

Abstract
Overexpression of peroxisome proliferator activator receptor γ (PPARγ) has been implicated in many types of cancer including cervical cancer. Radiation therapy remains the main nonsurgical modality for the treatment of cervical cancer. The present study reports the impact of pharmacological inhibition of PPARγ in enhancing the radiosensitization of cervical cancer cells in vitro. Three cervical cancer cell lines (HeLa, SiHa, and Me180) were treated with a PPARγ inhibitor, T0070907, and/or radiation. The changes in protein, cell cycle, DNA content, apoptosis, and cell survival were analyzed. The PPARγ is differentially expressed in cervical cancer cells with maximum expression in ME180 cells. T0070907 has significantly decreased the tubulin levels in a time-dependent manner in ME180 cells. The decrease in the tubulin levels after T0070907 in ME180 and SiHa cells was associated with significant increase in the cells at the G2/M phase. The changes in the tubulin and G2/M phase were not evident in HeLa cells. T0070907 reduced the protein levels of PPARγ; however, PPARγ silencing had no effect on the α-tubulin level in ME180 cells suggesting the PPARγ-dependent and -independent actions of T0070907. To ascertain the impact of synergistic effect of T0070907 and radiation, HeLa and ME180 cells were pretreated with T0070907 and subjected to radiation (4 Gy). Annexin V-fluorescein isothiocyanate analysis revealed increased apoptosis in cells treated with radiation and T0070907 when compared to control and individual treatment. In addition, T0070907 pretreatment enhanced radiation-induced tetraploidization reinforcing the additive effect of T0070907. Confocal analysis of tubulin confirmed the onset of mitotic catastrophe in cells treated with T0070907 and radiation. These results strongly suggest the radiosensitizing effects of T0070907 through G2/M arrest and mitotic catastrophe.
Keywords: PPARγ, T0070907, cervical cancer, cell cycle, radiation, mitotic catastrophe
Introduction
Despite improvements in screening, cervical cancer remains the third most common cancer and fourth leading cause of cancer death in the world.1 Epidemiological and laboratory-based studies have identified infection with human papillomavirus (HPV) as the major cause in cervical cancer development.2 Radiation therapy remains the main nonsurgical modality for the treatment of cervical cancer. Several observations suggest that inhibition of peroxisome proliferator activator receptor γ (PPARγ) function may be beneficial in treating neoplasms.3,4 Although PPARγ is overexpressed in many cancer cell types including cervical cancer,5 loss of function mutations are rare,6 which suggests that PPARγ is a tumor survival factor. Evidence that PPARγ function can contribute to carcinogenesis or cancer cell survival includes reports of a murine colon cancer model in which PPARγ activation leads to increased tumor formation.7,8 A significant correlation between high levels of PPARγ expression in pancreatic cancer cells and shorter overall survival time has been reported.9 In addition, PPARγ inhibitors decrease migration and invasion of HT-29 cells and reduces both metastasis number and size in a murine xenograft model.10 Collectively, these studies highlight the significance of endogenous inhibition of PPARγ actions in the treatment of several cancer cell types. Studies reporting the role of PPARγ inhibition in augmenting the actions of radiation in cancer cell are very limited and needs to be investigated.
T0070907 (PubChem SID: 53790303) was identified as a potent and selective antagonist which shows a more than 800-fold preference for PPARγ over PPARα and PPARδ.11 The suppression of PPARγ activity by T0070907 has been demonstrated in cell-based reporter gene and functional assays.10–12 Moreover, antitumor activity was observed when cancer cells were treated with T0070907.13–15 It has also been shown that inhibition of PPARγ by T0070907 suppressed pancreatic cell motility in vitro and invasion in vivo.12 Furthermore, T0070907 reduced tubulin protein levels resulting in cell cycle arrest, apoptosis and reduced metastasis of colorectal carcinoma cells.10 Cells in M and G2 phases are most radiosensitive16 than other phases and T0070907 treatment has been shown to induce G2/M. Hence, we have speculated that T0070907 can be exploited as a radiosensitizer. To date, there is no report available on the radiosensitizing role of T0070907.
In this study, the mechanisms underlying the synergistic effect of radiation and PPARγ antagonist treatment in cervical cancer cells were evaluated. The present study examined the changes in the tubulin, DNA content, and apoptosis to delineate the beneficial effects of radiation and T0070907. The data generated in the present study suggest enhanced apoptosis, G2/M arrest, and mitotic catastrophe in ME180 cervical cancer cells by T0070907 upon irradiation. The data also highlight the significance of pharmacological inhibition of endogenous activity of PPARγ by T0070907. T0070907 may provide a novel and effective therapeutic approach for enhancing the radio sensitization therapy in cervical cancer.
Materials and Methods
Cell Culture and Treatments
HeLa (HPV 18), SiHa (HPV 16), and ME-180 (HPV 68) cells with wild-type p53 were obtained from Korea cell line bank, Seoul, Korea, and were cultured in Dulbecco Modified Eagle Medium or Roswell Park Memorial Institute medium medium (WelGENE; Daegu, Korea) with 100 units/mL penicillin, 100 μg/mL streptomycin, and 10% fetal bovine serum (FBS) and incubated at 37°C in a humidified atmosphere of 95% air and 5% CO2. T0070907 (Cayman, Michigan) was dissolved in dimethyl sulfoxide (DMSO) vehicle. Cells were treated with 50 µmol/L at indicated time points with 0.1% FBS media and then cells were irradiated with a 6 MV linear accelerator (Siemens Mevatron M6700; Concord, CA), at a dose rate of 3 Gy/min and incubated at 37°C at indicated time points.
Cell Cycle Analysis Using Bromodeoxyuridine Fluorescein Isothiocyanate Assay
For cell cycle analysis, cells were pretreated with or without T0070907 at indicated time points and were pulse labeled with 10 µmol/L bromodeoxyuridine (BrdU) for 30 minutes and harvested using trypsinization. Cells were then fixed and stained using fluorescein isothiocyanate (FITC)-conjugated anti-BrdU and 7-aminoactinomycin D (7-AAD) following manufacturer’s instruction (BD BrdU Flow Kits #559619, BD Biosciences, CA). Finally, thecells were analyzed by BD FACS Calibur cytometry (BD Biosciences, California).
Western Blot Analysis
Cells pretreated with or without T0070907 were left untreated or irradiated and incubated at indicated time points at 37°C, 5% CO2. The cells were then washed with ice-cold phosphate-buffered saline (PBS) and lysed using cell lysis buffer (Cell Signaling: Beverly, MA), containing protease and phosphatase inhibitors (Roche, Mannheim, Germany). Cellular debris were cleared by centrifugation at 12000 rpm for 15 minutes at 4°C. The protein concentration in each sample was determined using BioRad reagent (BioRad; Hercules, CA). An equal amount of protein was subjected to sodium dodecyl sulfate-polyacyrlamide gel electrophoresis and transferred onto a polyvinylidene difluoride membrane (Millipore; Bedford, MA). The membrane was then blocked using blocking buffer and washed in 0.2% tris-buffered saline-containing Tween 20 (TBST) for 5 minutes. The membrane was incubated overnight with appropriate primary antibodies. Subsequently, the membranes were washed in 0.2% TBST for 10 minutes followed by incubation with appropriate secondary antibody for 50 minutes. The proteins were visualized using enhanced chemiluminescence (West-Zol plus, iNtRON Biotechnology; Seongnam, Korea).
Clonogenic Assay
Cells in growing phase were treated with either vehicle (DMSO) or T0070907 50 µmol/L at indicated time points and then irradiated. Varying number (100-2000) of cells for optimal colony counting was seeded in 60-mm dishes according to the presence or absence of T0070907 and radiation. After 14 days, the cells were stained with 0.1% crystal violet solution and colonies which are composed of at least 50 cells were counted.
Assessment of Apoptosis
Apoptosis was measured by flow cytometry using FITC Annexin V Apoptosis Detection Kit (BD Pharmingen). Cells were collected at indicated time points following radiation. Then, the cells were resuspended in binding buffer and 5 μL of FITC Annexin V and propidium iodide were added. The cells were then incubated for 15 minutes at room temperature (25°C) in the dark and then 400 μL of binding buffer was added to each sample and analyzed using flow cytometry (BD Biosciences) within 1 hour.
Confocal Microscopy
ME-180 cells were grown on coverslips placed in 35-mm dishes and pretreated with or without T0070907 were irradiated (4 Gy) as described earlier and incubated at indicated time points. After incubation time, cells were fixed with 4% paraformaldehyde in PBS and then permeabilized with 0.1% Triton X-100 and blocked with 10% FBS for 30 minutes followed by incubation with anti-α-tubulin antibody (1:100, Santa Cruz, CA) in 2% FBS/PBS overnight at 4°C. Cells were further incubated with Alexa-488 conjugated secondary antibody for 1 hour. Nuclei were stained with 4',6-diamidino-2-phenylindole (DAPI; 1 μg/mL, Sigma, St Louis, MO). Coverslips were mounted onto slides and images were captured and analyzed using confocal microscope (Leica DM-IRB; Mannheim, Germany).
Statistical Analysis
The significance of the observation was estimated by Student’s t test, using data from at least 3 independent replicates. The observation was deemed significant if the P value of accepting null hypothesis is < .05 or .01 (indicated by “*” and “**” in the figures).
Results
Peroxisome Proliferator Activator Receptor γ Is Differentially Expressed in Cervical Cancer Cells
Peroxisome proliferator activator receptor γ is overexpressed in many cancer cell types including cervical cancer5 suggesting that PPARγ is a tumor survival factor. Therefore, an attempt has been made to evaluate the expression of PPARγ in 3 different cervical cancer cells viz HeLa, ME180, and SiHa (Figure 1A). The expression of PPARγ was maximal in ME180 cells followed by SiHa cells. The expression of PPARγ is feeble in HeLa cells. These observations suggest that PPARγ may function as a survival factor in ME180 cells.
Figure 1.

A, Western blot experiment showing the differential expression of PPARγ in 3 cervical cancer cell lines HeLa, ME180, and SiHa. Actin is used as loading control. B, Western blot experiment showing the protein levels of α- and β-tubulin after 12, 24, and 48 hours treatment with T0070907 in 3 cervical cancer cell lines ME180, HeLa, and SiHa. Actin is used as loading control. C, Immunocytochemistry staining of α-tubulin and actin in the 3 cell lines after 24 hours. PPARγ indicates peroxisome proliferator activator receptor γ. (The color version of this figure is available in the online version at http://rs.sagepub.com/.)
T0070907 Reduces Tubulin Protein Level in ME180 Cells
Functioning of microtubule network requires the maintenance of critical threshold of tubulin proteins. T0070907 treatment has reduced the levels of α- and β-tubulin protein in a time-dependent manner in ME180 and SiHa cells; however, such a reduction was not observed in HeLa cells suggesting the cell type-specific effect of T0070907. The Western blot data on the reduction in tubulin proteins in ME180 and SiHa cells by T0070907 were corroborated with confocal microscopy analysis showing reduced α-tubulin levels. The changes in the levels of tubulin were not evidenced in HeLa cells (Figure 1B and C).
T0070907 Alters Cell Cycle Distribution
Cell cycle analysis was performed using flow cytometry to examine whether the cell cycle distribution profiles and DNA content were affected by T0070907 as a manifestation of its antiproliferative action. As illustrated in Figure 2, T0070907 treatment induced a significant G2/M phase arrest in ME180 and SiHa cells in a time-dependent manner (12, 24, and 48 hours). There was no difference observed at G2/M phase after T0070907 treatment in HeLa cells after 12, 24, and 48 hours. T0070907 treatment decreased the synthesis of DNA in SiHa and ME180 cervical cancer cells. (Figure 2).
Figure 2.

Flow cytometric analysis using BrdU showing the alterations in the cell cycle distribution after 12, 24, and 48 hours treatment with T0070907 (50 μmol/L) in 3 cervical cancer cell lines ME180, HeLa, and SiHa. BrdU indicates bromodeoxyuridine.
T0070907 Prevents the Radiation-Induced Alterations in the Cell Cycle Regulatory Proteins
Since T0070907 has promoted the apoptosis and induced cell cycle arrest in control and irradiated cancer cells, we wanted to delineate the role of T0070907 in the protein levels of cell division cycle (Cdc) 2, phospho-Cdc (p-Cdc) 2, Cdc25c, pCdc25c, and cyclin B1 (Figure 3A and B). Radiation treatment has resulted in a time-dependent increase in the expression of this protein, whereas T0070907 pretreatment has prevented the radiation-induced increase in these proteins in ME180 and HeLa cells. The induction of cyclin B1 and regulatory proteins suggests one of the mechanisms by which cells acquire radioresistance and the preventive actions of T0070907 suggest its radiosensitizing ability.
Figure 3.
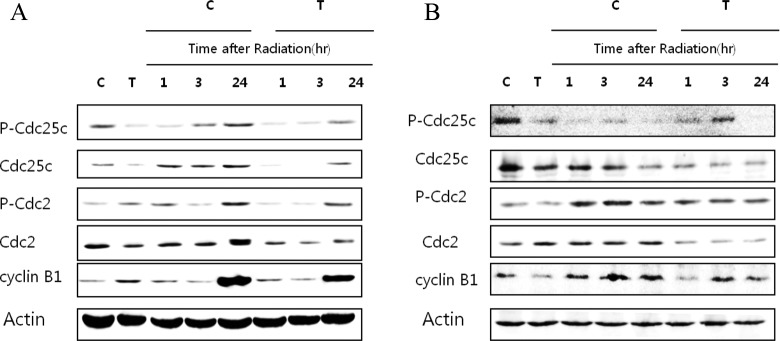
Western blot analysis showing the impact of radiation in a time-dependent manner in (A) ME180 and (B) HeLa cells treated with or without T0070907 (50 μmol/L) at indicated time points on the protein levels of Cdc2, pCdc2, Cdc25c, pCdc25c, and Cyclin B1. Cdc indicates cell division cycle; pCdc, phospho-cell division cycle.
T0070907 Reduces Colony Formation in Irradiated Cells
The ability of cells to proliferate and form colonies has been recognized generally as the single most reliable in vitro marker of premalignant or malignant transformation.17 Hence, an attempt has been made to delineate the synergistic effect of T0070907 (50 μmol/L, 24 hours) and radiation on colony formation. Significant reductions in the number of colonies were evidenced in cells treated with T0070907 after different dose of radiation in ME180 cells when compared with radiation alone. Such reduction in the ability to form colony after T0070907 was not evidenced in HeLa cells after different doses of radiation (Figure 4A and B).
Figure 4.
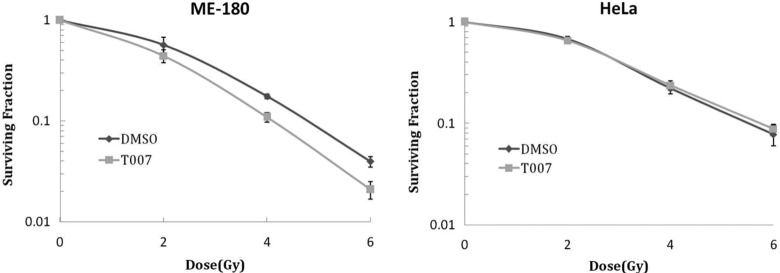
Clonogenic cell survival showing the impact of T0070907 (50 μmol/L) in reducing the ability of (A) ME180 and (B) HeLa cells to form colonies. * denotes statistical significance when compared with cells treated with similar dose of radiation alone without T0070907.
T0070907 Accelerates the Radiation-Induced Apoptosis
Radiation has been shown to induce apoptosis and decrease the survival of cancer cells. Hence, the impact of radiation on the induction of apoptosis was evaluated in ME180 (Figure 5A and B) and HeLa cells (Figure 5C and D) treated with or without T0070907. Radiation failed to significantly increase the apoptosis in both cell lines tested; however, T0070907 promoted apoptosis in ME180 cells and the apoptosis was maximum in both HeLa and ME180 cells treated with T0070907 and radiation. T0070907 has promoted the induction of protein levels of p53 by radiation suggesting the radiosensitizing effect of T0070907 in ME180 cervical cancer cells is p53 dependent (Figure 5E).
Figure 5.

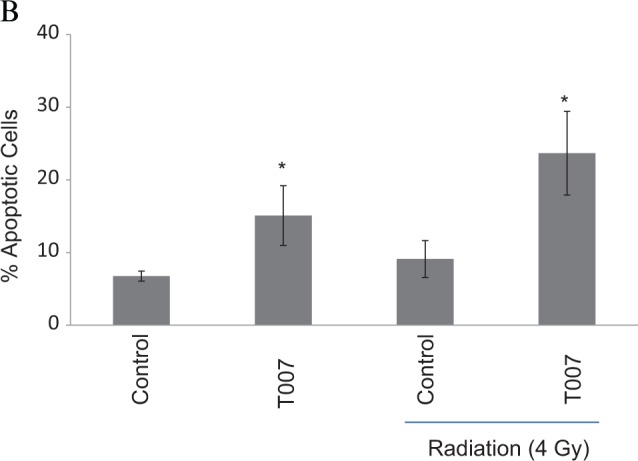


Annexin V binding assay showing the synergistic effect of radiation in cells treated with or without T0070907 (50 μmol/L) in (A and B) ME180 cells and (C and D) HeLa cells. * denotes statistical significance when compared with control at P < .05 level. E, Western blot data showing the effect of T0070907 and/or radiation on the protein levels of p53 in ME180 cells. Actin is used as loading control.
T0070907 and Radiation-Induced Mitotic Catastrophe
Mitotic catastrophe is another major modality of cell death in irradiated cells. Since T0070907 treatment has reduced the levels of tubulins significantly in ME180 cells and mitotic catastrophe is reported in cells with disrupted microtubules, we wanted to address the impact of T0070907 and radiation on the onset of mitotic catastrophe. To determine mitotic catastrophe, cell cycle analysis was measured following 4 Gy radiation in cells treated with or without T0070907. Increase in tetraploidization is evidenced after 24 and 48 hours in ME180 cells after radiation. Cells with 8 N DNA content were evidenced in ME180 cells treated with radiation and T0070907. There was no significant polyploidy cells increase in HeLa cells (Figure 6A and B).
Figure 6.

DNA content analysis displaying the abnormalities and tetraploidization in irradiated (A) ME180 cells and (B) HeLa cells treated with or without T0070907 (50 μmol/L) after 24 and 48 hours. C, Immunofluorescence staining with α-tubulin showing the impact of T0070907 (50 μmol/L) in control and irradiated after 48 hours in ME180 cells and the bar diagram (D) represents the average number of mitotic catastrophe after 24 and 48 hours irradiation in control and T0070907-treated ME180 cells. * and ** denote statistical significance when compared with control at P < .05 and P < .01 level, respectively. (The color version of this figure is available in the online version at http://rs.sagepub.com/.)
To further validate the findings of flow cytometric analysis, immunofluorescence was performed in ME180 cells treated with or without T0070907 alone or in combination with radiation. After treatment, cells were stained with anti-α-tubulin and DAPI, and cells with multinucleation or micronucleation were counted as mitotic catastrophe cells. T0070907 and radiation treatment alone induced a little increase in cells with mitotic catastrophe at 24 and 48 hours postirradiation (Figure 6C and D). Interestingly, the combined treatment significantly elevated mitotic catastrophe cells at both time points.
Discussion
DNA-damaging agents in the form of ionizing radiation and chemotherapeutic drugs are the main components of most current cancer treatment regimens.18 Antimitotic drugs that disrupt the function of mitotic spindle by altering polymerization dynamics of microtubules have been extensively used for cancer therapy. The present study evidenced a time-dependent significant reduction in the protein levels of α- and β-tubulin, suggesting the microtubule targeting actions of T0070907 as tubulins are the functional units of microtubules. The decreases in the tubulins observed in the present study are in agreement with earlier studies wherein T0070907 has accelerated the proteasomal degradation of tubulins. The decrease in the tubulins after T0070907 treatment was associated with cell cycle arrest at G2/M phase. Interestingly, the decrease in the level of tubulins and cell cycle arrest was evident only in ME180 and SiHa cells not in the other cell types studied. Therefore, it can be stated the actions of T0070907 are cell type specific. However, reduction in the synthetic phase is evident in both SiHa and ME180 cells. Differential expression pattern of PPARγ might be one of the potential mechanisms responsible for the cell type-specific effect of T0070907 on G2/M transition. T0070907 treatment has decreased the protein levels of PPARγ in a time-dependent manner suggesting the actions of T0070907 in ME180 involve PPARγ. However, PPARγ silencing did not result in the tubulin reduction in ME180 cells suggesting the possible involvement of other factors in the radiosensitizing effect of T0070907 in ME180 cells (Supplementary figure).
There is no appreciable cell death observed in ME180 cells following irradiation after 24 hours. Radiation treatment has upregulated the levels of total and phosphorylated Cdc25c and Cdc2 proteins in a time-dependent manner along with elevated cyclin B1 levels. Cyclin B1 forms with Cdc2 (cyclin-dependent kinase [Cdk] 1), a complex called mitosis promoting factor that is crucial for G2/M transition.19 Insufficient levels of cyclin B-Cdk1 complexes are related to cell cycle arrest at G2.20 Cyclin B1 is also considered as a potential prognostic marker in different cancers.21,22 T0070907 pretreatment has reduced the radiation-induced increase in the levels of cyclin B1 and the levels of total and phosphorylated Cdc25c and Cdc2. This can be attributed for augmented apoptosis and cell cycle arrest induced in cells treated with T0070907 and radiation.
An increase in the tetraploidization and multinucleation was noted in irradiated cells pretreated with T0070907 when compared with control and irradiation-treated cells suggesting the induction of chromosomal aberration. Often, irradiated cells adapt to the mitotic checkpoint and consequently do not die by apoptosis in metaphase. These cells exit the arrest but fail cytokinesis and enter the next G1 phase with a tetraploid DNA content.23,24 As a result, giant polyploid cells will be generated with aberrant nuclear morphology,25–27 multiple nuclei,25,28 and/or several micronuclei.29 These cells can survive for days but in the end, they die by delayed necrosis, delayed apoptosis, or induced senescence.30 Apoptosis in G1 occurs shortly after tetraploidization and is largely dependent on p53 activation of the Bax-dependent mitochondrial pathway.30 These cells can continue through several cycles of cell division, acquiring an increasing amount of chromosomal aberration, finally causing death via delayed apoptosis or delayed necrosis. Collectively, these observations point the significance of tetraploidization after irradiation in the execution of cell death.
Pretreatment with T0070907 has promoted radiation-induced apoptosis and mitotic catastrophe in ME180 cells suggesting the beneficial effects of synergistic treatment with T0070907 and radiation. Apoptosis is characterized morphologically by cytoplasmic shrinkage, chromatin condensation, and nuclear fragmentation with chromatinolysis,31 and biochemically by caspase activation and permeabilization of the outer mitochondrial membrane.32–34 Mitotic catastrophe is a special case of apoptosis and occurs during mitosis with a combination of deficient cell cycle checkpoints and cellular damage.35 DNA damage activates checkpoints to delay cell cycle progression. T0070907 induced increase in the protein levels of p53 after irradiation suggesting that the apoptotic effect of T0070907 is p53 dependent. p53 regulates G1/S transition (G1-checkpoint), while checkpoint kinase 1 (CHK1) prevents the entry of DNA-damaged cells into M-phase (G2-check point). In the case of chemoresistance, DNA damage induced by chemotherapeutic agents fails to arrest the cancer cells in the G1 phase and to promote apoptosis owing mainly to the deficient p53 signaling. However, DNA damage can activate the CHK1 pathway, induce S- and G2-checkpoints arrest,36,37 and facilitate DNA repair before entry into mitosis (M phase). It is therefore conceivable that agents capable of inducing conventional apoptosis (through DNA damage and p53 activation) and promoting mitotic catastrophe (via CHK1 inactivation and abrogation of G/M arrest) may offer potentially new therapeutic advantages. Previous investigations have shown that both PPARγ agonists and antagonists act as effective anticancer agents.38,39 The role of PPARγ agonists as anticancer agents has been well characterized in the treatment of colon, gastric, and lung cancer,40,41 whereas, PPARγ antagonists have been shown to induce potent antiproliferative effects in many hematopoietic and epithelial cancer cell lines.38,41 Results in the present study confirm and extend these previous findings. Dose–response studies showed that treatment with either PPARγ antagonist significantly inhibited the growth of human MCF-7 and MDA-MB-231 breast cancer cells in culture. Antiproliferative effects of PPARγ antagonist were found to be more pronounced in MDAMB-231 than that in MCF-7 breast cancer cells, and these results are similar to those reported previously.38,42 Also,in the present study, , the antiproliferative actions of T0070907 are more evident in ME180 cells than in other cell types studied. These findings suggest that antiproliferative action of PPARγ is cell type dependent and offers support to the differential effects of T0070907 in cervical cancer cells. More importantly, T0070907 has radiosensitized the ME180 and greatly reduced its ability to form colonies. Although increased apoptosis was noted in HeLa cells treated with radiation and T0070907, no significant reduction is observed in the ability of HeLa cells treated with radiation and T0070907 when compared with control cells to form colony suggesting that the induction of apoptosis and suppression of clonogenicity are independent processes. These observations are in line with an earlier study wherein cells that readily underwent apoptosis did not necessarily show a correlated loss of clonogenicity; for example, Me45 (human melanoma) cells showed the highest sensitivity to irradiation in clonogenic assays but much lower levels of apoptotic cells than R1 (rat rhabdomyosarcoma), or HL-60 cells (promyelocytic leukemia).43
Conclusion
The data generated in the present study report the alterations in the levels of tubulin, enhanced cell cycle arrest, tetraploidization, multinucleation, apoptosis, and reduced ability to form clones following T0070907 pretreatment and irradiation in cervical cancer ME180 cells. Taken together, these observations strongly support the induction of radiosensitization by T0070907 and offer new perspectives in the radiotherapy for cervical cancer. Emerging studies also point out the therapeutic efficacy of PPARγ agonist in the treatment of cancer and in this context the present study warrants extensive investigation before advocating PPARγ agonist therapy.
Acknowledgement
The authors appreciate the technical assistance of Ms. Kim, Hyun.
Footnotes
Authors’ Note: Zhengzhe An and Sridhar Muthusami contributed equally to the work.
Declaration of Conflicting Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship and/or publication of this article: This work was supported by the Korea Research Foundation (KRF) grant funded by the Korea government (MEST; No. NRF-2009-0075292).
Supplemental Material: The online supplemental figures are available at http://rs.sagepub.com/supplemental
References
- 1. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61(2):69–90 [DOI] [PubMed] [Google Scholar]
- 2. Woodman CB, Collins SI, Young LS. The natural history of cervical HPV infection: unresolved issues. Nat Rev Cancer. 2007;7(1):11–22 [DOI] [PubMed] [Google Scholar]
- 3. Martelli ML, Iuliano R, Le Pera I, et al. Inhibitory effects of peroxisome proliferator-activated receptor gamma on thyroid carcinoma cell growth. J Clin Endocrinol Metab. 2002;87(10):4728–4735 [DOI] [PubMed] [Google Scholar]
- 4. Panigrahy D, Shen LQ, Kieran MW, Kaipainen A. Therapeutic potential of thiazolidinediones as anticancer agents. Expert Opin Investig Drugs. 2003;12(12):1925–1937 [DOI] [PubMed] [Google Scholar]
- 5. Han S, Inoue H, Flowers LC, Sidell N. Control of COX-2 gene expression through peroxisome proliferator-activated receptor gamma in human cervical cancer cells. Clin Cancer Res. 2003;9(12):4627–4635 [PubMed] [Google Scholar]
- 6. Posch MG, Zang C, Mueller W, Lass U, von Deimling A, Elstner E. Somatic mutations in peroxisome proliferator-activated receptor-gamma are rare events in human cancer cells. Med Sci Monit. 2004;10(8):BR250–BR254 [PubMed] [Google Scholar]
- 7. Lefebvre AM, Chen I, Desreumaux P, et al. Activation of the peroxisome proliferator-activated receptor gamma promotes the development of colon tumors in C57BL/6J-APCMin/+ mice. Nat Med. 1998;4(9):1053–1057 [DOI] [PubMed] [Google Scholar]
- 8. Saez E, Tontonoz P, Nelson MC, et al. Activators of the nuclear receptor PPARgamma enhance colon polypformation. Nat Med. 1998;4(9):1058–1061 [DOI] [PubMed] [Google Scholar]
- 9. Kristiansen G, Jacob J, Buckendahl AC, et al. Peroxisome proliferator-activated receptor gamma is highly expressed in pancreatic cancer and is associated with shorter overall survival times. Clin Cancer Res. 2006;12(21):6444–6451 [DOI] [PubMed] [Google Scholar]
- 10. Schaefer KL, Takahashi H, Morales VM, et al. PPARgamma inhibitors reduce tubulin protein levels by a PPARgamma, PPARdelta and proteasome-independent mechanism, resulting in cell cycle arrest, apoptosis and reduced metastasis of colorectal carcinoma cells. Int J Cancer. 2007;120(3):702–713 [DOI] [PubMed] [Google Scholar]
- 11. Lee G, Elwood F, McNally J, et al. T0070907, a selective ligand for peroxisome proliferator-activated receptor gamma, functions as an antagonist of biochemical and cellular activities. J Biol Chem. 2002;277(22):19649–19657 [DOI] [PubMed] [Google Scholar]
- 12. Nakajima A, Tomimoto A, Fujita K, et al. Inhibition of peroxisome proliferator-activated receptor gamma activity suppresses pancreatic cancer cell motility. Cancer Sci. 2008;99(10):1892–900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Takahashi H, Fujita K, Fujisawa T, et al. Inhibition of peroxisome proliferator-activated receptor gamma activity in esophageal carcinoma cells results in a drastic decrease of invasive properties. Cancer Sci. 2006;97(9):854–860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Burton JD, Goldenberg DM, Blumenthal RD. Potential of peroxisome proliferator-activated receptor gamma antagonist compounds as therapeutic agents for a wide range of cancer types. PPAR Res. 2008;2008:494161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kim KR, Choi HN, Lee HJ, et al. A peroxisome proliferator-activated receptor gamma antagonist induces vimentin cleavage and inhibits invasion in high-grade hepatocellular carcinoma. Oncol Rep. 2007;18(4):825–832 [PubMed] [Google Scholar]
- 16. Hall EJ, Giaccia AJ. Radiosensitivity and cell age in the mitotic cycle. In: Lippincott Williams and Wilkins, eds. Radiobiology for the Radiologist. 7th ed. Philadelphia, PA: Lippincott Williams and Wilkins; 2011:54–66 [Google Scholar]
- 17. Tsao MS, Earp HS, Grisham JW. Gradation of carcinogen-induced capacity for anchorage-independent growth in cultured rat liver epithelial cells. Cancer Res. 1985;45(9):4428–4432 [PubMed] [Google Scholar]
- 18. Landsverk KS, Lyng H, Stokke T. The response of malignant B lymphocytes to ionizing radiation: cell cycle arrest, apoptosis and protection against the cytotoxic effects of the mitotic inhibitor nocodazole. Radiat Res. 2004;162(4):405–415 [DOI] [PubMed] [Google Scholar]
- 19. Pines J, Hunter T. Human cyclin A is adenovirus E1A-associated protein p60 and behaves differently from cyclin B. Nature. 1990;346(6286):760–763 [DOI] [PubMed] [Google Scholar]
- 20. Innocente SA, Abrahamson JL, Cogswell JP, Lee JM. p53 regulates a G2 checkpoint through cyclin B1. Proc Natl Acad Sci U S A. 1999;96(5):2147–2152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Niméus-Malmström E, Koliadi A, Ahlin C, et al. Cyclin B1 is a prognostic proliferation marker with a high reproducibility in a population-based lymph node negative breast cancer cohort. Int J Cancer. 2010;127(4):961–967 [DOI] [PubMed] [Google Scholar]
- 22. Kosacka M, Korzeniewska A, Jankowska R. The evaluation of prognostic value of cyclin B1 expression in patients with resected non-small-cell lung cancer stage I-IIIA--preliminary report [in Polish]. Pol Merkur Lekarski. 2010;28(164):117–121 [PubMed] [Google Scholar]
- 23. Weaver BA, Cleveland DW. Decoding the links between mitosis, cancer, and chemotherapy: The mitotic checkpoint, adaptation, and cell death. Cancer Cell. 2005;8(1):7–12 [DOI] [PubMed] [Google Scholar]
- 24. Yamada HY, Gorbsky GJ. Spindle checkpoint function and cellular sensitivity to antimitotic drugs. Mol Cancer Ther. 2006;5(12):2963–2969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Eriksson D, Joniani HM, Sheikholvaezin A, et al. Combined low dose radio- and radioimmunotherapy of experimental HeLa Hep 2 tumours. Eur J Nucl Med Mol Imaging. 2003;30(6):895–906 [DOI] [PubMed] [Google Scholar]
- 26. Eriksson D, Löfroth PO, Johansson L, Riklund KA, Stigbrand T. Cell cycle disturbances and mitotic catastrophes in HeLa Hep2 cells following 2.5 to 10 Gy of ionizing radiation. Clin Cancer Res. 2007;13(18 pt 2):5501s–5508s [DOI] [PubMed] [Google Scholar]
- 27. Castedo M, Kroemer G. Mitotic catastrophe: a special case of apoptosis [in French]. J Soc Biol. 2004;198(2):97–103 [PubMed] [Google Scholar]
- 28. Erenpreisa J, Kalejs M, Ianzini F, et al. Segregation of genomes in polyploid tumour cells following mitotic catastrophe. Cell Biol Int. 2005;29(12):1005–1011 [DOI] [PubMed] [Google Scholar]
- 29. Roninson IB, Broude EV, Chang BD. If not apoptosis, then what? Treatment-induced senescence and mitotic catastrophe in tumor cells. Drug Resist Updat. 2001;4(5):303–313 [DOI] [PubMed] [Google Scholar]
- 30. Rieder CL, Maiato H. Stuck in division or passing through: what happens when cells cannot satisfy the spindle assembly checkpoint. Dev Cell. 2004;7(5):637–651 [DOI] [PubMed] [Google Scholar]
- 31. Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26(4):239–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ferri KF, Kroemer G. Organelle-specific initiation of cell death pathways. Nat Cell Biol. 2001;3(11):E255–E263 [DOI] [PubMed] [Google Scholar]
- 33. Brenner C, Kroemer G. Apoptosis. Mitochondria–the death signal integrators. Science. 2000; 289(5482):1150–1151 [DOI] [PubMed] [Google Scholar]
- 34. Ravagnan L, Roumier T, Kroemer G. Mitochondria, the killer organelles and their weapons. J Cell Physiol. 2002;192(2):131–137 [DOI] [PubMed] [Google Scholar]
- 35. Castedo M, Perfettini JL, Roumier T, Andreau K, Medema R, Kroemer G. Cell death by mitotic catastrophe: a molecular definition. Oncogene. 2004;23(16):2825–2837 [DOI] [PubMed] [Google Scholar]
- 36. Xiao Z, Chen Z, Gunasekera AH, et al. Chk1 mediates S and G2 arrests through Cdc25A degradation in response to DNA-damaging agents. J Biol Chem. 2003;278(24):21767–2173 [DOI] [PubMed] [Google Scholar]
- 37. Ashwell S, Zabludoff S. DNA damage detection and repair pathways—recent advances with inhibitors of checkpoint kinases in cancer therapy. Clin Cancer Res. 2008;14(13):4032–4037 [DOI] [PubMed] [Google Scholar]
- 38. Burton JD, Castillo ME, Goldenberg DM, Blumenthal RD. Peroxisome proliferator-activated receptor-gamma antagonists exhibit potent antiproliferative effects versus many hematopoietic and epithelial cancer cell lines. Anticancer Drugs. 2007;18(5):525–534 [DOI] [PubMed] [Google Scholar]
- 39. Kim KY, Kim SS, Cheon HG. Differential anti-proliferative actions of peroxisome proliferator-activated receptor-gamma agonists in MCF-7 breast cancer cells. Biochem Pharmacol. 2006;72(5):530–540 [DOI] [PubMed] [Google Scholar]
- 40. Tachibana K, Yamasaki D, Ishimoto K, Doi T. The role of PPARs in cancer. PPAR Res. 2008;2008:102737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lea MA, Sura M, Desbordes C. Inhibition of cell proliferation by potential peroxisome proliferator-activated receptor (PPAR) gamma agonists and antagonists. Anticancer Res. 2004;24(5A):2765–2771 [PubMed] [Google Scholar]
- 42. Zaytseva YY, Wallis NK, Southard RC, Kilgore MW. The PPARgamma antagonist T0070907 suppresses breast cancer cell proliferation and motility via both PPARgamma-dependent and -independent mechanisms. Anticancer Res. 2011;31(3):813–823 [PubMed] [Google Scholar]
- 43. Kumala S, Niemiec P, Wideł M, Hancock R, Rzeszowska-Wolny J. Apoptosis and clonogenic survival in three tumour cell lines exposed to gamma rays or chemical genotoxic agents. Cell Mol Biol Lett. 2003;8(3):655–665 [PubMed] [Google Scholar]