

Abstract
Immunosuppression of tumor-infiltrating lymphocytes (TIL) is a common feature of advanced cancer, but its biological basis has remained obscure. We demonstrate here a molecular link between epithelial-to-mesenchymal transition (EMT) and CD8+ TIL immunosuppression, two key drivers of cancer progression. We show that microRNA-200 (miR-200), a cell-autonomous suppressor of EMT and metastasis, targets PD-L1. Moreover, ZEB1, an EMT activator and transcriptional repressor of miR-200, relieves miR-200 repression of PD-L1 on tumor cells, leading to CD8+ T cell immunosuppression and metastasis. These findings are supported by robust correlations between the EMT score, miR-200 levels and PD-L1 expression in multiple human lung cancer datasets. In addition to revealing a link between EMT and T cell dysfunction, these findings also show that ZEB1 promotes metastasis through a heretofore unappreciated cell non-autonomous mechanism, and suggest that subgroups of patients in whom malignant progression is driven by EMT activators may respond to treatment with PD-L1 antagonists.
Introduction
Lung cancer is a leading cause of cancer-related mortality, and metastasis is the primary cause of death 1. Thus, successful prevention of lung cancer mortality requires a thorough understanding of the biological process of metastasis. K-rasLA1/+p53R172HΔg/+ (KP) mice develop lung cancer that faithfully recapitulates the clinical features of human disease and have been widely used to study lung cancer initiation and progression. We previously showed in this model that microRNA-200 (miR-200) expression is repressed in highly metastatic cancer cells and that forced miR-200 expression reversed the epithelial-mesenchymal transition (EMT) phenotype, abrogating tumor invasion and metastasis 2,3. The significance of these findings is supported by the evidence that low miR-200 expression is part of a microRNA expression signature in primary tumors that predicts disease recurrence in patients with early-stage lung cancer 4. The miR-200 family includes five members arranged in two genomic clusters (miR-200a/200b/429 and 200c/141) that directly target EMT-inducing transcription factors, including zinc-finger E-box-binding homeobox 1 (ZEB1) 5. In turn, ZEB1 can directly repress the transcription of both miR-200 loci. In cancer cells, the miR-200/ZEB1 axis controls the expression of multiple genes involved in migration, invasion, and metastatic growth at distant sites. Thus, miR-200 and ZEB1 form a double-negative feedback loop and function as a key regulatory axis of the EMT program 6-10.
The infiltration of lymphocytes into tumor tissues was initially believed to solely reflect attempts by the immune system to reject cancer cells. However, increasing evidence indicates that immune responses can exert either anti- or pro-tumorigenic effects 11,12. Immune cells within the tumor microenvironment, such as tumor–associated macrophages, myeloid-derived suppressor cells, and regulatory T cells, may contribute to the formation of a suppressive milieu that protects cancer cells from immune destruction 13,14. Furthermore, although cytotoxic CD8+ tumor infiltrating lymphocytes (CD8+TILs) can effectively kill cancer cells 15, prolonged exposure of CD8+TILs to cancer cells in the tumor bed can lead to complete or partial loss of their effector function, producing a state commonly referred to as exhaustion 16. How the tumor microenvironment controls CD8+TIL function is poorly understood. A growing number of studies indicate that co-inhibitory receptor signaling may play a key role in driving CD8+ T cell exhaustion 16-18. Indeed, blocking the interaction between the co-inhibitory receptor PD-1 and its ligand PD-L1 (also known as B7-H1, CD274) with antibodies has profound therapeutic effects in some patients with advanced non-small cell lung cancer, renal cancer, and melanoma 19-21. Furthermore, the clinical responses to anti-PD-1 antibody therapy appear to correlate with higher PD-L1 expression on cancer cells 21. Nevertheless, PD-L1 is also expressed on various other cell types in the tumor stroma, can be induced by various factors22,23, and its high expression has been associated with poor survival 17,24,25. However, the interplay of tumor-intrinsic and -extrinsic mechanisms controlling PD-L1 expression and consequent CD8+ T cell dysfunction remains poorly defined.
In this study, we investigate the regulation of tumor cell PD-L1 expression by the miR-200/ZEB1 axis, and the subsequent suppression of CD8+ T cells in the tumor microenvironment. We uncover a novel mechanism of tumor-intrinsic regulation of PD-L1 via the miR-200/ZEB1 axis, thus linking an EMT regulatory program to CD8+TIL exhaustion and further find that immunosuppression is critical to tumor metastasis.
Results
Tumor-intrinsic PD-L1 correlates with miR-200 and EMT
We recently described a 76 gene EMT signature from mRNA expression in non-small cell lung cancer, which is predictive for response of cell lines and patient tumors to targeted and chemotherapeutic agents 26. Application of this signature to score the EMT status of samples in The Cancer Genome Atlas (TCGA) lung adenocarcinoma dataset 27(n = 230) revealed robust separation of samples along an EMT spectrum, with significant anti-correlation to the expression of the five microRNA-200 family members, the mRNA levels of ZEB1/2, and the EMT markers E-cadherin, N-cadherin, vimentin, and β-catenin, along with the protein levels of multiple EMT markers (E-cadherin, claudin-7, α-catenin, β-catenin, fibronectin) (Fig. 1a). The expression of PD-L1 was strongly up-regulated in tumors with a high mesenchymal score (high EMT score) (Fig. 1b) (Spearman rho=0.527, p<2E-16). Analysis of all the microRNAs that correlate with PD-L1 expression in the TCGA dataset ranked the miR-200 family as the top microRNAs with statistically significant negative correlation, e.g. for miR-200b, Spearman rho= −0.454, p=1.55E-13 (Fig. 1c, Supplementary Fig. 1, and Supplementary Table 1). Application of the EMT signature to a separate validation set of surgically resected specimens showed statistically higher PD-L1 expression in the samples categorized as mesenchymal (n=92) versus epithelial (n=92) (p=3.11E-5) (Fig. 1d). These findings suggest that a significant number of primary human non-small cell lung cancers express PD-L1 and that the expression correlates with the mesenchymal phenotype of the tumor, as determined by the miR-200 levels.
Figure 1. EMT correlates with miR-200 and PD-L1 expression in clinical lung cancer datasets.
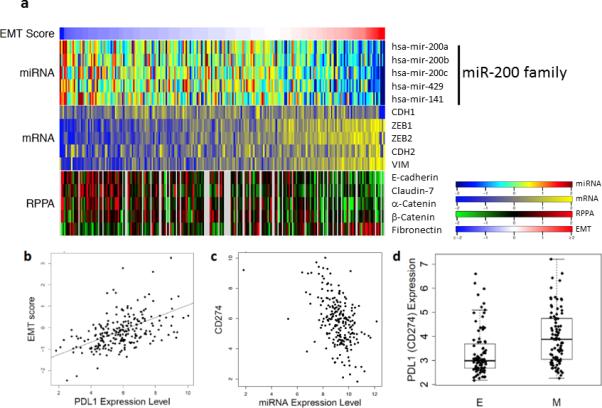
(a) Application of the EMT score (red (right) represents mesenchymal and blue (left) is epithelial) to the lung adenocarcinoma samples from the TCGA dataset (n=230) stratifies the samples by miR-200 family levels (top), EMT marker expression by RNA sequencing (middle), and protein markers (bottom). (b) Dot plot of the concordant EMT score and PD-L1 mRNA expression levels from the samples in the LUAD TCGA. The Spearman correlation is 0.527, p < 0.0001. (c) Dot plot of the concordant PD-L1 (CD274) and miR-200b expression levels from the TCGA samples. The Spearman correlation is −0.454, p < 0.0001. (d) Application of the EMT score to the samples from the PROSPECT dataset. Samples in the top 1/3 were designated mesenchymal (M) and in the bottom 1/3 designated as epithelial (E). Each group has n=92 and is represented in the box plot format. The PD-L1 mRNA expression level is graphed for the samples in each group. t-test, p < 0.0001.
Tumor cell PD-L1 is regulated by miR-200/ZEB1 axis in NSCLC
When transplanted into immunocompetent syngeneic mice, lung adenocarcinoma cell lines derived from KrasLA1/+p53R172HΔG/+ mice (KP cell lines) produce either highly metastatic, mesenchymal tumors (344SQ and 531LN3) or poorly metastatic, epithelial tumors (393P), properties that are manipulable by ectopic expression of ZEB1 or miR-200b/a/429 2,28. To further test the association of PD-L1 with EMT status and the miR-200/ZEB1 axis, we first evaluated the concordant reciprocal changes between PD-L1 and miR-200/ZEB1 expression in vitro. Stable constitutive, inducible or transient miR-200 expression (or conversely, ZEB1 knockdown) suppressed PD-L1 expression on murine (344SQ, 531LN2, 531LN3, and 393P_ZEB1) and human (H157, H1155, H1299, and H460) mesenchymal lung cancer cell lines, while PD-L1 expression was up-regulated in non-metastatic epithelial cells (murine 393P and human HCC827) upon constitutive ZEB1 expression (Fig. 2a and Supplementary Figs. 2, 3a, b). TGF-β is a potent EMT inducer that can disrupt the normal ZEB1-miR-200 double-negative feedback loop to drive a mesenchymal phenotype. In both human and murine lung cancer cells treated with TGF-β, the levels of ZEB1 and PD-L1 were significantly up-regulated (Supplementary Fig. 3c).
Figure 2. PD-L1 levels on human and murine NSCLC cell lines are regulated by the miR-200/ZEB1 axis.
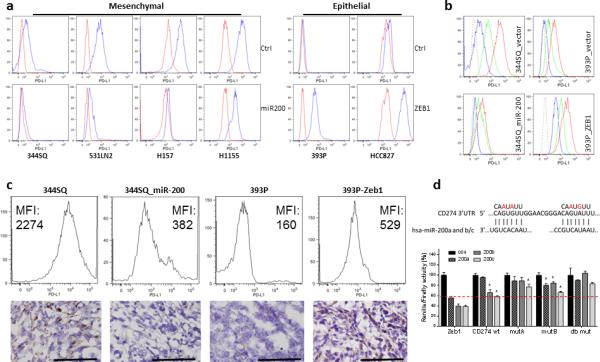
(a) Representative FACS histogram of PD-L1 expression on murine (344SQ, 531LN2, and 393P) and human (H157, H1155, and HCC827) lung cancer cells by overexpression of miR-200 and Zeb1. The analysis was independently repeated three times. (b) The representative FACS histogram of PD-L1 expression on murine lung cancer cells co-cultured with 129/Sv murine splenocytes with or without IFN-γ neutralizing antibody. Long dash is isotype control staining; blue line is anti-PD-L1 staining without splenocytes; green line is anti-PD-L1 staining with splenocytes blocked by the neutralizing antibody to IFN-γ (20 μg ml−1, 48 hrs); red line is anti-PD-L1 staining with splenocytes (48 hrs). The assay was independently performed three times. (c) The representative FACS histogram of PD-L1 (MFI, mean fluorescence intensity) expression in primary subcutaneous tumors grown in syngeneic 129/Sv mice (n = 3) injected with the indicated cell lines shown in the upper panel. The representative PD-L1 IHC staining of each tumor type shown in the bottom panel. Samples were obtained 2 weeks post-cell injection. Scale bar, 100 μm. (d) 3’-UTR luciferase reporter assay for wild-type PD-L1, single (mutant A or B) and double mutants (A and B) versus a Zeb1 3’-UTR control in murine 344SQ cells. The reporters were transiently co-transfected with synthetic miR-200 precursors (200a, 200b, or 200c) or control oligomers (con) into 344SQ cells. Values (p < 0.05) were normalized based on renilla luciferase and expressed as the mean values (±S.D.) of triplicate wells relative to that of controls co-transfected with empty reporter and empty expression vector or scrambled precursors, which were set at 1.0. The target sequences are shown in the top panel, with the introduced mutations highlighted in red.
Interferon γ (IFN-γ) is a pleiotropic cytokine that plays a central role in promoting innate and adaptive mechanisms of host defense against a variety of tumors and infectious agents 29,30. Previous reports have established that PD-L1 is up-regulated in response to IFN-γ produced by NK, T cells and macrophages; hence the infiltration of these cells is thought to drive tumor, stromal and myeloid cells to up-regulate PD-L1, protecting the tumor from immune attack 25,31. We found that upon in vitro IFN-γ stimulation in a co-culture system, the tumor cell expression of PD-L1 was up-regulated. Strikingly, the mesenchymal tumor cells (344SQ and 393P_ZEB1) were more responsive to IFN-γ than epithelial tumor cells (344SQ_miR-200 and 393P) (Fig. 2b). The consistent changes in PD-L1 expression upon miR-200 or ZEB1 expression observed in vitro were also found in syngeneic KP tumors grown in vivo (Fig. 2c). These findings clearly demonstrate that the miR-200/ZEB1 axis plays a dominant role in regulating the tumor cell expression of PD-L1 in either the presence or absence of IFN-γ.
The 3’-UTR of PD-L1 contains two very closely approximated sites that are predicted to bind the miR-200 family seed sequences (miR-200a and miR-200b/c) (Fig. 2d, Supplementary Fig. 4a, and Supplementary Table 2), leading us to postulate that PD-L1 is a miR-200 target. Transfection of a wild-type PD-L1 3’-UTR luciferase reporter construct into murine (344SQ) or human (H157 or H1299) lung cancer cells with low endogenous miR-200 levels revealed luciferase reporter activity that was suppressed upon co-transfection of miR-200b or −200c pre-miRs (Fig. 2d and Supplementary Fig. 4b), demonstrating a direct regulation of PD-L1 by the microRNA-200 family members. Mutation of each of the sites partially abrogated the pre-miR recognition, while the double mutant returned the reporter activity to control levels (Fig. 2d and Supplementary Fig. 4c).
Metastatic phenotype is dependent upon CD8+ T cell function
Initially, we found that lung tissues from the genetically engineered KrasLA1 mice, which develop non-metastatic lung adenocarcinomas, had significantly more CD8+ T cells than lung tissues from the Kras/p53 (KP) mice (Fig. 3a), in which miR-200 loss drives EMT and metastasis 2,3,32. Following subcutaneous or orthotopic intra-pulmonary injection into syngeneic, immunocompetent mice, KP cell lines (393P, 344SQ, 393LN, 531LN2) formed tumors with CD8+ T cell abundances that inversely associated with their metastatic potential (Fig. 3b and Supplementary Fig. 5a-d). To examine whether intratumoral CD8+ T cell suppression promotes tumor growth and metastasis, mice bearing high-miR-200 tumors (393P) were treated with control IgG or anti-CD8 antibody to immunodeplete CD8+ T cells, which enhanced tumor growth and metastatic capacity (Fig. 3c and Table 1). As a second approach, 393P or 344SQ cells were injected into syngeneic wild-type or lymphocyte-deficient Rag2−/− mice; metastases were significantly more abundant in Rag2−/− mice than they were in wild-type mice (Fig. 3d and Table 1), and adoptive transfer of CD8+ T cells into Rag2−/− mice reduced primary tumor growth and suppressed metastasis (Fig. 3e and Table 1). Interestingly, the epithelial and mesenchymal cell types retained their differential metastatic abilities in the Rag2−/− animals, suggesting an additional role for other cell types, such as NK cells. Although it warrants additional investigation, we did not explore this observation further in the current work.
Figure 3. CD8+TILs determine the metastatic potential in lung adenocarcinoma models.
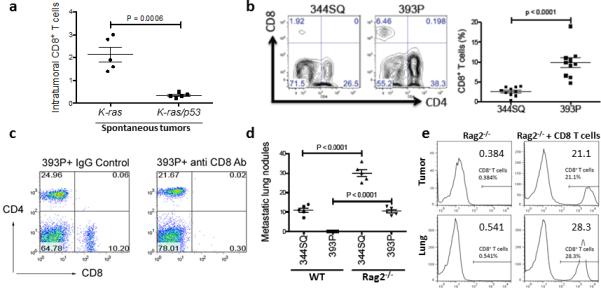
(a) CD8+ T cells measured by flow cytometric analysis in single-cell suspensions prepared from tumor-bearing lungs of 8- to 12- month-old K-rasLA1/+ (K-ras) and K-rasLA1/+p53R172HΔG/+ (KP) spontaneous mouse model (n = 5). The analysis was independently repeated at least three times. t-test, p = 0.0006. (b) The results of a FACS analysis of CD8+TILs in 344SQ and 393P primary tumors isolated 2 weeks after subcutaneous tumor cell injection into 129/Sv mice (n = 10). The results contain the data from two independent experiments. t-test, p < 0.0001. (c) Representative flow cytometric plot of CD8+TIL numbers from 393P tumor-bearing 129/Sv mice (n = 5) treated with anti-CD8 or IgG control antibodies (200 μg, intraperitoneally; twice weekly for 2 weeks beginning on day 1 after a subcutaneous cancer cell injection). (d) Lung metastases in WT or 129/SvRag2−/− mice (n = 5) injected subcutaneously with 344SQ or 393P cells and necropsied 5 weeks later. The analysis was independently repeated twice. t-test, p < 0.0001. (e) CD8+ T cells isolated from 129/Sv mice were adoptively transferred to syngeneic 129/SvRag2−/− mice (n = 5). Representative FACS histograms depict the levels of CD8+ T cells in 344SQ tumors and lungs of the reconstituted mice with reconstituted CD8+ T cells versus those in the controls.
Table 1.
Primary subcutaneous tumor mass and lung metastases.
| Treatment | Tumor mass (mg) | Metastasis |
|---|---|---|
| 393P + IgG control | 154 ± 19 | 0 |
| 393P + anti-CD8 Ab | 738 ± 60 | 5.6 ± 0.93 |
| 344SQ in Rag2−/− | 2160 ± 191 | 27.6 ± 0.51 |
| 344SQ in Rag2−/− + CD8 T cells | 740 ± 147 | 6.0 ± 1.52 |
In anti-CD8 antibody treatment group, 129/Sv mice (n = 5) treated with anti-CD8 or IgG control antibodies (200 μg, intraperitoneally; twice weekly beginning on day 1 after the subcutaneous 393P cell injection.). Analysis was done 6 weeks after tumor cell injection; In CD8 T cell adoptive transfer group, 4 × 106 CD8+ T cells isolated from splenocytes of 129/Sv mice were injected via the tail vein into 129/SvRag2−/− mice (n = 5) 48 hr prior to tumor inoculation. Analysis was done 5 weeks after tumor cell injection. Data from two independent experiments are shown as mean ± s.e.m. t-test, *P < 0.05.
miR-200/ZEB1 in tumor cells regulates CD8+TIL phentoype
Tumors of 393P cells expressing ZEB1 demonstrated significantly reduced total CD8+TILs, an increased percentage of exhausted CD8+ T cells (PD1+TIM3+) (Fig. 4a, b), and both increased overall tumor burden (391 ± 24 mg vs 1194 ± 161 mg) and metastatic lung nodules (0 vs 2.4 ± 0.5). Conversely, ectopic miR-200b/a/429 expression in highly metastatic KP cells (344SQ or 531LN2) increased the numbers of proliferating and granzyme B+ CD8+ T cells, decreased the exhausted CD8+ T cells (PD1+TIM3+) and subsequently suppressed metastases (Fig. 4a-d and Supplemental Fig. 5e). These effects of ectopic miR-200b/a/429 were reversed by treatment with anti-CD8 antibody (Fig. 4e, f) or growth in Rag2−/− mice (Fig. 4g).
Figure 4. The miR-200/ZEB1 axis controls tumor metastasis through regulating CD8+TILs.
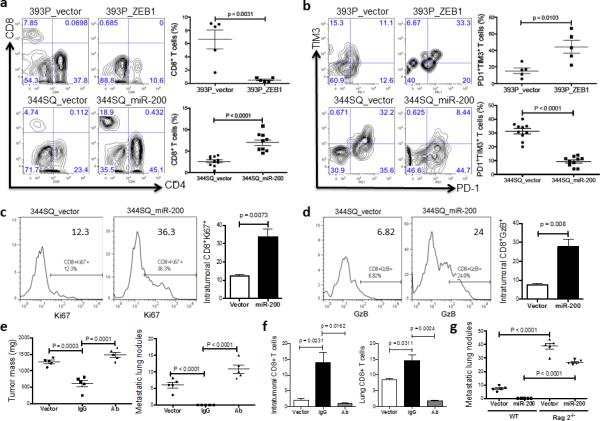
(a, b) FACS analysis of (a) CD8+TIL frequency; (b) PD1 and TIM3 marker expression on CD8+ T cells from 393P_vector and 393P_ZEB1 (n = 5), as well as 344SQ_vector and 344SQ_miR-200 (n = 10) primary tumors. Analysis was done 2 weeks post-cancer cell injection. (c, d) (c) Intratumoral Ki67+CD8+ T cells; (d) granzyme B (GzB)+CD8+ T cells in 344SQ_vector or 344SQ_miR-200 primary tumors 6 weeks post-subcutaneous injection of cancer cells into 129/Sv mice. Representative Ki67 or GzB staining in an individual tumor sample is shown on the left, and mean Ki67+ or GzB+ populations of gated CD8+ T cells in total T cells are shown on the right (n = 5). (e) CD8+ T cell depletion results in tumor growth and metastasis in mice (n = 5) that received subcutaneous tumor cell injections. No treatment (344SQ_vector (Vector)), IgG (344SQ_miR-200 + IgG control), or Ab (344SQ_miR-200 + anti-CD8 Ab). The analysis was done 6 weeks post-injection. (f) Relative abundance of CD8+ T cells in the tumor (left) or lung (right) from 129/Sv mice (n =5) with syngeneic control 344SQ tumors (Vector), 344SQ_miR-200 tumors with control IgG treatment (IgG) or anti-CD8 antibody treatment (Ab). (g) Lung metastases of 344SQ_vector (Vector) and 344SQ_miR-200 (miR-200) tumors in wild-type (WT) or 129/SvRag2−/− (Rag2−/−) mice (n = 5). The analysis was done 6 weeks post-tumor cell subcutaneous injection. All the analyses were independently repeated twice. Data are shown as mean ± s.e.m. t-test was used to analyze, with P values shown in the graphs.
Because CD8+ T cell infiltration affected not only metastasis but also the growth of primary tumors (Fig. 4e and Table 1), we examined whether the effects of CD8+ T cells on metastasis depend on primary tumor size. Adjusted numbers of 344SQ, 344SQ_miR-200 and 393P tumor cells were injected into 129/SV mice to allow the primary subcutaneous tumors to grow to a similar size. The mice were sacrificed 6 weeks post-injection when the primary tumor masses reached 800-1000 mg in size, and lung metastases were examined. Significant differences in lung metastasis were observed, even though the primary tumors were of similar size (Supplementary Fig. 6a,b). In a complementary experiment, we found that the number of CD8+ T cells and the percentage of PD1+TIM3+ exhausted T cells were strictly dependent on metastatic potential, not on primary tumor size (Supplementary Fig. 6c-e). In addition, metastatic potentials of the different cell lines following injection with the same cell number via the tail vein were the same as with subcutaneous injection (Supplementary Fig. 6f), further indicating that metastatic potential was not dependent upon the primary tumor mass. Furthermore, miR-200 levels in tumor cells did not affect the numbers of CD8+ T cells in the spleen and peripheral blood, nor did it affect CD8+ T cell migration in vitro, suggesting that miR-200 controls the local abundance of CD8+TILs in the tumor microenvironment without affecting CD8+ T cell recruitment. Collectively, our data demonstrated that metastatic potential is dependent upon the miR-200 levels in KP tumors, which determine the abundance and function of CD8+TILs, independent of primary tumor size, growth rate, or route of inoculation.
miR-200/ZEB1 regulating CD8+TIL function is PD-L1-dependent
To determine whether cancer cell-autonomous PD-L1 directly induces CD8+TIL exhaustion and tumor metastasis, we depleted PD-L1 in 344SQ cells by shRNA-mediated knockdown (Fig. 5a) and tested them in syngeneic mice. Primary tumor size and number of lung metastases in 344SQ-shPD-L1-bearing mice were significantly reduced relative to control transfectants (Fig. 5b), along with a four-fold increase in CD8+ T cells and a decrease in the percentage of exhausted CD8+TILs (PD1+TIM3+ and PD1+LAG3+) (Fig. 5c, d). Similarly, PD-L1 neutralization in mice bearing implanted 344SQ and 393P_ZEB1 tumors by intra-peritoneal injection of an anti-PD-L1 antibody reduced tumor size, metastasis and exhausted CD8+ T cells relative to control IgG (Fig. 6a-c), and increased infiltrating CD8+ T cells, intratumoral Ki-67+ and granzyme B+ CD8+ T cells (Fig. 6d and Supplementary Fig. 7). On the other hand, the anti-PD-L1 antibody treatment did not show significant effects on tumors with constitutively high miR-200 levels (344SQ_miR-200 and 393P) (Fig. 6). Consistent with these findings, when PD-L1 was genetically reconstituted into 344SQ_miR-200 and 344SQ-shPD-L1 cells, the primary subcutaneous tumors grew larger, had less CD8+TILs and more exhausted T cells, and produced more metastatic lungs nodules (Supplementary Fig. 8). We thus conclude that miR-200 down-regulation increases PD-L1 tumor cell expression, which drives CD8+ T cell suppression in the tumor microenvironment and facilitates metastasis.
Figure 5. Genetic targeting of PD-L1 expression on cancer cells reverses the CD8+TIL dysfunction and suppresses metastasis.
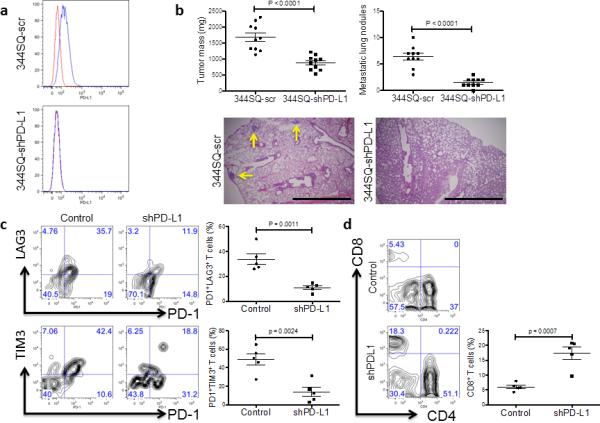
(a) Cell surface expression of PD-L1 on 344SQ PD-L1 knockdown (344SQ-shPD-L1) vs 344SQ scramble control (344SQ-scr) cells by FACS (red line, isotype control staining; blue line, anti-PD-L1 staining). The measurement was independently repeated at least three times. (b) Primary tumor mass (top left) and lung metastases (top right) in 129/Sv mice (n = 10) injected subcutaneously with 344SQ-shPD-L1 or 344SQ-scr cancer cells. Micrometastases (bottom) observed in hematoxylin and eosin-stained lung tissue sections are indicated by yellow arrows. Scale bar, 2mm. Samples were obtained 6 weeks post-injection. The data from two independent experiments were pooled. Data are shown as mean ± s.e.m. t-test was used to analyze. P values are shown in the graphs. (c, d) FACS analysis of (c) surface PD1, LAG3, and TIM3 marker expression levels on CD8+ T cells; (d) CD8+TIL frequency for primary tumors in 129/Sv mice (n = 5) injected subcutaneously with 344SQ-shPD-L1 (shPD-L1) or 344SQ-scr control (Control) cancer cells and necropsied 2 weeks later. The analyses were independently repeated three times. Data are shown as mean ± s.e.m. t-test was used to analyze. P values are shown in the graphs.
Figure 6. Pharmacologic targeting of PD-L1 with antibody treatment prevents tumor growth and metastasis.

(a and b) The tumor burden (a) and lung metastases (b) 6 weeks post-cancer cell injection into 129/Sv mice (n = 5) with IgG control (IgG) or anti-PD-L1 antibody (Ab) treatment. Anti-PD-L1 antibody or IgG control was injected (100 μg, intraperitoneally) twice a week for 6 weeks after the subcutaneous tumor cell injection. Following euthanasia, mice were necropsied to harvest primary tumors and lungs, which were weighed, and to quantify distant metastases. (c and d) The CD8+ T cell function and the abundance of CD8+ T cells in tumors (n = 5) after anti-PD-L1 antibody treatment. All the analyses were independently repeated twice. Data are shown as mean ± s.e.m. t-test was used to analyze. P values are shown in the graphs.
Intratumoral CD8+ T cell activity is dynamically modulated through diverse mechanisms including loss of tumor cell antigenicity, changes in the expression of B7 family T cell co-inhibitory molecules (PD-L1 and PD-L2) and co-stimulatory molecules (CD80, CD86, and MHC Class II) on tumor cells and the recruitment and activation of a variety of intratumoral immune cells 19-21. Flow cytometric analysis of high- versus low-miR-200 tumors demonstrated no evidence of enhanced recruitment of myeloid-derived suppressor cells (CD11b+ Gr-1+) or tumor-associated macrophages (CD11b+ CD11c+ Gr-1+). Constitutive miR-200 expression in 344SQ cells did not increase MHC Class I expression or enhance CD8+ T cell-mediated tumor cell cytotoxicity in cell killing assays, arguing against altered tumor cell antigenicity affecting CD8+TIL-mediated tumor rejection.
Based on the extensive studies of IFN-γ, it is well known that this cytokine plays a key role in tumor surveillance 29 and is a potent inducer of PD-L1 expression, which was proposed as a cause of tumor immune evasion 25. We questioned whether the miR-200/ZEB1 axis modulates the IFN-γ pathway through regulation of JAK/STAT signaling, but found no signaling pathway changes to support this (Supplementary Fig. 9a). We then tested the impact of IFN-γ blockade on tumor growth in vivo. Upon treatment with a neutralizing IFN-γ antibody, PD-L1 expression on mesenchymal (344SQ) tumors dramatically decreased (Supplementary Fig. 9b), while both the mesenchymal (344SQ) and epithelial (344SQ_miR-200) syngeneic tumors showed increased growth, with a dramatic decrease in total CD8+TILs (Supplementary Fig. 9c, d). However, within the IFN-γ blocking antibody group, there was still a significant difference between miR-200high and miR-200low tumors in growth, CD8+TILs and exhausted T cells (Supplementary Fig. 9c-e). Additionally, with IFN-γ blockade, there was a significant decrease in exhausted T cells in mesenchymal tumors while there was no significant decrease in epithelial tumors (Supplementary Fig. 9e), again emphasizing that mesenchymal tumors are more sensitive to IFN-γ. Taken overall, the data demonstrates that the miR-200/ZEB1 axis is a downstream regulator of PD-L1 that can modulate the effects of the IFN-γ pathway.
Repression of anti-tumor immunity requires tumor cell PD-L1
Diverse cell types in the tumor microenvironment (e.g., myeloid cells, fibroblasts, and endothelial cells) express PD-L1, which could suppress anti-tumor immunity in the absence of tumor cell PD-L1 expression. However, when multiple different cancer cell types (B16 melanoma, KPC pancreas, or MC38 colon) were implanted into PD-L1 wild-type (WT) and knockout (KO) mice, there was no significant difference in tumor growth between WT and KO mice (Supplementary Fig. 10). To determine whether expression on other cell types rescues PD-L1 deficiency on tumor cells, we utilized the Lewis lung carcinoma (LLC) model in which, on the basis of treatment with anti-PD-L1 antibody, PD-L1 promotes primary tumor growth and metastasis (Supplementary Fig. 11). In addition, miR-200 represses PD-L1 expression in the LLC model as in the KP model (Supplementary Fig. 12a). We generated LLC cell lines with intermediate or very low level PD-L1 expression using shRNA to PD-L1 (Fig. 7a), injected these cells into wild-type or PD-L1-knockout mice (Fig. 7b), and measured tumor growth rate (Fig. 7c), intratumoral CD8+ T cells (Fig. 7d), mouse survival, final tumor weight, number of metastases (Supplementary Fig. 12b-d). PD-L1 deficiency in tumor cells suppressed tumor growth and metastasis and increased intratumoral CD8+ T cells, which were reversed by stable reconstitution of PD-L1 in the shPDL1 LLC cells (Fig. 7e, f). In contrast, PD-L1 deficiency in the host had no detectable effect on PDL1-deficient or –replete LLC tumors (Fig. 7a-f and Supplementary Fig. 12b-d). We conclude that PD-L1 expression on tumor cells plays a dominant role in the regulation of CD8+ T cell-mediated anti-tumor immunity.
Figure 7. PD-L1 expression on tumor cells is critical to the repression of anti-tumor immunity.
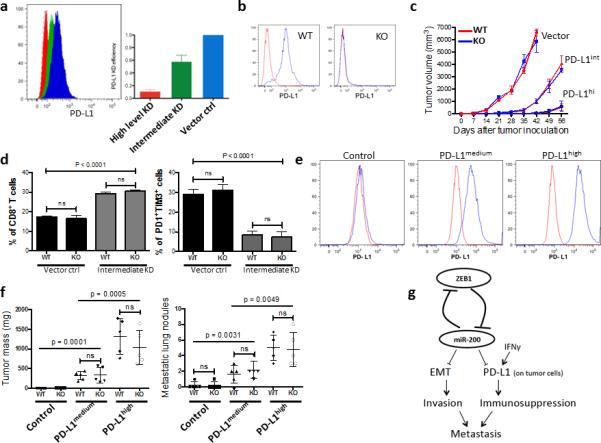
(a) The knockdown (KD) efficiency of PD-L1 in LLC-JSP murine lung cancer cells measured by FACS. Representative histograms (left), and statistical analysis (right). The measurement was independently repeated at least three times. (b) Representative FACS histogram of PD-L1 expression on myeloid cells (CD11b+) in PD-L1 KO or WT mice (n = 3). (c) Tumor growth in PD-L1 KO or WT mice (n = 6) of subcutaneously injected LLC-JSP cells (10, 000 cells with 100 μl of PBS per mouse) with differing PD-L1 knockdown (Vector, PD-L1 KD vector control; PD-L1int, PD-L1 intermediate KD; PD-L1hi, PD-L1 high level KD). The data is shown from two independent experiments. Data are shown as mean ± s.e.m. (d) FACS analysis of CD8+TIL frequency and T cell exhaustion marker expression levels on CD8+ T cells in subcutaneous primary tumors of LLC-JSP vector control (Vector ctrl) and LLC-JSP intermediate PD-L1 knockdown (Intermediate KD) in PD-L1 WT (WT) and PD-L1 KO (KO) mice (n = 5, from two independent experiments). Analysis was done 3 weeks post-tumor cell injection (20, 000 cells with 100 μl of PBS per mouse). t-test was used to analyze the data. p < 0.0001. (e) PD-L1 expression levels on LLC-JSP-shPD-L1 cells (the high knockdown efficiency cells) after reconstitution of PD-L1. The measurement was independently repeated at least three times. Control, LLC-JSP-shPD-L1 + vector control; PD-L1medium, LLC-JSP-shPD-L1 + intermediate overexpression of PD-L1; PD-L1high, LLC-JSP-shPD-L1 + high overexpression of PD-L1. (f) Primary tumor masses and lung metastases in PD-L1 KO or WT mice (n = 5) injected subcutaneously with the different reconstituted cell lines. Analysis was done 4 weeks post-subcutaneous cancer cell injection (10, 000 cells with 100 μl of PBS per mouse). The analyses were independently repeated twice. Data are shown as mean ± s.e.m. t-test was used to analyze the data. ns, p > 0.05. (g) The working model of the mutual regulation of EMT and immune suppression by miR-200/ZEB1 axis and their complementary role in metastasis.
Discussion
During the initial stages of metastasis, epithelial tumor cells undergo EMT, causing a loss of cell-cell contacts, increased motility, and invasion into the surrounding stroma 33. Occurring in parallel are changes in the tumor microenvironment that suppress anti-tumor immunity 11. Although the synchronicity of these distinct biological processes implies the possibility of a common genetic driver, the existence of a molecular linkage between them has not been reported. Here, we provide evidence that EMT is tied to suppression of anti-tumor immunity through miR-200/ZEB1, an EMT regulatory axis that controls PD-L1 expression on tumor cells.
Due to the double-negative feedback loop between ZEB1 and miR-200 that governs a reversible switch between epithelial and mesenchymal states 7,9,10, we previously investigated the role of the miR-200/ZEB1 axis in metastasis of lung adenocarcinoma and found that the ZEB1-induced suppression of miR-200 promotes tumor cell invasion and metastasis 2,28,32. Since its discovery as a master regulator of EMT, the miR-200 family was subsequently found to regulate a number of complementary cellular functions relevant to tumor growth and metastasis, including stem cell properties, sensitivity to chemotherapeutic agents, reduction in angiogenesis, and responsiveness to pro-apoptotic signals. The target genes identified to date through which miR-200 regulates these biologic properties are in most cases non-overlapping 34-41. Similarly, studies on miR-31 have demonstrated that multi-tasking is an essential feature of its capacity to promote metastasis; concurrent re-expression of three target genes (ITGA5, RDX, and RHOA) is required to abrogate miR-31-imposed metastasis suppression 42. Among its pleiotropic targets controlling complementary tumor cell functions, here we demonstrate that PD-L1 is a downstream target of the miR-200/ZEB1 axis and contributes to immunosuppression in the primary tumor tissue. Although the miR-200/ZEB1 axis regulation of PD-L1 may not entirely account for the immunosuppressive effects observed, PD-L1 blockade significantly improves CD8+ T cell infiltration, reverses the exhausted T cell phenotype, reduces tumor burden and metastases only with mesenchymal tumors (344SQ, 531LN2, 393P_ZEB1, and LLC-JSP) and not with epithelial tumors (344SQ_miR-200 and 393P) (Fig. 6, Fig. 7, and Supplementary Figs. 7, 11, 12c, d), while the reconstitution of PD-L1 expression reverses the phenotype of epithelial tumors (344SQ_miR-200, 344SQ-shPD-L1, and LLC-JSP-shPD-L1) (Fig. 7e, f, Supplementary Fig. 8). We additionally observed that even in the absence of IFN-γ mesenchymal tumors produce T cell exhaustion due to miR-200/ZEB1-regulated change of PD-L1 expression (Supplementary Fig. 9b-e), indicating that mesenchymal tumor cells are hardwired to prevent immune attack. However, there was a synergy between IFN-γstimulation and miR-200 repression, illustrating how different modes of PD-L1 regulation can converge in the same tumor cells. These findings represent a remarkable departure from the tumor cell-autonomous processes regulated by the pleiotropic miR-200 family and are, to our knowledge, the first example of a microRNA expressed in tumor cells that regulates metastasis by modulating intratumoral immune cell function. The convergence of these biological processes upon a single microRNA presents a clear selective advantage to tumor cells in which this microRNA is silenced, as shown in the model (Fig. 7g).
The dynamic interaction of immune and cancer cells in tumor tissues plays a critical role in controlling cancer progression. Among the various immune cell infiltrates into tumor tissues, CD8+TILs appear to be the key immune cells showing strong cytotoxic activity that is direct and selective for cancer cells 43. Survival duration correlates positively with intratumoral CD8+ T cell abundance and negatively with intratumoral expression of the T cell co-inhibitory molecule PD-L1 and its receptor PD1, providing strong clinical relevance of prognostic value in patients with various epithelial tumor types, including lung cancer 44-46. Furthermore, PD-L1/PD1 axis blockade in preclinical models, including those reported here, suppresses tumor growth and metastasis 44,45. However, PD-L1 is expressed on multiple intratumoral cell types that may cooperate to effect CD8+ T cell suppression. Here, we addressed the relative importance of different cellular sources of PD-L1 to assess whether PD-L1 up-regulation on tumor cells alone is sufficient to cause CD8+ T cell immunosuppression (Fig. 7, Supplementary Figs. 8, 12b-d). We found that intratumoral CD8+ T cell exhaustion is mediated primarily by tumor cell PD-L1 expression. PD-L1 on tumor cells, rather than other cell types, results in CD8+ T cell exhaustion and diminution of CD8+TIL number in a chronic fashion. The finding that other cell types exerted no measurable effect was surprising but may not be unique to the models studied in depth here as multiple different tumor models show a similar effect (Figs. 5, 7 and Supplementary Figs. 8, 12), which serves to illustrate the point that miR-200/ZEB1 controls metastasis in part through tumor cell non-autonomous mediators.
Patients with advanced melanoma experience a survival advantage following CD8+ T cell reactivation by treatment with antibodies targeting CTLA-4, another B7-CD28 family member 47. Evidence from lung cancer preclinical models presented here and elsewhere 48 that PD-L1-induced CD8+ T cell exhaustion promotes tumor growth and metastasis raises the possibility that treatment with PD-L1 inhibitors might provide therapeutic benefit to lung cancer patients. Clinical trials with anti-PD-L1 or PD1 neutralizing antibodies are ongoing in patients with a variety of solid tumors, including lung cancer, with preliminary data indicating significant benefit for subsets of lung cancer patients with established metastatic disease 49. Such strategies might be combined with anti-CD25 antibodies that deplete CD4+Foxp3+ regulatory T cells, which enhance the anti-tumor efficacy of cytotoxic chemotherapy in lung cancer preclinical models 50. On the basis of evidence presented here that PD-L1 is up-regulated in early-stage lung adenocarcinomas that have a mesenchymal expression pattern and low miR-200 expression, representing up to one-third of patients, it is tempting to speculate that PD-L1 inhibitors will be particularly efficacious in this group of patients.
Methods
Human samples
The initial set of samples analyzed consisted of lung adenocarcinoma cases from The Cancer Genome Atlas (TCGA) project (http://cancergenome.nih.gov/)27, which represent early-stage surgical resection specimens and were collected in accordance with The Cancer Genome Atlas (TCGA) Human Subjects Protection and Data Access Policies. . The second set of samples was from the Department of Thoracic/Head and Neck Medical Oncology at MD Anderson. The Profiling of Resistance patterns and Oncogenic Signaling Pathways in Evaluation of Cancers of the Thorax (PROSPECT) trial was developed in 2006 with the purpose of investigating molecular mechanisms of therapeutic resistance, and was recently completed. After MDACC Institutional Review Board approval of the protocol, retrospectively-identified samples were collected from two hundred and fifteen patients enrolled in the PROSPECT trial. Stage distribution varied, but almost all patients had locoregionally confined disease, as expected from a set of specimens from surgical resection. For each dataset an EMT score for each sample was computed using an averaging scheme based on mRNA expression of 76 EMT signature genes previously published by our group26. The scores were calculated as the average expression level of mesenchymal genes minus the average expression level of epithelial genes.
Plasmids and reagents
pLenti4.1-miR-200 was obtained from Dr. Gregory Goodall (University of Adelaide) and used as described elsewhere2,7,28. A modified doxycycline inducible pTRIPz-RFP vector expressing miR-200a, miR-200b, or in combination (miR-200ab) was generously provided to us by Dr. Gregory Goodall at the University of Adelaide. Mouse ZEB1 was subcloned into pcDNA3.1/His C (Invitrogen) to generate the ZEB1 overexpression vector. shRNA to mouse ZEB1 and PD-L1 and scrambled controls in the pGFP-V-RS vector were obtained from OriGene. Mouse PD-L1 expression vector pUNO1-mCD274 was purchased from Invivogen. Pre-microRNAs miR-200c and negative control (scrambled oligos) (Life Technologies) were reverse transfected into cells with Lipofectamine 2000 (Life Technologies) at a final concentration of 50 nM.
siRNA knockdown for PD-L1 expression analysis
Cells seeded in a 6-well plate were transfected with siRNA of ZEB1 (40 nM; OriGene) as well as their scrambled controls. The cells were harvested 48 hr after transfection to examine PD-L1 expression.
PD-L1 3’-UTR reporter assay
miRNA-binding sites were predicted by TargetScan (release 5.1; http://www.targetscan.org). Human CD274 (4.6kb) was cloned by PCR and ligated into the pCI-neo-hRL vector. The ZEB1 3’-UTR (1.9 kb) subcloned into the same vector was used as a control. Mutation of the individual miR-200 sites (Mut A or Mut B) or a double mutant at both sites was made and cloned into the same vector. These reporters were co-transfected with individual synthetic miR-200 family precursors (5 nM; Ambion) into either murine 344SQ, or human H1299 or H157 cells (1.0 × 105 cells/well) in 24 well plates. pGL3-basic (50 ng; Promega) was used as an internal control. Forty-eight hours post-transfection dual luciferase activity was measured with the Dual-Luciferase Reporter Assay System (Promega). Results were normalized on the basis of firefly luciferase according to the manufacturers protocol.
Cells and mice
The epithelial 393P and 393LN as well as mesenchymal 344P, 344SQ, 531LN2, and 531LN3 cell lines were derived from KP mice 27,51. B16 melanoma, KPC pancreatic, and MC38 colon, LLC-JSP Lewis lung cancer cell lines were maintained in our laboratories. Stable cell lines 344SQ_miR-200, 531LN2_miR-200, and H1155_miR-200 were generated by pLenti4.1-miR-200 lentivirus transduction. H157 cells were transduced by lentiviral delivery of the doxycycline-inducible pTRIPz-miR-200 or control vector (miR-200a, b or ab). Stable cell lines 344SQ-shPD-L1 and LLC-JSP-shPD-L1 were generated by transfection with shRNA expression vectors. Stable cell lines 393P_ZEB1 and HCC827_ZEB1 were generated by transfection with the corresponding expression vectors. Stable cell lines PD-L1-expressing 344SQ_miR-200, PD-L1-overexpressed 344SQ-shPD-L1, and PD-L1-overexpressed LLC-JSP-shPD-L1 were generated with mouse PD-L1 expression vector pUNO1-mCD274 (Invivogen). Stable cell line LLC-JSP-t2 was generated in our laboratory. Human lung cancer H322, H1299, H441, H157, H1155, HCC827, and H460 cell lines were purchased from the American Type Culture Collection. The H460 and H1299 cells with stable shRNA-mediated knockdown of ZEB1 were kindly provided by Dr. Raymond E Meyn (Professor, Department of Experimental Radiation Oncology, Division of Radiation Oncology, The University of Texas MD Anderson Cancer Center, Houston, TX).
Six- to 8-week-old 129/Sv or C57BL/6 male mice were obtained from Charles River Laboratories; 129/SvRag2−/− mice were obtained from Taconic and 6- or 8-week-old male mice were used for the experiments. PD-L1-deficient C57/BL6 mice were obtained from L. Chen lab and maintained as a homozygous line and 6- or 8-week-old male mice were used for the experiments. Spontaneous K-ras and K-ras/p53 mice were generated by our laboratory52. 8- to 12- month-old K-ras and K-ras/p53 mice were used for the experiments. All animal procedures were reviewed and approved by The University of Texas MD Anderson Cancer Center Animal Care and Use Committee.
Tumor models
For examining intratumoral lymphocyte populations, 2 × 106 cancer cells in 100 μl of 50% Growth Factor Reduced BD Matrigel™ Matrix (BD Biosciences) were subcutaneously injected into the flanks of 129/Sv mice. If not specified otherwise, tumors were harvested and analyzed 2 weeks after injection. For studying the growth rate of the primary tumor mass and formation of lung metastases, if not indicated, 1 × 106 cancer cells in 100 μl of phosphate-buffered saline (PBS) were injected subcutaneously into the mouse flank. For the orthotopic model, 1 × 106 cancer cells were mixed with a 1:10 dilution of Matrigel and injected into the lung as described elsewhere53. Mice were monitored regularly and euthanized 5-6 weeks after the tumor cell injections. At end points, mice were sacrificed to examine the primary and metastatic tumors. For studying the relationship of the primary subcutaneous tumor and the lung metastasis, 1 × 106 344SQ cells, 2 × 106 344SQ_miR-200 cells, or 3 × 106 393P cells in 100 μl of PBS were injected subcutaneously into 129/Sv mice. Mice with comparable sizes of primary tumors were chosen for examining lung metastasis at necropsy 6 weeks after injection. For experimental pulmonary metastasis, 5 × 105 cancer cells were injected through the tail vein. Lungs were removed to measure metastatic nodules 6 weeks later.
For lung metastasis measurement, except for LLC-JSP-t2-bearing lungs, in which the metastatic lung nodules were counted under the fluorescent microscope, the lungs were removed and immersed in cold PBS for 2-3 hrs, the gray nodules on the surface of lung were counted as described previously 52,53.
Antibody-mediated cell depletion
Anti-CD8 antibody (2.43; BioXCell) or an immunoglobulin G (IgG) control was injected into the mice (200 μg, intraperitoneally) twice weekly for 2 weeks beginning on day 1 after a subcutaneous cancer cell injection.
CD8 T cell adoptive transfer
CD8+ T cells were isolated from splenocytes of 129/Sv mice by MACS technology according to the manufacturer's instructions (Miltenyi Biotec). CD8+ T cells (4 × 106) were injected via the tail vein into 129/Sv Rag2−/− mice 48 hr prior to tumor inoculation.
anti-PD-L1 and anti-IFN-γ antibody neutralization
Anti-PD-L1 (9G2; BioXcell) antibody or an isotype matched IgG control was injected into 129/Sv or C57BL/6 mice (100 μg, intraperitoneally) twice a week for 3 or 6 weeks beginning on day 1 after the subcutaneous indicated tumor cell injection. 129/Sv mice were treated on days −1, +2 and +5 with intraperitoneal injections of 250 μg of neutralizing antibody to murine IFN-γ (XMG1.2; BioXcell) as described 29.
Flow cytometry
Single-cell suspensions were prepared and stained according to standard protocols for flow cytometry with the following antibodies: mouse CD3-PerCP (BD Biosciences, 560527 1:100 dilution), CD4-APC (mouse; BD Biosciences, 553051, 1:100 dilution), CD4-FITC (mouse; BD Biosciences, 553729, 1:50 dilution), CD8b-APC (mouse; eBioscience, 17-0083, 1:100 dilution), CD8b-PE (mouse; eBioscience, 12-0083-83, 1:200 dilution), CD11b-APC (mouse; eBioscience, 17-0112-83, 1:100 dilution), Ki67-PE (eBioscience, 12-5698-82, 1:100 dilution), GzB-PE (eBioscience, 12-8898-82, 1:100 dilution), PD1-PE (mouse; eBioscience, 12-9985-83, 1:150 dilution), PD1-FITC (mouse; eBioscience, 11-9985-82, 1:150 dilution), PD-L1-PE (mouse; BD Biosciences, 558091, 1:200 dilution), PD-L1-PE (human; eBioscience, 12-5983-42, 1:200 dilution), TIM3-PE (mouse; eBioscience, 12-5870-83, 1:150 dilution), and LAG3-PE (mouse; eBioscience, 12-2231-83, 1:100 dilution). For intracellular staining, cells were fixed and permeabilized with BD Cytofix/Cytoperm (BD Biosciences). The data were acquired on a Fortessa or Calibur platform (BD Biosciences) and analyzed with FlowJo software (version 7.6; Tree Star). For analyzing the abundance and the function of CD4+ or CD8+ TILs, single-cell suspensions were prepared from tumors and inguinal lymph nodes and stained, the staining of inguinal lymph node cells was used as the reference of lymphocyte gating, then CD3+ cells were gated, and then CD4+ or CD8+ population was analyzed.
RNA and protein analyses
Total RNA was isolated from cultured cells with TRIzol (Invitrogen). cDNA was synthesized by using the SuperScript III kit (Invitrogen), and qPCR was done with the SYBR Green PCR Master Mix (Applied Biosystems). Primers were designed with National Center for Biotechnology Information (NCBI) primer design software. Relative expression levels were normalized by B2M and calculated by the 2−ΔΔCt method 54. The primers used for qPCR are shown in Supplementary Table 3 (mZEB1, hZEB1, mPD-L1, hPD-L1, hNcad, hEcad, hVim).
miR-200 expression assays were performed using 10 ng of total RNA according to the manufacturer's instructions (Applied Biosystems). Values were normalized on the basis of control miR-16 levels.
Total protein was extracted using NP40 lysis buffer (0.5% NP40, 250 mM NaCl, 50 mM Hepes, 5 mM ethylenediaminetetraacetic acid, 0.5 mM egtazic acid) supplemented with protease inhibitors cocktails (Sigma-Aldrich). Lysates was centrifuged at 12,000 rpm for 10 min, and the supernatant was collected for experiments. Protein lysates (40 μg) were resolved on denaturing gels with 4% to 20% sodium dodecyl sulfate (SDS)-polyacrylamide and transferred to nitrocellulose membranes (Biorad Laboratories). Membranes were probed with the following antibodies: primary antibodies anti-ZEB1 (Santa Cruz, sc-25388, 1:500 dilution), anti-Ncad (BD Biosciencses, 610921, 1:500 dilution), anti-Ecad (BD Biosciences, 610182, 1:10000 dilution), anti-Vim (Cell Signaling, 3932s, 1:500 dilution), anti-STAT1 (Abcam, ab2415, 1:2000 dilution), anti-p-STAT1 (Tyr 701) (Santa Cruz, sc-7988, 1:1000 dilution), anti-p-STAT1 (Ser 727) (Abcam, ab109461, 1:1000 dilution), anti-Jak1 (Abcam, ab75744, 1:1000 dilution), anti-p-Jak1 (Tyr 1022/1023) (Santa Cruz, sc-16773, 1:1000 dilution), anti-IFNGR1 (Santa Cruz, sc-702, 1:3000 dilution), anti-Actin (Abcam, ab8227, 1:5000 dilution), and secondary antibody labeled by horseradish peroxidase (Amersham GE Healthcare). The secondary antibody was visualized using a chemiluminescent reagent Pierce ECL kit (Thermo Scientific). The original photos of western blots are shown in Supplementary Fig. 13.
Histologic analysis
Tissues were fixed in 10% paraformaldehyde and embedded in paraffin. Hematoxylin and eosin-stained sections were examined to identify micrometastases. For IHC, cryosections (8 μm) of tumor tissues were fixed with acetone and stained with antibody against mouse PD-L1 (Abcam, ab58810, dilution 1:100) and horseradish peroxidase-conjugated secondary antibody. Images (20×) were acquired with an Olympus BX41 microscope.
Statistics
All statistical analyses were performed with GraphPad Prism 5.0 software. Significant differences (p < 0.05) between two groups were identified by Student t tests.
Supplementary Material
Acknowledgments
We thank Dr. Ying Ma, Nasser Kazimi, and Jessica Cabrejos for expert advice and technical assistance. This work was supported by ACS RSG LIB-117155, 5-P50-CA70907-12 PP-3b, NSFC Grant (no. 31170832), and Guangdong Innovation Research Team Program (no. 201001Y0104687244), MOST PPG (2014CB745203) and RSFPWIH Grant (201302018) to F.X-F.Q.; 5-R01-CA132608-03, 5-P50-CA70907-12 PP-3 to J.M.K. and J.V.H.; 5-R01-CA168484-04 (J.V.H.); ACS RSG-10-056-01-LIB (S.E.U.); NIH Cancer Center Support Grant (CA016672) to MDACC core facilities; Rexanna's Foundation for Fighting Lung Cancer, The Stading Lung Cancer Research Fund, MD Anderson Cancer Center Physician Scientist Award and K08-CA151651 to D.L.G.; MD Anderson Cancer Center Physician Scientist Award, Sidney Kimmel Scholar Award and the Cancer Clinical Investigator Team Leadership Award (P30CA016672) to L.A.B.; Training Program in Molecular Genetics of Cancer, NCI T32 CA009299-32 (C.U.). D.L.G. is a R. Lee Clark Fellow of the University of Texas MD Anderson Cancer Center, supported by the Jeane F. Shelby Scholarship Fund. Supported by the generous philanthropic contributions to The University of Texas MD Anderson Lung Cancer Moon Shots Program. We would like to thank the members of our labs and Dr. James Allison and Dr. Padmanee Sharma for critical reading of the manuscript.
Footnotes
Author contributions
L.C., D.L.G., J.V.H., J.M.K., and F.X.-F.Q designed research; L.C., S.G., M.A.C., Y.-H.A., X.Z., X.Y., D.D., W.L., J.R., M.P., C.U., D.P., M.M.-V., B.E., D.P., and S.W. performed research; L.C., D.L.G., L.A.B., L.D., J.W., S.A., G.R., M.S., J.W.W., L.C., I.I.W., S.E.U., J.V.H., J.M.K., and F.X.-F.Q contributed new reagents/analytic tools; L.C., D.L.G., S.G., M.A.C., X.Z., L.D., J.W., M.M.-V., and G.R. analyzed data; L.C., D.L.G., S.E.U., J.V.H., J.M.K., and F.X.-F.Q. wrote the manuscript. All authors reviewed and edited the manuscript.
Supplementary Information accompanies this paper.
Competing financial interests: The authors declare no competing financial interests.
How to cite this article: Chen L, et al. Tumor metastasis is regulated via microRNA-200/ZEB1 axis control of PD-L1 expression on tumor cells and intratumoral immunosuppression.
References
- 1.Herbst RS, Heymach JV, Lippman SM. Lung cancer. N Engl J Med. 2008;359:1367–1380. doi: 10.1056/NEJMra0802714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gibbons DL, et al. Contextual extracellular cues promote tumor cell EMT and metastasis by regulating miR-200 family expression. Genes Dev. 2009;23:2140–2151. doi: 10.1101/gad.1820209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gibbons DL, et al. Expression signatures of metastatic capacity in a genetic mouse model of lung adenocarcinoma. PLoS One. 2009;4:e5401. doi: 10.1371/journal.pone.0005401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Patnaik SK, Kannisto E, Knudsen S, Yendamuri S. Evaluation of microRNA expression profiles that may predict recurrence of localized stage I non-small cell lung cancer after surgical resection. Cancer Res. 2010;70:36–45. doi: 10.1158/0008-5472.CAN-09-3153. [DOI] [PubMed] [Google Scholar]
- 5.Lamouille S, Subramanyam D, Blelloch R, Derynck R. Regulation of epithelialmesenchymal and mesenchymal-epithelial transitions by microRNAs. Curr Opin Cell Biol. 2013;25:200–207. doi: 10.1016/j.ceb.2013.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bracken CP, et al. A double-negative feedback loop between ZEB1-SIP1 and the microRNA-200 family regulates epithelial-mesenchymal transition. Cancer Res. 2008;68:7846–7854. doi: 10.1158/0008-5472.CAN-08-1942. [DOI] [PubMed] [Google Scholar]
- 7.Gregory PA, et al. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol. 2008;10:593–601. doi: 10.1038/ncb1722. [DOI] [PubMed] [Google Scholar]
- 8.Park SM, Gaur AB, Lengyel E, Peter ME. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008;22:894–907. doi: 10.1101/gad.1640608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Korpal M, Lee ES, Hu G, Kang Y. The miR-200 family inhibits epithelial-mesenchymal transition and cancer cell migration by direct targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2. J Biol Chem. 2008;283:14910–14914. doi: 10.1074/jbc.C800074200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Burk U, et al. A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep. 2008;9:582–589. doi: 10.1038/embor.2008.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. 2012;21:309–322. doi: 10.1016/j.ccr.2012.02.022. [DOI] [PubMed] [Google Scholar]
- 12.Yoshihara K, et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun. 2013;4:2612. doi: 10.1038/ncomms3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang Y, Sparwasser T, Figlin R, Kim HL. Foxp3+ T cells inhibit antitumor immune memory modulated by mTOR inhibition. Cancer Res. 2014 doi: 10.1158/0008-5472.CAN-13-2928. [DOI] [PubMed] [Google Scholar]
- 14.Stromnes IM, et al. Targeted depletion of an MDSC subset unmasks pancreatic ductal adenocarcinoma to adaptive immunity. Gut. 2014 doi: 10.1136/gutjnl-2013-306271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Restifo NP, Dudley ME, Rosenberg SA. Adoptive immunotherapy for cancer: harnessing the T cell response. Nat Rev Immunol. 2012;12:269–281. doi: 10.1038/nri3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wherry EJ. T cell exhaustion. Nat Immunol. 2011;12:492–499. doi: 10.1038/ni.2035. [DOI] [PubMed] [Google Scholar]
- 17.Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol. 2013;13:227–242. doi: 10.1038/nri3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Youngblood B, Wherry EJ, Ahmed R. Acquired transcriptional programming in functional and exhausted virus-specific CD8 T cells. Curr Opin HIV AIDS. 2012;7:50–57. doi: 10.1097/COH.0b013e32834ddcf2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brahmer JR, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366:2455–2465. doi: 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hamid O, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med. 2013;369:134–144. doi: 10.1056/NEJMoa1305133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Topalian SL, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dong H, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002;8:793–800. doi: 10.1038/nm730. [DOI] [PubMed] [Google Scholar]
- 23.Taube JM, et al. Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci Transl Med. 2012;4:127ra137. doi: 10.1126/scitranslmed.3003689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Freeman GJ, Wherry EJ, Ahmed R, Sharpe AH. Reinvigorating exhausted HIV-specific T cells via PD-1-PD-1 ligand blockade. J Exp Med. 2006;203:2223–2227. doi: 10.1084/jem.20061800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thompson RH, et al. Tumor B7-H1 is associated with poor prognosis in renal cell carcinoma patients with long-term follow-up. Cancer Res. 2006;66:3381–3385. doi: 10.1158/0008-5472.CAN-05-4303. [DOI] [PubMed] [Google Scholar]
- 26.Byers LA, et al. An Epithelial-Mesenchymal Transition Gene Signature Predicts Resistance to EGFR and PI3K Inhibitors and Identifies Axl as a Therapeutic Target for Overcoming EGFR Inhibitor Resistance. Clin Cancer Res. 2013;19:279–290. doi: 10.1158/1078-0432.CCR-12-1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.The Cancer Genome Atlas Network Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014 doi: 10.1038/nature13385. DOI: 10.1038/nature13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ahn YH, et al. ZEB1 drives prometastatic actin cytoskeletal remodeling by downregulating miR-34a expression. J Clin Invest. 2012;122:3170–3183. doi: 10.1172/JCI63608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kaplan DH, et al. Demonstration of an interferon gamma-dependent tumor surveillance system in immunocompetent mice. Proc Natl Acad Sci U S A. 1998;95:7556–7561. doi: 10.1073/pnas.95.13.7556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dighe AS, Richards E, Old LJ, Schreiber RD. Enhanced in vivo growth and resistance to rejection of tumor cells expressing dominant negative IFN gamma receptors. Immunity. 1994;1:447–456. doi: 10.1016/1074-7613(94)90087-6. [DOI] [PubMed] [Google Scholar]
- 31.Schoenborn JR, Wilson CB. Regulation of interferon-gamma during innate and adaptive immune responses. Adv Immunol. 2007;96:41–101. doi: 10.1016/S0065-2776(07)96002-2. [DOI] [PubMed] [Google Scholar]
- 32.Yang Y, et al. The Notch ligand Jagged2 promotes lung adenocarcinoma metastasis through a miR-200-dependent pathway in mice. J Clin Invest. 2011;121:1373–1385. doi: 10.1172/JCI42579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tsai JH, Yang J. Epithelial-mesenchymal plasticity in carcinoma metastasis. Genes Dev. 2013;27:2192–2206. doi: 10.1101/gad.225334.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ali S, et al. Gemcitabine sensitivity can be induced in pancreatic cancer cells through modulation of miR-200 and miR-21 expression by curcumin or its analogue CDF. Cancer Res. 2010;70:3606–3617. doi: 10.1158/0008-5472.CAN-09-4598. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 35.Iliopoulos D, et al. Loss of miR-200 inhibition of Suz12 leads to polycomb-mediated repression required for the formation and maintenance of cancer stem cells. Mol Cell. 2010;39:761–772. doi: 10.1016/j.molcel.2010.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kong D, et al. Epithelial to mesenchymal transition is mechanistically linked with stem cell signatures in prostate cancer cells. PLoS One. 2010;5:e12445. doi: 10.1371/journal.pone.0012445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Leskela S, et al. The miR-200 family controls beta-tubulin III expression and is associated with paclitaxel-based treatment response and progression-free survival in ovarian cancer patients. Endocr Relat Cancer. 2011;18:85–95. doi: 10.1677/ERC-10-0148. [DOI] [PubMed] [Google Scholar]
- 38.Schickel R, Park SM, Murmann AE, Peter ME. miR-200c regulates induction of apoptosis through CD95 by targeting FAP-1. Mol Cell. 2010;38:908–915. doi: 10.1016/j.molcel.2010.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wellner U, et al. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat Cell Biol. 2009;11:1487–1495. doi: 10.1038/ncb1998. [DOI] [PubMed] [Google Scholar]
- 40.Roybal JD, et al. miR-200 Inhibits lung adenocarcinoma cell invasion and metastasis by targeting Flt1/VEGFR1. Mol Cancer Res. 2011;9:25–35. doi: 10.1158/1541-7786.MCR-10-0497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pecot CV, et al. Tumour angiogenesis regulation by the miR-200 family. Nat Commun. 2013;4:2427. doi: 10.1038/ncomms3427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Valastyan S, et al. A pleiotropically acting microRNA, miR-31, inhibits breast cancer metastasis. Cell. 2009;137:1032–1046. doi: 10.1016/j.cell.2009.03.047. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 43.Fridman WH, Pages F, Sautes-Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer. 2012;12:298–306. doi: 10.1038/nrc3245. [DOI] [PubMed] [Google Scholar]
- 44.Mu CY, Huang JA, Chen Y, Chen C, Zhang XG. High expression of PD-L1 in lung cancer may contribute to poor prognosis and tumor cells immune escape through suppressing tumor infiltrating dendritic cells maturation. Med Oncol. 2011;28:682–688. doi: 10.1007/s12032-010-9515-2. [DOI] [PubMed] [Google Scholar]
- 45.Zhang Y, Huang S, Gong D, Qin Y, Shen Q. Programmed death-1 upregulation is correlated with dysfunction of tumor-infiltrating CD8+ T lymphocytes in human non-small cell lung cancer. Cell Mol Immunol. 2010;7:389–395. doi: 10.1038/cmi.2010.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhuang X, et al. A high number of CD8+ T cells infiltrated in NSCLC tissues is associated with a favorable prognosis. Appl Immunohistochem Mol Morphol. 2010;18:24–28. doi: 10.1097/PAI.0b013e3181b6a741. [DOI] [PubMed] [Google Scholar]
- 47.Hodi FS, et al. Immunologic and clinical effects of antibody blockade of cytotoxic T lymphocyte-associated antigen 4 in previously vaccinated cancer patients. Proc Natl Acad Sci U S A. 2008;105:3005–3010. doi: 10.1073/pnas.0712237105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Akbay EA, et al. Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer Discov. 2013 doi: 10.1158/2159-8290.CD-13-0310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brahmer JR. Harnessing the immune system for the treatment of non-small-cell lung cancer. J Clin Oncol. 2013;31:1021–1028. doi: 10.1200/JCO.2012.45.8703. [DOI] [PubMed] [Google Scholar]
- 50.Ganesan AP, et al. Tumor-infiltrating regulatory T cells inhibit endogenous cytotoxic T cell responses to lung adenocarcinoma. J Immunol. 2013;191:2009–2017. doi: 10.4049/jimmunol.1301317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang Y, et al. ZEB1 sensitizes lung adenocarcinoma to metastasis suppression by PI3K antagonism. J Clin Invest. 2014;124:2696–2708. doi: 10.1172/JCI72171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zheng S, El-Naggar AK, Kim ES, Kurie JM, Lozano G. A genetic mouse model for metastatic lung cancer with gender differences in survival. Oncogene. 2007;26:6896–6904. doi: 10.1038/sj.onc.1210493. [DOI] [PubMed] [Google Scholar]
- 53.Onn A, et al. Development of an orthotopic model to study the biology and therapy of primary human lung cancer in nude mice. Clin Cancer Res. 2003;9:5532–5539. [PubMed] [Google Scholar]
- 54.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.