed for social sciences but has since been applied to several fields of biology. The ‘agents’ of ABM are autonomous decision-makers which can be used to represent biomolecules such as proteins or metabolites or, on a larger scale, cells. The rules governing the system are found on the agent level — each class of agent has certain rules which all instances of the agent follow (e.g. all instances of ATP follow the same set of rules). ABMs can account for time and three-dimensional space and although the rules are assigned on the agent-level, those actions can describe emergent properties on a larger scale. Few examples of applying ABM to molecular modeling exist in the literature to date — one example is the ABM of the NF-κB pathway [77].
Constraint-based models are able to address the issues of long computational time with respect to large networks and lack of genome-wide reaction rates normally found with kinetic models. Rather than seeking one immediate solution they start by restraining all possibilities down to a biologically relevant solution space, by the addition of various types of constraints. For example enzymatic capacity and thermodynamic restraints place an upper bound on the flux for a reaction. The greater challenges of this approach are generating the constraints to apply, and developing methods to probe the solution space for interesting phenotypes. Omics data may be used alongside these models [78] or can be integrated into them as constraints to introduce the context of that state [79]. One popular toolbox for implementing constraint-based models is COBRA, developed on MatLab but also available as a package for Python [80].
3.5. Multi-omics analysis
The more of the system that is directly measured the more easy it should be to discern the pathways and structures involved. Conversely the more of the system directly measured the more information must be integrated and the more complex the analysis. This describes both the promise and challenges of multi-omics studies. The most obvious way to integrate various omics experimental datasets is simply to analyze each set separately and retain only the positives from each set for further downstream analysis.
The alternative to this set-by-set approach is to integrate the data prior to analysis. Generally, this can be done in one of two ways — either simply adding all datasets into one large matrix, or identifying biological relationships between the molecules and analyzing the resulting network.
Specialized tools for this type of analysis are currently limited and are only beginning to emerge. Ondex is one tool designed to aid in multi-omics analysis, however it lacks novel multi-omics statistical approaches [81]. Mixomics is an R package which uses correlation between molecules to identify groups of related molecules corresponding to e.g. a disease state [82]. It can be used to analyze two omics sets simultaneously and the edges between nodes must be user-defined, rather than relying on an underlying database.
4. Applications
4.1. Normal function — why modeling?
The high number and diversity of experimental data, especially those covered by the life sciences domain never was so easy to access. Ranging from the description of simple to more complex processes, from isolated enzymatic reactions or temporal processes within metabolic networks to patterns of gene expression and regulation. Nonetheless, it is not conceivable to predict the behavior of complex or even simple systems with enough precision and accuracy based only on empirical data. Thus, the major advantage of modeling relies on the use of computational power within a set of predefined axioms to simulate a particular environment, as e.g. knockout of one or a group of system components and then predict the network outcome. Another benefit is that the algorithms of computer programs can be reused on several systems. Additionally, the costs associated with modeling are much lower than for experiments, therefore organism-dependent experiments can be reduced.
The process by which bacteria sense changes in the surrounding environment and direct their motility efforts toward favorable stimuli and therefore away from unfavorable stimuli is called chemotaxis [83]. Here the author describes the actual state of the art of the mathematical approaches for modeling this process within an individual bacterium cell. The best studied model within this process is Escherichia coli, that makes use of successive rotation/spinning changes of flagella from counter-clockwise to clockwise, in order to produce runs and reorientation, respectively. This process is controlled through a well-defined set of intracellular protein–protein interactions also sometimes referred to “molecular machines” and has been explored successfully by mathematical modeling of the intracellular signaling in bacteria [83].
The pursuit of building a whole-cell model involves the formulation and application of new modeling approaches and in particular the integration of models for each type of cellular process [84]. Apart from the modeling of a particular event as cell cycle to the whole-cell modeling, Karr et al. [85,86] used a hybrid methodology relying on the combination of ordinary differential equations (ODEs) into frameworks, and constraint-based and Boolean methods, modeled individual biological processes and merged the outputs in order to compute the overall state of the cell. Thus, they were able to virtually simulate the life cycle of Mycoplasma genitalium cells for every molecule and then representing the function of every annotated gene in a single computational model. This type of approach gives new insights for the prediction of phenome based on genome, providing improved and comprehensive models of cellular physiology, hence allowing researchers to prioritize experiments and constrict their lines of research [85,86].
Another major chalenge within the systems biology field is the understanding of gene regulatory networks involved in development, especially during the initial establishment of germ layers. Besides this subject being well characterized among a high number of model organisms, the advantage of axolotl over other amphibian embryology models (e.g. Xenopus laevis) is that it has only a single Mix and Nodal gene needed for the specification of the mesoderm layer [87]. There the authors proposed mathematical models based on ODEs for the description of the subjacent mechanism underlying the axolotl mesoderm and anterior mesoendoderm specification [87], where they used both in vitro and in vivo models to firstly stimulate Nodal signaling through the use of Activin and secondly the presence of maternal transcriptional factor β-catenin that activates Nodal signaling and therefore regulates the expression of the down-stream targets [87].
4.2. Human disease
It seems natural that most diseases are not solely the consequence of an abnormality in a single gene, taking into account the existent interdependencies between molecular components in a human cell, but instead reflects the perturbations of the complex intracellular and intercellular network that links tissue and organ systems [88]. Thus, moving from the reductionist approach, which evaluates only individual components within a system, to more complex interactions between biomolecules and the human -environment interplay, seems the proper approach for solving several health disorders and consequently improve the general population healthcare [89]. Consequently, a new subfield is emerging, the network medicine that focuses on applying systems biology to pharmacology by “understanding the molecular systems and its perturbations as a whole” to unravel relationships among disease processes [90] enabling the development of specific drugs and pave the road toward personalized medicine.
In the following section we will briefly describe some studies related with the medical field using systems biology approaches, highlighting the contrast between existing and candidate biomarkers for disease diagnosis and progression as well as probable druggable targets.
4.3. Disease description and epidemiology
Neurodegenerative (ND) diseases are a large class of complex diseases that includes Alzheimer's disease (AD), Parkinson's disease (PD), Huntington's disease (HD), and prion disease [91,92]. These diseases have the accumulation of protein fibrillar aggregates as a consequence of protein misfolding in common [91]. ND diseases are chronic, progressively debilitating, and to date incurable [92]. Their prevalence has increased globally, markedly in the elderly populations of developed countries [92].
AD is the commonest cause of dementia and is characterized by progressive loss of memory and other cognitive functions [93] and is considered a major epidemic worldwide, where currently more than 35 million people live with this disease, being estimated that by 2050 it will reach 115 million [94].
The main pathological hallmark of AD is the development of amyloid plaques and their accumulation in neurons, and whether they cause AD or whether they are a by-product of the disease process is still largely unknown [95].
4.4. Current disease diagnostic and prognostic
The clinical diagnostic of AD is often made during the mild stage of the disease, taking in consideration a list of cognitive-behavioral signs and symptoms [96,97]. Biopsy samples are out of “equation” for the diagnosis, so the current approach is based on the combination of cognitive and psychiatric assessment, with genetic profiling and imaging studies [92,95] as structural magnetic resonance imaging (sMRI) that is able to measure morpho-anatomical changes of brain e.g. loss of neural cells, axons and expansion of CSF space [92].
4.5. Genetic determinants and causative mutations
The most relevant genetic determinants of AD are mutations in the amyloid precursor protein (APP), presenilin (PS)-1 and PS-2 known for triggering early-onset (< 60 years) autosomal dominant AD [93]. On the other hand, the apolipoprotein E type 4 variant (apoE4) has been implicated in the familial late-onset (> 60 years) and the apoE2 seems to confer protection in AD [93].
Novel gene candidates of familial late-onset AD susceptibility have been described, essentially derived from gene-wide analysis studies (GWAS) as previously reviewed by Kim et al. [98]. Briefly, CLU, CR1, PICALM, BIN1, SORL1, GAB2, ABCA7, MS4A, CD2AP, CD33, EPHA1 and HLA-DRB1/5 can potentially modulate the risk of late-onset AD, but still to a lesser extent than apoE4 [93].
Additionally, two novel susceptibility loci of AD were found via an alternative reanalysis of a GWAS dataset, where they searched for patterns of association within genes [99]. They identified the TP53INP1 (chromosome 8), that curiously encodes a pro-apoptotic tumor suppressor, given the fact that epidemiological data exists supporting an inverse association between AD and cancer [100,101]. Furthermore, the IGHV1-67 (chromosome 14) was also identified in this study and though no known function is associated with this gene, it's known that neighbor genes are involved in IgG heavy-chain somatic recombinations [99].
Recently, Leduc et al. [102] found a single-nucleotide polymorphism (rs3846662) in the HMGCR gene in the context of AD, which exhibited the same protective effect as that of apoE2, and as a result subjects showed delayed age of onset and a significant reduced risk of AD.
4.6. Epigenetic events
At the epigenetic level, several studies have been reporting altered molecular events such as DNA methylation and hydroxymethylation in AD brain tissue, which are respectively linked with decreased and increased patterns of gene expression [103]. Additionally, histone H3 acetylation in the PS-1 and beta-secretase 1 (BACE1) promoter regions was found increased in an in vitro study, using neuroblastoma N2a murine cells transfected with Swedish human mutated APP, which leads to an enhanced activation of transcription and ultimately causes elevated expression of both AD related genes [104]. Remarkably, Frost et al. [104] found that heterochromatin loss and overexpression of the piwi-like protein 1 (PIWIL1) are molecular events conserved across Drosophila, mouse and human in tau-induced neurodegeneration (tauopathy), therefore chromatin structure could have a potential role as a therapeutic target for AD.
4.7. MicroRNAs (miRNAs)
MicroRNAs have been proposed as potential molecular markers for the diagnosis of several diseases, due to their stability and ease of quantification in biofluids [98,105]. A set of three miRNAs: miR-125b, -9 and -181c has been linked to AD, and their expression has been found decreased in serum, in CSF and in brain tissues [105–107]. Therefore, the proposed gene targets for miR-125b are CDKN2A, SYN-2 and 15-LOX, which are associated with key processes as glial proliferation, synaptic and neurotrophic deficits, respectively [107]. Additionally, the miR-9 family targets SYNJ1 and SYNPR, both associated with synaptic dysfunction; GMEB2 with neuronal trafficking and TGFBI with TGF signaling [108]. Furthermore, miR-181c targets SIRT1 that is associated with inflammation and response to stress; BTBD3 that is associated with anti-apoptosis and TRIM2, which has a role in Aβ degeneration [108].
4.8. Proteins of interest
The major constituent of the extracellular senile plaques is the amyloid-beta (Aβ) peptides (Aβ42/Aβ40), derived from the APP metabolism by secretase processing [92]. The BACE1 cleaves the APP into C-terminal fragment (C99), releasing soluble APPβ. In turn, the retained C99 is then cleaved by the γ-secretase complex, generating Aβ and APPβ intracellular domain (AICD). These key proteins involved in Aβ production have gained interest as AD biomarkers. Some studies reported changed levels of these proteins in the CSF of AD patients, such as decreased levels of Aβ42/Aβ40, increased levels of APP isoforms and also of BACE1 [92]. On the other hand, the neurofibrillary tangles (NFTs) are mainly made up of hyperphosphorylated insoluble forms of tau protein, which have high resistance to enzymatic proteolysis, resulting in accumulation in neurons. Their use as an indicative parameter helps in both the diagnosis and prognosis of AD, and also in the distinction from other tauopathies [92,95].
Recently, Kim et al. [108] identified that the leukocyte immunoglobulin-like receptor 2 (LilrB2) act as a receptor for the Aβ in human brain, and their binding promotes the activation of cofilin, resulting in actin filament disassembly and spine loss, which could contribute to synaptic loss and cognitive impairment in AD progression [108]. This suggests that immune receptors may have a role in AD, and therefore a selective block of LilrB2 function could be a potential therapy for AD treatment [108].
4.9. Metabolomics and lipidomics
Despite the high number of studies searching for candidate biomarkers such as e.g. tau or Aβ protein, still no fluid based biomarkers have been fully validated for use in clinical evaluation of AD progression [92,95]. Nevertheless, in a recent publication [109], a set of ten plasma lipids was discovered and validated and were able to predict the phenoconversion of cognitively normal individuals to either amnestic mild cognitive impairment or even AD within a temporal frame of two till three years with more than 90% accuracy. The identified biomarker panel features targets such as phosphatidylcholine and acylcarnitine that have roles in both cell membrane integrity and functionality. Moreover, they could be sensitive to early neurodegeneration of preclinical cases of AD, however external validation is needed before further advancement in clinical use [109].
Furthermore, Gonzalez-Dominguez et al. [110] observed changes in serum levels of phospholipids, prostaglandins and diacyglycerols that can be linked with metabolic disorders seen in AD. Moreover, serum levels of metabolites such as oleamine, guanine, arginine, histidine, imidazole and putrescine displayed altered expression in patients recently diagnosed with sporadic AD, and thus they could arise as potential biomarker candidates [110].
The most widely reported marker candidates are protein-based biomarkers, particularly the aggregation-prone proteins central to ND disease pathology and also proteins associated with oxidative stress and inflammation. Therapeutical value has been attributed to etifoxine which shows to be effective toward the treatment of peripheral nerve injuries and axonal neuropathies for the treatment of ND disorders [111]. While other small molecules comprising dimebon, piracetam, and simvastatin target Aβ, mitochondrial membranes, and anti-apoptotic proteins (Bcl-2) respectively, they have been identified to be effective in the symptomatic treatment of AD [112,113].
4.10. Systems biology approach toward AD biomarker and drug discovery
Several studies aiming for the identification of disease biomarkers have failed to prove their specificity and sensitivity, because in majority, they were unable to distinguish between the true biological signals from all the noise. Then the use of a systems approach could provide a set of tools from statistical, computational and biological areas that could amplify the signal and therefore allow and ameliorate mapping of the involved biological networks.
We above presented the state of art of AD with several independent studies ranging from genetic determinants to disease traits. Next, we will present studies that merged and integrated data through systems biology approaches in order to yield biological meaning of all de novo generated information.
The integration of genomic data through a network approach has been used to highlight cellular pathways underlying clinical traits and ultimately to pinpoint genes that are probable key regulators in biological processes [114]. An example of this type of approach was the identification of molecular interactions that are disrupted in patients with late AD onset, where the authors [115] built co-expression networks based on gene expression and genotyping data from postmortem brain tissues of hundreds of patients and healthy individuals. They identified several modules of distinct functional categories and cellular specificity. After, they applied an integrative network analysis to first reorganize by rank the most relevant modules of late-onset AD and then identify by Bayesian inference the main causal gene regulators of these altered networks. As a result, they acknowledged the TYROBP as a key driver of the immune/microglia module that was found reconfigured in the disease state, highlighting the benefit of the use of network analysis to better identify and prioritize pathways and gene targets involved in complex diseases. Similarly, Miller et al. [116] reported that microglial activation occurs early in AD progression, where they also identified the TYROBP as the main hub of the microglia module via weighted gene co-expression network analysis. Furthermore, they assessed two brain areas CA1 and CA3 in the disease context, through microarray gene expression and found that the hippocampal region CA3 has genes associated with disease protection: ABCA1, MT1H, PDK4 and RHOBTB3, and the genes FAM13A1, LINGO2 and UNC13C with disease vulnerability [116], thus, reinforcing the in silico finding of microglia activation occurring early during AD progression and also their association with NFT formation in addition to amyloid deposition [116].
Epidemiological screen studies of human populations have identified novel associations between specific metabolites and disease traits [92,117]. Sertbas et al. [117] developed a stoichiometric model of brain metabolism, covering several cellular pathways and molecular interactions in astrocytes and neurons. Then, they applied the developed framework to analyze transcriptional modifications associated with six neurodegenerative diseases, including AD and identified the fatty acid synthesis, and phosphatidylcholine and inositol metabolism that have been associated with AD and CDP-diacylglycerol biosynthesis as a pathway that could allow the distinction of AD from other neurodegenerative diseases.
In order to fulfill the need of curated data of AD to build enhanced AD pathway maps, the “AlzPathway” was developed [118]. It handles a comprehensive map of signaling pathways linked with AD [119] and could therefore be a major contribution toward the implementation of novel pipelines for AD drug discovery [120].
A proper phenotype-based diagnostic within ND diseases is challenging, because of the overlap of several clinical conditions among the different types of ND diseases. Thus, obtaining well-characterized cohort samples either for biomarker discovery or for performing clinical trials for drug development and implementation is a task that needs to be overcome. However, with the advent of new high-throughput technologies, we can be closer to the discovery of numerous novel biomarker candidates for each ND disease subtype [92]. Moreover, all de novo generated data should be integrated, in order to provide the whole picture of the disease, than single research studies are able to provide [121]. Therefore, for a proper evaluation of progression in complex diseases we should apply systems biology approaches that will enable us to look into the biology inside of the “black box” [122].
5. Summary and outlook
Preprocessing methods for omics data are for the most part well defined, however the identifiers to which the data are assigned are often erroneous or missing which can negatively affect downstream analysis. Greater efforts into database curation must be taken and tools must select appropriate identifiers for their inputs to mitigate these problems.
With the ever-reducing cost of omics technologies there is great potential in multi-omics studies. One of the greatest challenges currently is to further study and develop methods of downstream data analysis. Ideally these methods should be flexible to any number of omics sets, and should be trained and tested against well understood systems where possible. One challenge which may be insurmountable for some time is to have one analysis platform which has all the various algorithms available which might be useful to multi-omics analysis rather than coercing and exporting data between disparate analysis platforms.
To facilitate these multi-omics methods and assist systems biology in general it would be extremely useful to have a database linking all biological molecules through various processes such as transcription, translation, protein–protein interactions and so forth, and containing a variety of ID types to map between them. Such a database should contain biomolecules in clusters so that the user could select the level of complexity required. For example two proteins may come from the same gene and may either be splice variants or different protein modifications. These differences may cause great or little difference in function and in downstream analysis output. Therefore the user may wish to summarize (i.e. average) to one value, retain the different sets of information or calculate both the summarization and a ratio of the most common variants.
In order to better model complex organisms, samples from multiple tissues of the same set of individuals should be studied simultaneously using omics data, which will require the development of novel analysis methods. Acquiring the relevant tissues from humans can of course be difficult however depending on the tissues involved. Comparative systems biology may help identify which organisms may be similar enough in each particular aspect for use as models — of course the positive identification of a useful model cannot be totally assured prior to deriving the system itself; however negative identifications can help rule out those organisms which seem extremely unlikely to be helpful.
It is sometimes suggested that omics technologies and systems biology have failed to deliver many breakthrough enhancements to the treatment of complex disease [123]. In some cases it may be that in fact such diseases are not truly one disease from a systems or reductionist point-of-view, but several with the same or similar phenotypic end-points — i.e. with the current terminology they are unknown subtypes of disease. If this is the case then the overlap between the systems is poor and statistical methods which the approach relies on require very large cohorts for identification of these subtypes and subsequent description of each system. Other possibilities are that longitudinal data or samples from different tissues are required.
Acknowledgments
The research leading to these results has received funding from the European Union's Seventh Framework Programme FP7/2007–2013 under grant agreement FP7-PEOPLE-2013-ITN-608332 and European Union-MASCARA (HEALTH-2011-278249).
Scott Robinson and Marco Fernandes have contributed equally.
Contributor Information
Scott W. Robinson, Email: Scott.Robinson@glasgow.ac.uk.
Marco Fernandes, Email: m.fernandes.1@research.gla.ac.uk.
Holger Husi, Email: Holger.Husi@glasgow.ac.uk.
References
- 1.Crick F.H. On protein synthesis. Symp Soc Exp Biol. 1958;12:138–163. [PubMed] [Google Scholar]
- 2.Baluska F., Witzany G. At the dawn of a new revolution in life sciences. World J Biol Chem. 2013;4:13–15. doi: 10.4331/wjbc.v4.i2.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brenner S. History of science. The revolution in the life sciences. Science. 2012;338:1427–1428. doi: 10.1126/science.1232919. [DOI] [PubMed] [Google Scholar]
- 4.Fuks F. DNA methylation and histone modifications: teaming up to silence genes. Curr Opin Genet Dev. 2005;15:490–495. doi: 10.1016/j.gde.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 5.Smith Z.D., Meissner A. DNA methylation: roles in mammalian development. Nat Rev Genet. 2013;14:204–220. doi: 10.1038/nrg3354. [DOI] [PubMed] [Google Scholar]
- 6.Laurent L., Wong E., Li G., Huynh T., Tsirigos A., Ong C.T. Dynamic changes in the human methylome during differentiation. Genome Res. 2010;20:320–331. doi: 10.1101/gr.101907.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schwartz S., Meshorer E., Ast G. Chromatin organization marks exon–intron structure. Nat Struct Mol Biol. 2009;16:990–995. doi: 10.1038/nsmb.1659. [DOI] [PubMed] [Google Scholar]
- 8.Sharma A. Transgenerational epigenetic inheritance: focus on soma to germline information transfer. Prog Biophys Mol Biol. 2013;113:439–446. doi: 10.1016/j.pbiomolbio.2012.12.003. [DOI] [PubMed] [Google Scholar]
- 9.Saletore Y., Meyer K., Korlach J., Vilfan I.D., Jaffrey S., Mason C.E. The birth of the Epitranscriptome: deciphering the function of RNA modifications. Genome Biol. 2012;13:175. doi: 10.1186/gb-2012-13-10-175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tanzer A., Stadler P.F. Molecular evolution of a microRNA cluster. J Mol Biol. 2004;339:327–335. doi: 10.1016/j.jmb.2004.03.065. [DOI] [PubMed] [Google Scholar]
- 11.McManus C.J., Graveley B.R. RNA structure and the mechanisms of alternative splicing. Curr Opin Genet Dev. 2011;21:373–379. doi: 10.1016/j.gde.2011.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Prabakaran S., Lippens G., Steen H., Gunawardena J. Post-translational modification: nature's escape from genetic imprisonment and the basis for dynamic information encoding. Wiley Interdiscip Rev Syst Biol Med. 2012;4:565–583. doi: 10.1002/wsbm.1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Todeschini A.L., Georges A., Veitia R.A. Transcription factors: specific DNA binding and specific gene regulation. Trends Genet. 2014;30:211–219. doi: 10.1016/j.tig.2014.04.002. [DOI] [PubMed] [Google Scholar]
- 14.Jaskiewicz L., Filipowicz W. Role of Dicer in posttranscriptional RNA silencing. Curr Top Microbiol Immunol. 2008;320:77–97. doi: 10.1007/978-3-540-75157-1_4. [DOI] [PubMed] [Google Scholar]
- 15.Shyamsundar R., Kim Y.H., Higgins J.P., Montgomery K., Jorden M., Sethuraman A. A DNAmicroarray survey of gene expression in normal human tissues. Genome Biol. 2005;6:R22. doi: 10.1186/gb-2005-6-3-r22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baquero F., Nombela C. The microbiome as a human organ. Clin Microbiol Infect. 2012;18(Suppl. 4):2–4. doi: 10.1111/j.1469-0691.2012.03916.x. [DOI] [PubMed] [Google Scholar]
- 17.Maga G., Hubscher U. Proliferating cell nuclear antigen (PCNA): a dancer with many partners. J Cell Sci. 2003;116:3051–3060. doi: 10.1242/jcs.00653. [DOI] [PubMed] [Google Scholar]
- 18.He X., Zhang J. Toward a molecular understanding of pleiotropy. Genetics. 2006;173:1885–1891. doi: 10.1534/genetics.106.060269. [1044] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Laoide B.M., Foulkes N.S., Schlotter F., Sassone-Corsi P. The functional versatility of CREM is determined by its modular structure. EMBO J. 1993;12:1179–1191. doi: 10.1002/j.1460-2075.1993.tb05759.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chintapalli V.R., Wang J., Dow J.A. Using FlyAtlas to identify better Drosophila melanogaster models of human disease. Nat Genet. 2007;39:715–720. doi: 10.1038/ng2049. [DOI] [PubMed] [Google Scholar]
- 21.Downing T.L., Soto J., Morez C., Houssin T., Fritz A., Yuan F. Biophysical regulation of epigenetic state and cell reprogramming. Nat Mater. 2013;12:1154–1162. doi: 10.1038/nmat3777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bizzarri M., Palombo A., Cucina A. Theoretical aspects of Systems Biology. Prog Biophys Mol Biol. 2013;112:33–43. doi: 10.1016/j.pbiomolbio.2013.03.019. [DOI] [PubMed] [Google Scholar]
- 23.Price M.E., Cotton A.M., Lam L.L., Farre P., Emberly E., Brown C.J. Additional annotation enhances potential for biologically-relevant analysis of the Illumina Infinium HumanMethylation450 BeadChip array. Epigenetics Chromatin. 2013;6:4. doi: 10.1186/1756-8935-6-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harbig J., Sprinkle R., Enkemann S.A. A sequence-based identification of the genes detected by probesets on the Affymetrix U133 plus 2.0 array. Nucleic Acids Res. 2005;33:e31. doi: 10.1093/nar/gni027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Husi H., Skipworth R.J., Fearon K.C., Ross J.A. LSCluster, a large-scale sequence clustering and aligning software for use in partial identity mapping and splice-variant analysis. J Proteome. 2013;84:185–189. doi: 10.1016/j.jprot.2013.04.006. [DOI] [PubMed] [Google Scholar]
- 26.Kogenaru S., Qing Y., Guo Y., Wang N. RNA-seq and microarray complement each other in transcriptome profiling. BMC Genomics. 2012;13:629. doi: 10.1186/1471-2164-13-629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dettmer K., Aronov P.A., Hammock B.D. Mass spectrometry-based metabolomics. Mass Spectrom Rev. 2007;26:51–78. doi: 10.1002/mas.20108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Griffin T.J., Aebersold R. Advances in proteome analysis bymass spectrometry. J Biol Chem. 2001;276:45497–45500. doi: 10.1074/jbc.R100014200. [DOI] [PubMed] [Google Scholar]
- 29.Matsumoto K., Satoh K., Maniwa T., Araki A., Maruyama R., Oda T. Noticeable decreased expression of tenascin-X in calcific aortic valves. Connect Tissue Res. 2012;53:460–468. doi: 10.3109/03008207.2012.702818. [DOI] [PubMed] [Google Scholar]
- 30.Cairns D.A., Thompson D., Perkins D.N., Stanley A.J., Selby P.J., Banks R.E. Proteomic profiling using mass spectrometry—does normalising by total ion current potentially mask some biological differences? Proteomics. 2008;8:21–27. doi: 10.1002/pmic.200700598. [DOI] [PubMed] [Google Scholar]
- 31.Alfassi Z.B. On the normalization of amass spectrum for comparison of two spectra. J Am Soc Mass Spectrom. 2004;15:385–387. doi: 10.1016/j.jasms.2003.11.008. [DOI] [PubMed] [Google Scholar]
- 32.Nakamura Y., Cochrane G., Karsch-Mizrachi I. International Nucleotide Sequence Database C. The International Nucleotide Sequence Database Collaboration. Nucleic Acids Res. 2013;41:D21–D24. doi: 10.1093/nar/gks1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.UniProt C. Update on activities at the Universal Protein Resource (UniProt) in 2013. Nucleic Acids Res. 2013;41:D43–D47. doi: 10.1093/nar/gks1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wishart D.S., Jewison T., Guo A.C., Wilson M., Knox C. HMDB 3.0—The Human Metabolome Database in 2013. Nucleic Acids Res. 2013;41:D801–D807. doi: 10.1093/nar/gks1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vizcaino J.A., Cote R.G., Csordas A., Dianes J.A., Fabregat A., Foster J.M. The Proteomics Identifications (PRIDE) database and associated tools: status in 2013. Nucleic Acids Res. 2013;41:D1063–D1069. doi: 10.1093/nar/gks1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Altman T., Travers M., Kothari A., Caspi R., Karp P.D. A systematic comparison of the MetaCyc and KEGG pathway databases. BMC Bioinforma. 2013;14:112. doi: 10.1186/1471-2105-14-112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cerami E.G., Gross B.E., Demir E., Rodchenkov I., Babur O. Pathway Commons, a web resource for biological pathway data. Nucleic Acids Res. 2011;39:D685–D690. doi: 10.1093/nar/gkq1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cline M.S., Smoot M., Cerami E., Kuchinsky A., Landys N. Integration of biological networks and gene expression data using Cytoscape. Nat Protoc. 2007;2:2366–2382. doi: 10.1038/nprot.2007.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Edgar R., Domrachev M., Lash A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Parkinson H., Sarkans U., Shojatalab M., Abeygunawardena N., Contrino S., Coulson R. ArrayExpress — a public repository for microarray gene expression data at the EBI. Nucleic Acids Res. 2005;33:D553–D555. doi: 10.1093/nar/gki056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brazma A. Minimum Information About a Microarray Experiment (MIAME) — successes, failures, challenges. Sci World J. 2009;9:420–423. doi: 10.1100/tsw.2009.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Steinbeck C., Conesa P., Haug K., Mahendraker T., Williams M., Maguire E. MetaboLights: towards a new COSMOS of metabolomics data management. Metabolomics. 2012;8:757–760. doi: 10.1007/s11306-012-0462-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ludman E.J., Fullerton S.M., Spangler L., Trinidad S.B., Fujii M.M., Jarvik G.P. Glad you asked: participants' opinions of re-consent for dbGaP data submission. J Empir Res Hum Res Ethics. 2010;5:9–16. doi: 10.1525/jer.2010.5.3.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu C., Orozco C., Boyer J., Leglise M., Goodale J., Batalov S. BioGPS: an extensible and customizable portal for querying and organizing gene annotation resources. Genome Biol. 2009;10 doi: 10.1186/gb-2009-10-11-r130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kapushesky M., Emam I., Holloway E., Kurnosov P., Zorin A., Malone J. Gene Expression Atlas at the European Bioinformatics Institute. Nucleic Acids Res. 2010;38:D690–D698. doi: 10.1093/nar/gkp936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Peck D., Crawford E.D., Ross K.N., Stegmaier K., Golub T.R., Lamb J. A method for highthroughput gene expression signature analysis. Genome Biol. 2006;7:R61. doi: 10.1186/gb-2006-7-7-r61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ashburner M., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Federhen S. The NCBI Taxonomy database. Nucleic Acids Res. 2012;40:D136–D143. doi: 10.1093/nar/gkr1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rebhan M., Chalifa-Caspi V., Prilusky J., Lancet D. GeneCards: integrating information about genes, proteins and diseases. Trends Genet. 1997;13:163. doi: 10.1016/s0168-9525(97)01103-7. [DOI] [PubMed] [Google Scholar]
- 50.Kelder T., van Iersel M.P., Hanspers K., Kutmon M., Conklin B.R., Evelo C.T. WikiPathways: building research communities on biological pathways. Nucleic Acids Res. 2012;40:D1301–D1307. doi: 10.1093/nar/gkr1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.van Iersel M.P., Kelder T., Pico A.R., Hanspers K., Coort S., Conklin B.R. Presenting and exploring biological pathways with PathVisio. BMC Bioinforma. 2008;9 doi: 10.1186/1471-2105-9-399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gentleman R.C., Carey V.J., Bates D.M., Bolstad B., Dettling M., Dudoit S. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Suarez-Farinas M., Pellegrino M., Wittkowski K.M., Magnasco M.O. Harshlight: a “corrective make-up” program for microarray chips. BMC Bioinforma. 2005;6:294. doi: 10.1186/1471-2105-6-294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Irizarry R.A., Hobbs B., Collin F., Beazer-Barclay Y.D., Antonellis K.J., Scherf U. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 55.Titulaer M.K., Siccama I., Dekker L.J., van Rijswilk A.L.C.T., Heeren R.M.A., Smitt P.A.S. A database application for pre-processing, storage and comparison of mass spectra derived from patients and controls. BMC Bioinforma. 2006;7 doi: 10.1186/1471-2105-7-403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kandasamy K., Pandey A., Molina H. Evaluation of several MS/MS search algorithms for analysis of spectra derived from electron transfer dissociation experiments. Anal Chem. 2009;81:7170–7180. doi: 10.1021/ac9006107. [DOI] [PubMed] [Google Scholar]
- 57.Wenger C.D., Coon J.J. A proteomics search algorithm specifically designed for high resolution tandem mass spectra. J Proteome Res. 2013;12:1377–1386. doi: 10.1021/pr301024c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cox J., Matic I., Hilger M., Nagaraj N., Selbach M., Olsen J.V. A practical guide to the MaxQuant computational platform for SILAC-based quantitative proteomics. Nat Protoc. 2009;4:698–705. doi: 10.1038/nprot.2009.36. [DOI] [PubMed] [Google Scholar]
- 59.Creek D.J., Jankevics A., Burgess K.E., Breitling R., Barrett M.P. IDEOM: an Excel interface for analysis of LC–MS-based metabolomics data. Bioinformatics. 2012;28:1048–1049. doi: 10.1093/bioinformatics/bts069. [DOI] [PubMed] [Google Scholar]
- 60.Scheltema R.A., Jankevics A., Jansen R.C., Swertz M.A., Breitling R. PeakML/mzMatch: a file format, Java library, R library, and tool-chain for mass spectrometry data analysis. Anal Chem. 2011;83:2786–2793. doi: 10.1021/ac2000994. [DOI] [PubMed] [Google Scholar]
- 61.Smith C.A., Want E.J., O'Maille G., Abagyan R., Siuzdak G. XCMS: processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification. Anal Chem. 2006;78:779–787. doi: 10.1021/ac051437y. [DOI] [PubMed] [Google Scholar]
- 62.Sammon J.W. A nonlinear mapping for data structure analysis. IEEE Trans Comput. 1969;C 18:401. [Google Scholar]
- 63.Pearson K. On lines and planes of closest fit to systems of points in space. Philos Mag. 1901;2:559–572. [Google Scholar]
- 64.Kaiser H.F. The varimax criterion for analytic rotation in factor-analysis. Psychometrika. 1958;23:187–200. [Google Scholar]
- 65.Shah S.H., Hauser E.R., Bain J.R., Muehlbauer M.J., Haynes C., Stevens R.D. High heritability of metabolomic profiles in families burdened with premature cardiovascular disease. Mol Syst Biol. 2009;5:258. doi: 10.1038/msb.2009.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Slonim N., Atwal G.S., Tkacik G., Bialek W. Information-based clust ring. Proc Natl Acad Sci U S A. 2005;102:18297–18302. doi: 10.1073/pnas.0507432102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wagner C.D., Persson P.B. Chaos in the cardiovascular system: an update. Cardiovasc Res. 1998;40:257–264. doi: 10.1016/s0008-6363(98)00251-x. [DOI] [PubMed] [Google Scholar]
- 68.Hochberg Y. A sharper Bonferroni procedure for multiple tests of significance. Biometrika. 1988;75:800–802. [Google Scholar]
- 69.Smyth G.K. Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3 doi: 10.2202/1544-6115.1027. [Article3] [DOI] [PubMed] [Google Scholar]
- 70.Bindea G., Mlecnik B., Hackl H., Charoentong P., Tosolini M. ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics. 2009;25:1091–1093. doi: 10.1093/bioinformatics/btp101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bindea G., Galon J., Mlecnik B. CluePedia Cytoscape plugin: pathway insights using integrated experimental and in silico data. Bioinformatics. 2013;29:661–663. doi: 10.1093/bioinformatics/btt019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Arakelyan A., Nersisyan L. KEGGParser: parsing and editing KEGG pathway maps in Matlab. Bioinformatics. 2013;29:518–519. doi: 10.1093/bioinformatics/bts730. [DOI] [PubMed] [Google Scholar]
- 73.Wu G., Feng X., Stein L. A human functional protein interaction network and its application to cancer data analysis. Genome Biol. 2010;11:R53. doi: 10.1186/gb-2010-11-5-r53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gao J., Tarcea V.G., Karnovsky A., Mirel B.R., Weymouth T.E. Metscape: a Cytoscape plug-in for visualizing and interpreting metabolomic data in the context of human metabolic networks. Bioinformatics. 2010;26:971–973. doi: 10.1093/bioinformatics/btq048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Marbach D., Costello J.C., Kuffner R., Vega N.M., Vega N.M., Prill R.J., Camacho DM. Wisdomof crowds for robust gene network inference. Nat Methods. 2012;9:796–804. doi: 10.1038/nmeth.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gillespie D.T. Exact stochastic simulation of coupled chemical-reactions. J Phys Chem. 1977;81:2340–2361. [Google Scholar]
- 77.Pogson M., Smallwood R., Qwarnstrom E., Holcombe M. Formal agent-based modelling of intracellular chemical interactions. Biosystems. 2006;85:37–45. doi: 10.1016/j.biosystems.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 78.Hyduke D.R., Lewis N.E., Palsson B.O. Analysis of omics data with genome-scale models of metabolism. Mol BioSyst. 2013;9:167–174. doi: 10.1039/c2mb25453k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Becker S.A., Palsson B.O. Context-specific metabolic networks are consistent with experiments. PLoS Comput Biol. 2008;4:e1000082. doi: 10.1371/journal.pcbi.1000082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ebrahim A., Lerman J.A., Palsson B.O., Hyduke D.R. COBRApy: COnstraints-Based Reconstruction and Analysis for Python. BMC Syst Biol. 2013;7:74. doi: 10.1186/1752-0509-7-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kohler J., Baumbach J., Taubert J., Specht M., Skusa A., Ruegg A. Graph-based analysis and visualization of experimental results with ONDEX. Bioinformatics. 2006;22:1383–1390. doi: 10.1093/bioinformatics/btl081. [DOI] [PubMed] [Google Scholar]
- 82.Le Cao K.A., Boitard S., Besse P. Sparse PLS discriminant analysis: biologically relevant feature selection and graphical displays for multiclass problems. BMC Bioinforma. 2011;12:253. doi: 10.1186/1471-2105-12-253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tindall M.J., Porter S.L., Maini P.K., Gaglia G., Armitage J.P. Overview of mathematical approaches used to model bacterial chemotaxis I: the single cell. Bull Math Biol. 2008;70:1525–1569. doi: 10.1007/s11538-008-9321-6. [DOI] [PubMed] [Google Scholar]
- 84.Covert M.W., Xiao N., Chen T.J., Karr J.R. Integrating metabolic, transcriptional regulatory and signal transduction models in Escherichia coli. Bioinformatics. 2008;24:2044–2050. doi: 10.1093/bioinformatics/btn352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Macklin D.N., Ruggero N.A., Covert M.W. The future ofwhole-cellmodeling. Curr Opin Biotechnol. 2014;28:111–115. doi: 10.1016/j.copbio.2014.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Karr J.R., Sanghvi J.C., Macklin D.N., Gutschow M.V., Bolival B., Jr. A whole-cell com- putational model predicts phenotype from genotype. Cell. 2012;150:389–401. doi: 10.1016/j.cell.2012.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Brown L.E., King J.R., Loose M. Two different network topologies yield bistability in models of mesoderm and anterior mesendoderm specification in amphibians. J Theor Biol. 2014;353:67–77. doi: 10.1016/j.jtbi.2014.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Barabasi A.L., Gulbahce N., Loscalzo J. Network medicine: a network-based approach to human disease. Nat Rev Genet. 2011;12:56–68. doi: 10.1038/nrg2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Roukos D.H. Networks medicine: from reductionism to evidence of complex dynamic biomolecular interactions. Pharmacogenomics. 2011;12:695–698. doi: 10.2217/pgs.11.28. [DOI] [PubMed] [Google Scholar]
- 90.Chen J.Y., Piquette-Miller M., Smith B.P. Network medicine: finding the links to personalized therapy. Clin Pharmacol Ther. 2013;94:613–616. doi: 10.1038/clpt.2013.195. [DOI] [PubMed] [Google Scholar]
- 91.Hardy J., Revesz T. The spread of neurodegenerative disease. N Engl J Med. 2012;366:2126–2128. doi: 10.1056/NEJMcibr1202401. [DOI] [PubMed] [Google Scholar]
- 92.Lausted C., Lee I., Zhou Y., Qin S., Sung J. 2014. Systems approach to neurodegenerative disease biomarker discovery; pp. 457–481. [DOI] [PubMed] [Google Scholar]
- 93.Huang Y., Mucke L. Alzheimer mechanisms and therapeutic strategies. Cell. 2012;148:1204–1222. doi: 10.1016/j.cell.2012.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hampel H., Lista S. Alzheimer disease: from inherited to sporadic AD—crossing the biomarker bridge. Nat Rev Neurol. 2012;8:598–600. doi: 10.1038/nrneurol.2012.202. [DOI] [PubMed] [Google Scholar]
- 95.Anderson H. Alzheimer disease. 2014. http://emedicine.medscape.com/article/1134817
- 96.Knopman D.S., DeKosky S.T., Cummings J.L., Chui H., Corey-Bloom J., Relkin N. Practice parameter: diagnosis of dementia (an evidence-based review). Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2001;56:1143–1153. doi: 10.1212/wnl.56.9.1143. [DOI] [PubMed] [Google Scholar]
- 97.Jack C.R., Jr., Albert M.S., Knopman D.S., McKhann G.M., Sperling R.A., Carrillo M.C. Introduction to the recommendations from the National Institute on Aging—Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7:257–262. doi: 10.1016/j.jalz.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kim D.H., Yeo S.H., Park J.M., Choi J.Y., Lee T.H., Park S.Y. Genetic markers for diagnosis and pathogenesis of Alzheimer's disease. Gene. 2014;545:185–193. doi: 10.1016/j.gene.2014.05.031. [DOI] [PubMed] [Google Scholar]
- 99.Escott-Price V., Bellenguez C., Wang L.S., Choi S.H., Harold D., Jones L. Gene-wide analysis detects two new susceptibility genes for Alzheimer's disease. PLoS One. 2014;9:e94661. doi: 10.1371/journal.pone.0094661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Driver J.A., Beiser A., Au R., Kreger B.E., Splansky G.L., Kurth T. Inverse association between cancer and Alzheimer's disease: results from the Framingham Heart Study. BMJ. 2012;344:e1442. doi: 10.1136/bmj.e1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Liu T., Ren D., Zhu X., Yin Z., Jin G., Zhao Z. Transcriptional signaling pathways inversely regulated in Alzheimer's disease and glioblastomamultiform. Sci Rep. 2013;3:3467. doi: 10.1038/srep03467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Leduc V., De Beaumont L., Theroux L., Dea D., Aisen P., Petersen R.C. HMGCR is a genetic mod- ifier for risk, age of onset and MCI conversion to Alzheimer's disease in a three cohorts study. Mol Psychiatry. 2014 doi: 10.1038/mp.2014.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Coppieters N., Dieriks B.V., Lill C., Faull R.L., Curtis M.A., Dragunow M. Global changes in DNA 1274 methylation and hydroxymethylation in Alzheimer's disease human brain. Nat Neurosci. 2014;35:1334–1344. doi: 10.1016/j.neurobiolaging.2013.11.031. [DOI] [PubMed] [Google Scholar]
- 104.Lu X., Deng Y., Yu D., Cao H., Wang L., Liu L. Histone acetyltransferase p300 mediates histone acetylation of PS1 and BACE1 in a cellular model of Alzheimer's disease. PLoS One. 2014;9:e103067. doi: 10.1371/journal.pone.0103067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kiko T., Nakagawa K., Tsuduki T., Furukawa K., Arai H., Miyazawa T. MicroRNAs in plasma and cerebrospinal fluid as potential markers for Alzheimer's disease. J Alzheimers Dis. 2014;39:253–259. doi: 10.3233/JAD-130932. [DOI] [PubMed] [Google Scholar]
- 106.Schonrock N., Humphreys D.T., Preiss T., Gotz J. Target gene repression mediated by miRNAs miR-181c and miR-9 both of which are down-regulated by amyloid-beta. J Mol Neurosci. 2012;46:324–335. doi: 10.1007/s12031-011-9587-2. [DOI] [PubMed] [Google Scholar]
- 107.Tan L., Yu J.T., Liu Q.Y., Tan M.S., Zhang W., Hu N. CirculatingmiR-125b as a biomarker of Alzheimer's disease. J Neurol Sci. 2014;336:52–56. doi: 10.1016/j.jns.2013.10.002. [DOI] [PubMed] [Google Scholar]
- 108.Kim T., Vidal G.S., Djurisic M., William C.M., Birnbaum M.E., Garcia K.C. Human LilrB2 is a beta-amyloid receptor and its murine homolog PirB regulates synaptic plasticity in an Alzheimer's model. Science. 2013;341:1399–1404. doi: 10.1126/science.1242077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Mapstone M., Cheema A.K., Fiandaca M.S. Plasma phospholipids identify antecedent memory impairment in older adults. 2014;20:415–418. doi: 10.1038/nm.3466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Gonzalez-Dominguez R., Garcia-Barrera T., Gomez-Ariza J.L. Metabolomic study of lipids in serumfor biomarker discovery in Alzheimer's disease using direct infusion mass spectrometry. J Pharm Biomed Anal. 2014;98c:321–326. doi: 10.1016/j.jpba.2014.05.023. [DOI] [PubMed] [Google Scholar]
- 111.Girard C., Liu S., Cadepond F., Adams D., Lacroix C., Verleye M. Etifoxine improves peripheral nerve regeneration and functional recovery. Proc Natl Acad Sci. 2008;105:20505–20510. doi: 10.1073/pnas.0811201106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Eckert G.P., Renner K., Eckert S.H., Eckmann J., Hagl S., Abdel-Kader R.M. Mitochondrial dysfunction a pharmacological target in Alzheimer's disease. Mol Neurobiol. 2012;46:136–150. doi: 10.1007/s12035-012-8271-z. [DOI] [PubMed] [Google Scholar]
- 113.Vlasblom J., Jin K., Kassir S., Babu M. Exploringmitochondrial system properties of neu-rodegenerative diseases through interactome mapping. J Proteome. 2014;100:8–24. doi: 10.1016/j.jprot.2013.11.008. [DOI] [PubMed] [Google Scholar]
- 114.Civelek M., Lusis A.J. Systems genetics approaches to understand complex traits. Nat Rev Genet. 2014;15:34–48. doi: 10.1038/nrg3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zhang B., Gaiteri C., Bodea L.G., Wang Z., McElwee J., Podtelezhnikov A.A. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer's disease. Cell. 2013;153:707–720. doi: 10.1016/j.cell.2013.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Miller J.A., Woltjer R.L., Goodenbour J.M., Horvath S., Geschwind D.H. Genes and pathways underlying regional and cell type changes in Alzheimer's disease. Genome Med. 2013;5:48. doi: 10.1186/gm452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Sertbas M., Ulgen K., Cakir T. Systematic analysis of transcription-level effects of neurodegenerative diseases on human brainmetabolism by a newly reconstructed brain-specific metabolic network. FEBS Open Biol. 2014;4:542–553. doi: 10.1016/j.fob.2014.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Mizuno S., Iijima R., Ogishima S., Kikuchi M., Matsuoka Y., Ghosh S. AlzPathway: a comprehensive map of signaling pathways of Alzheimer's disease. BMC Syst Biol. 2012;6:52. doi: 10.1186/1752-0509-6-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ogishima S., Mizuno S., Kikuchi M., Miyashita A., Kuwano R., Tanaka H. A map of Alzheimer's disease-signaling pathways: a hope for drug target discovery. Clin Pharmacol Ther. 2013;93:399–401. doi: 10.1038/clpt.2013.37. [DOI] [PubMed] [Google Scholar]
- 120.Bennett D.A., Yu L., De Jager P.L. Building a pipeline to discover and validate novel therapeutic targets and lead compounds for Alzheimer's disease. Biochem Pharmacol. 2014;88:617–630. doi: 10.1016/j.bcp.2014.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Meng Q., Mäkinen V.P., Luk H., Yang X. Systems biology approaches and applications in obesity, diabetes, and cardiovascular diseases. Curr Cardiovasc Risk Rep. 2013;7:73–83. doi: 10.1007/s12170-012-0280-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.MacKay T.F.C., Stone E.A., Ayroles J.F. The genetics of quantitative traits: challenges and prospects. Nat Rev Genet. 2009;10:565–577. doi: 10.1038/nrg2612. [DOI] [PubMed] [Google Scholar]
- 123.Raybould H.E., Holzer P., Reddy S.N., Yang H., Tache Y. Capsaicin-sensitive vagal afferents contribute to gastric acid and vascular responses to intracisternal TRH analog. Peptides. 1990;11:789–795. doi: 10.1016/0196-9781(90)90196-c. [DOI] [PubMed] [Google Scholar]