

Abstract
Objective
Ceramides are precursors of complex sphingolipids (SLs), which are important for normal functioning of both the developing and mature brain. Altered SL levels have been associated with many neurodegenerative disorders, including epilepsy, although few direct links have been identified between genes involved in SL metabolism and epilepsy.
Methods
We used quantitative real-time PCR, Western blotting, and enzymatic assays to determine the mRNA, protein, and activity levels of ceramide synthase 2 (CERS2) in fiibroblasts isolated from parental control subjects and from a patient diagnosed with progressive myoclonic epilepsy (PME). Mass spectrometry and fluorescence microscopy were used to examine the effects of reduced CERS2 activity on cellular lipid composition and plasma membrane functions.
Results
We identify a novel 27 kb heterozygous deletion including the CERS2 gene in a proband diagnosed with PME. Compared to parental controls, levels of CERS2 mRNA, protein, and activity were reduced by ˜50% in fibroblasts isolated from this proband, resulting in significantly reduced levels of ceramides and sphingomyelins containing the very long-chain fatty acids C24:0 and C26:0. The change in SL composition was also reflected in a reduction in cholera toxin B immunofluorescence, indicating that membrane composition and function are altered.
Interpretation
We propose that reduced levels of CERS2, and consequently diminished levels of ceramides and SLs containing very long-chain fatty acids, lead to development of PME.
Introduction
Progressive myoclonic epilepsy (PME) is a heterogeneous group of disorders characterized by myoclonus, tonic-clonic seizures, and progressive neurological dysfunction, including cognitive impairment and ataxia. Disease onset and symptoms vary greatly among these disorders. A greater understanding of the underlying pathophysiological processes would help both diagnosis and treatment.1–3
Sphingolipids (SLs) are abundant in nervous tissue and are especially enriched in myelin. Most typical myelin lipids are cerebrosides (glucosylceramide [GlcCer] and galactosylceramide [GalCer]), which are approximately 10-fold more abundant in white compared to gray matter.4 GalCer is the most abundant cerebroside in the brain, and is especially enriched in C22-24-fatty acids (FAs).4,5 Sphingomyelin (SM) is another major myelin lipid and is highly enriched in C18- and C24-SLs. During development, C18-SM decreases from 80% to 30% of the total SM content in myelin, while C24-SM increases from 4% to 33%. Oligodendrocytes show a SL composition similar to myelin and are also enriched in C24-SLs.4,5 Gangliosides, acidic glycosphingolipids, are predominantly comprised of C18-SLs, and are especially enriched in gray matter.6
Ceramides are the building blocks of all complex SLs (Fig.1). In mammals, Ceramides are synthesized by a family of six enzymes (CERS1-6), which all display distinct tissue expression levels and utilize acyl-CoAs of defined chain length (reviewed in7,8). Ceramide synthase 2 (CERS2) is the most widely expressed and is especially abundant in liver and kidney, and primarily uses C22-C24-acyl-CoAs for ceramide synthesis.9 CERS1 predominantly uses C18:0- and C18:1-acyl-CoAs is the most abundant ceramide synthase in the central nervous system, and more abundant in gray matter than in white matter; however, CERS2 is also highly expressed in white matter.10 Knockout of CerS2 in mice results in decreased levels of C22-24-ceramides and -SLs in the brain, whereas long-chain ceramides, for example, C18-ceramide and sphinganine, increase. In addition to impaired liver function,11 CerS2 null mice exhibit a number of nervous system dysfunctions, including myelin sheath defects and cerebellar degeneration, which consequently results in abnormal motor function, including generalized and symmetrical myoclonic jerks and sensitivity to auditory stimuli.9,12,13 CerS2 null mice also have altered biophysical membrane properties and show elevated levels of reactive oxygen species in the liver due to impaired activity of mitochondrial complex IV.14–16
Figure 1.
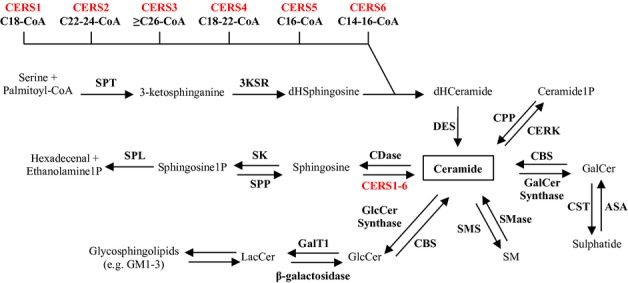
Overview of sphingolipid metabolism. Ceramide is at the central hub of sphingolipid metabolism and is synthesized de novo from serine and palmitoyl-CoA. In the third step of this four-step process, ceramide synthases acylate dihydro-sphingosine (dHSphingosine) to form dihydro-ceramide (dHCeramide). Six mammalian ceramide synthases (CERS1-6) (shown in red) have been identified and each of these utilizes a unique subset of acyl-CoAs (indicated beneath each CERS). Once formed, ceramide can be metabolized in five different ways: (1) Ceramide can be phosphorylated by ceramide kinase (CERK). (2) Ceramide can be glycosylated by galactosylceramide (GalCer) synthase producing GalCer which can be further metabolized into sulfatides. (3) Attachment of phosphocholine to ceramide yields sphingomyelin (SM) in a reaction catalyzed by SM synthase (SMS). (4) Glycosylation of ceramide by glucosylceramide (GlcCer) synthase followed by galactosyltransferase I (GalTI) produces GlcCer and lactosylceramide (LacCer), respectively. LacCer constitute the foundation for the synthesis of more complex glycosphingolipids including gangliosides GM1 and GM3. (5) Ceramide can be degraded to sphingosine, which in turn can be phosphorylated to sphingosine 1-phosphate (sphingosine1P) and further degraded into hexadecanal and ethanolamine 1-phosphate (ethanolamine1P). The last step is the only known exit route from the sphingolipid pathway. SPT, serine palmitoyltransferase; 3KSR, 3-ketosphinganine reductase; DES, dihydroceramide desaturase; CPP, ceramide phosphatase; CBS, cerebrosidase; ASA, arylsulfatase A; CST, cerebroside sulfotransferase; SMase, sphingomyelinase; SK, sphingosine kinase; SPP, sphingosine phosphate phosphatase; SPL, sphingosine1P lyase.
Previously, homozygous loss of GM3 synthase (also known as lactosylceramide α-2,3 sialyltransferase) has been linked to infantile-onset, symptomatic epilepsy syndrome,17 and more recently two CerS1 knockout mice strains have been shown to display degeneration of cerebellar Purkinje neurons and accumulation of lipofuscin.18 As altered SL levels are found in many neurodegenerative disorders and in a variety of other diseases,19 understanding the consequences of these alterations and deciphering SL regulation, as well as its impact on cellular properties, may provide novel therapeutic targets.
Case Report
The proband is a 30-year-old man born to unrelated Caucasian parents with no family history of known chromosomal abnormalities, epilepsy, or developmental delay. Pregnancy and delivery were normal. He had no neonatal problems, weighed 2.9 kg, and had a length of 50 cm at birth. Before the age of 2 years, he had three febrile seizures (FS) and two afebrile seizures, presumably generalized tonic-clonic seizures (GTCS). Initial electroencephalography (EEG) recordings and computerized tomography (CT) scans were normal. To prevent seizures, he was treated with valproic acid (VPA), which rendered him seizure-free. When he was 6 years old, VPA was withdrawn and he remained seizure-free without antiepileptic treatment until the age of 10 years, when VPA was reintroduced due to recurrence of GTCS. He attended a regular school until he was 12 years old, at which time he was diagnosed with learning disabilities and therefore moved to a special class. At the age of 13 years, developmental delay was noticed along with tremor of the hands, and gait disturbances were observed but regarded as side effects of VPA. At the age of 14, he was diagnosed with severe myoclonus. When VPA was substituted by Lamotrigine, cognitive function appeared to improve, but due to unacceptable myoclonia, VPA was reintroduced at the proband's demand. A combination of high dose VPA and oxcarbazepine kept him seizure-free with almost no myoclonia for nearly 2 years. At the age of 20, the frequency of GTCS (predominantly during sleep) and myoclonia increased, and despite administration of several antiepileptic drugs, such as clonazepam, clobazam, levetiracetam, piracetam, and zonisamide, his seizures could not be controlled. Furthermore, withdrawal of oxcarbazepine severely increased the frequency of tonic-clonic seizures. For a period of time, he was is reluctant to go outside due to extreme photosensitivity, which caused frequent falls.
Laboratory investigations revealed normal hematological indices as well as normal liver and kidney function. Prior to Simvastatin treatment, the plasma level of cholesterol was >10 mmol/L (normal level <5.2 mmol/L) and after treatment, high-density lipoprotein, low-density lipoprotein, and triglyceride levels were 6.6, 1.4, and 1.5 mmol/L, respectively. Hematoxylin and eosin staining of a muscle biopsy demonstrated discrete myopathic abnormalities with increased fiber variability and an increased number of central nuclei. The inherited mitochondrial disorders, Mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS), Myoclonic Epilepsy with Ragged Red Fibers (MERRF), and Neuropathy, ataxia, and retinitis pigmentosa (NARP) were excluded, and genetic tests for the lipid storage disease, Niemann Pick Type C (sequencing of NPC1 and NPC2), were normal. In addition, whole exome sequencing was performed without detecting any disease-causing mutations. A variety of other tests were performed, and the only abnormality detected was a small reduction in the respiratory chain complex I/citrate synthase ratio (0.18 compared to the normal range of 0.19–0.54) (Table1).
Table 1.
Case report: list of test values
| Test | Value | Normal range |
|---|---|---|
| Hemoglobin | >10 | 10–11.1 |
| Lactate | 1.2 | 0.7–2.1 |
| Alkaline phosphatase | 117–125 | 35–105 |
| Glut1 deficiency | 4.3 | – |
| β-Galactosidase | 201 | 130–340 |
| Arylsulfatase A | 8.0 | 3.5–15 |
| Hexosaminidase A + B | 2225 | 1200–3500 |
| Hexosaminase A | 211 | 140–410 |
| Galactocerebrosidase | 1.01 | 0.5–3.4 |
| α-Fucosidase | 59 | 20–110 |
| Palmitoyl-protein thioesterase | 22 | 15–90 |
| Hexosaminidase A + B (plasma) | 596 | 400–1800 |
| Chitotriosidase I (plasma) | 13 | 0–115 |
| Cerotic acid | 98 | 40–115 |
| Complex I | 63 | 25–164 |
| Complex II | 130 | 39–171 |
| Complex II + III | 134 | 33–216 |
| Complex III | 467 | 117–794 |
| Complex IV | 1225 | 286–1852 |
| Citrate synthase (CS) | 351 | 127–477 |
| Complex I/CS ratio | 0.18 | 0.19–0.54 |
| Complex II/CS ratio | 0.37 | 0.24–0.50 |
| Complex II + III/CS ratio | 0.38 | 0.19–0.72 |
| Complex III/CS ratio | 1.33 | 0.82–2.14 |
| Complex IV/CS ratio | 3.5 | 2.2–5.0 |
| Complex I/II ratio | 0.48 | 0.45–1.33 |
| Complex II + III/II ratio | 1.03 | 0.48–1.71 |
| Complex III/II ratio | 3.6 | 1.6–5.8 |
| Complex IV/II ratio | 9.4 | 7.3–13.6 |
| PCR analysis of whole mitochondria gene on DNA isolated from muscle biopsy is normal | ||
| MELAS (3243A G) | Not detected | – |
| MERRF (8834A G) | Not detected | – |
| NARP (993T G) | Not detected | – |
EEG recordings at disease onset did not reveal any abnormalities. When the proband was 19 years of age, alpha background activity was preserved, with intermittent and irregular beta and theta activity without an altered pattern during intermittent photic stimulation. Over the years, the EEG background activity was slightly reduced to an activity of 7½–8 Hz, with sharp waves followed by 2–3 Hz slow waves in the left temporal region. During photostimulation and during sleep, myoclonic jerks without EEG correlation were seen, predominantly in the upper limbs and on the left side of the body. EEG-EMG polygraphic recordings were not performed.
Sensory evoked potentials (SEP) analysis showed a cortical response with increased amplitude. Moreover, visual evoked potentials (VEP) showed abnormal cortical potentials and high amplitude spikes in the mid-occipital region. Magnetic resonance imaging (MRI) of the brain at the age of 15 years indicated no abnormalities. However, at 27 years of age, an MRI scan showed a minor lesion, probably a minimal heterotropia in the left temporal lobe, and discrete atrophy bilaterally of the frontoparietal lobes and cerebellum.
Upon neurological examination at 30 years of age, the proband appeared moderately intellectually disabled with dysarthria and ataxia. He was slim (weight 62 kg, height 167 cm), myopic, and had normal hearing. He now lives in residential care, cohousing with younger mentally disabled persons. Within the last year, his condition has worsened and he has been compelled to give up his sheltered employment at the local supermarket. At present he is having 4–8 GTCS per month and consistently experiences myoclonus. He is treated with 2300 mg VPA, 900 mg oxcarbazepine, and 4800 mg piracetam per day.
Subjects/Material and Methods
Study oversight
This study was approved by the Ethics Committee at Western Sealand and written informed consent was obtained from the patient and his parents.
Genetic studies
DNA samples were typed for 1.8 million probe sets on the Affymetrix Genome-Wide Human SNP Array 6.0 (Affymetrix, Santa Clara, CA). CNV (copy number variation) analysis was performed by the algorithm implemented in the Affymetrix Genotyping Console version 4.0. (Affymetrix, Santa Clara, CA). The presumed pathogenic CNV was verified in the index patient, and also tested in the parents with TaqMan qPCR (Life Technologies, Carlsbad, CA) according to manufacturer's instructions. Seven specific primer pairs were designed to amplify the 10 coding exons of CERS2 and the adjacent intron-exon boundaries. PCR amplicons were sequenced using Applied BiosystemsTM according to the supplier's recommendations. Paternity was confirmed by genotyping 15 short tandem repeat (STR) markers located on 10 different chromosomes.
Control cohort
The control cohort comprised 1075 unselected individuals provided by the PopGen biobank.
Cell culture
A skin biopsy excised from the upper arm was transferred to a tissue culture flask. The biopsy was cultured in Roswell Park Memorial Institute media-1640, 20% Fetal calf serum (supplemented with 4 mmol/L l-glutamine, 0.017 mg/mL benzylpenicillin) and grown at 37°C, with 5% CO2. Primary fibroblasts were grown in Dulbecco's modified Eagle's medium supplemented with high glucose, 1 mmol/L sodium pyruvate (Invitrogen, Carlsbad, CA), 44.04 mmol/L sodium hydrogen carbonate, 33 μmol/L biotin, 34 μmol/L pantothenic acid, 20% fetal bovine serum (Sigma-Aldrich, St. Louis, MO), 100 units penicillin/0.1 mg streptomycin per liter (Sigma-Aldrich), and GlutaMAX supplement (Invitrogen) at 37°C, with 5% CO2.
Quantitative real-time PCR
Total RNA was harvested from muscle biopsies and human primary fibroblasts using ice-cold Trizol (Invitrogen) according to manufacturer's instructions. cDNA and quantitative real-time PCR were performed as described.20 Expressions were normalized to TATA-binding protein (TBP) and/or β-actin. Statistical analyses were performed in GraphPad Prism version 6 (GraphPad Software, La Jolla, CA).
Western blotting
Total cell extracts were prepared from cultured fibroblasts, and proteins were separated by sodium dodecyl sulfate (SDS) polyacrylamide gel electrophoresis and subsequently transferred to a Polyvinylidene Difluoride membrane by electroblotting. CERS2 was probed using a rabbit anti-human CERS2 antibody (Abcam, Cambridge, England). TBP was probed using a rabbit anti-human TFIIB antibody (Santa Cruz Biotechnology, Inc., Dallas, TX), which served as loading control. All immunoprobed proteins were detected by enhanced chemiluminescence.
Ceramide synthase activity
Ceramide synthase activities were determined in cell extracts as previously described.21
Lipid analysis by mass spectrometry
Analysis of ceramides and SLs in isolated fibroblasts were carried out by Avanti Polar Lipids, Inc., Alabama, AL. Results were normalized to cell number.
GM1 staining
Fibroblasts were washed three times in M1 medium (150 mmol/L NaCl, 5 mmol/L KCl, 1 mmol/L CaCl2, 1 mmol/L MgCl2, 5 mmol/L glucose, 20 mmol/L Hepes, pH 7.3) and stained with Cholera Toxin B-Alexa488 (10 μg/mL in M1 medium) (Invitrogen) for 20 min at 37°C. Wide-field fluorescence microscopy and digital image acquisition were performed using a Leica DMIRBE microscope with a 63×, 1.4 NA oil immersion objective (Leica Lasertechnik GmbH, Wetzlar, Germany). All pictures were taken within 20 min after CTxB staining using a standard fluorescein filter set (470-nm, [20-nm bandpass] excitation filter, 510-nm longpass dichromatic filter, and 537-nm [23-nm] bandpass emission filter). All images were acquired using identical settings. Images were analyzed using the freeware ImageJ [National Institute of Health (NIH), Bethesda, MD].
Light sensitivity of CerS2 null mice
Mice were maintained in a specific pathogen-free and temperature-controlled (22 ± 1°C) mouse facility on a reverse 12 h light/dark cycle (lights on at 20:00) according to institutional guidelines. Food and water were given ad libitum. All experimental protocols were approved by the Institutional Animal Care and Use Committee of The Weizmann Institute of Science. Light sensitivity was assessed by measuring freezing behavior in a circular open field (Ø = 56.5 cm) under two illumination conditions, dark (10 lux, 5 min) and light (120 lux, 5 min). An overhead camera (Sony DCR-SR30E HANDYCAM, Tokyo, Japan) recorded mouse behavior. Freezing was determined by off-line analyses of the video tracks using an automated tracking system (Ethovision, Noldus, Wageningen, the Netherlands).
Results
Genetic analysis
We performed genome-wide SNP 6.0 array analysis on DNA isolated from the proband and identified a heterozygous 27 kb de novo deletion on chromosome 1q21 containing the entire CERS2 gene (Fig.2A). To exclude a recessive condition, we sequenced the remaining allele of CERS2, but no further mutations were identified. The deletion also included the distal part of SETDB1 (exon 15–22), a histone methyltransferase. To examine the expression levels of both genes, we performed quantitative PCR on total RNA isolated from muscle biopsies from the proband and six unrelated controls. As predicted, the level of CERS2 mRNA was reduced to ˜50% in the proband (Fig.2B). Moreover, expression of the longest predicted transcripts of SETDB1 was reduced, while expression of shorter transcripts was unaffected (Fig.2B), consistent with the identified deletion. To obtain further genetic evidence for CERS2 pathogenicity, we performed SNP 6.0 array analysis on 1075 healthy controls, and no deletions or duplications involving CERS2 were detected. In addition, we performed a mutation analysis of all 10 coding exons and intron-exon boundaries of CERS2 using bidirectional sequencing in a cohort of 100 probands with progressive ataxia and/or epilepsy. No CERS2 mutations were detected in this cohort.
Figure 2.
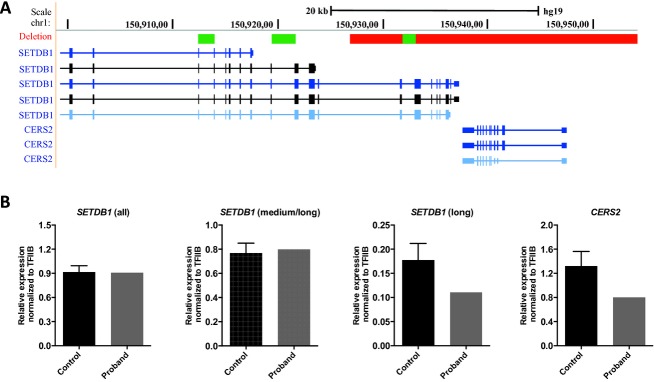
Genomic location of the CERS2 deletion. (A) The deletion is indicated by the red bar. The figure is based on the UCSC Genome Browser (http://www.genome.ucsc.edu, assembly hg19) showing the genomic positions from the Affymetrix Genome-Wide Human SNP Array 6.0. The deletion is a heterozygous 27 kb deletion of 1q21 containing the entire CERS2 gene and part of the SETDB1 gene (exon 15–22). Regions of SETDB1 amplified by quantitative real-time PCR are indicated by the green bars. (B) Total RNA was isolated from muscle biopsies from the proband and six unrelated controls as described in the experimental section and expression levels of CERS2 and SETDB1 isoforms were determined by quantitative real-time PCR. Three primer sets were used for the detection of SETDB1; all detects all isoforms, medium/long detects the medium to long isoforms, and long detects the three long isoforms. Mean ± SD is shown, N (control) = 6, N (proband) = 1.
Biochemical analysis of primary fibroblasts isolated from proband
In order to characterize the biochemical phenotypes associated with the heterozygous deletion of CERS2, we characterized primary fibroblasts generated from skin biopsies from the proband and the healthy parents. Parental fibroblasts were used as controls, and after establishing paternal consanguinity, we confirmed that both parents carry two functional CERS2 alleles (results not shown). Compared to parental cells, expression levels of CERS2 and the long SETDB1 transcripts were reduced by more than 50% in proband cells (Fig.3A; data not shown). We observed similar decreases in protein levels of CERS2 and high molecular weight isoforms of SETDB1 (Fig.3B; data not shown). The mRNA expression levels of CERS1, CERS4, and CERS6 varied between proband and parents, with no obvious tendency to be either increased or decreased in proband fibroblasts compared to parental fibroblasts (Fig.3A). Analysis of CERS2 activity using C22-CoA as a substrate showed a 50% reduction compared to control fibroblasts (Fig.3C). However, no changes in ceramide synthase activity were observed in proband cells using C24- or C24:1-CoAs as substrate. We next examined ceramide and SL composition by mass spectrometry (Figs.4, 5); ceramides containing very long-chain FAs (i.e., C24–C26) were significantly reduced in proband cells compared to controls, while long-chain ceramides (i.e., C16–C18) increased (Fig.4A and B). Additionally, the abundance of SMs containing very long-chain FAs was significantly reduced in proband fibroblasts compared to parental control cells, while the abundance of SMs with long-chain FAs was unchanged (Fig.4C and D). Despite these changes in ceramide and SM composition and levels, levels of the very long-chain glycosylated ceramides lactosylceramide (LacCer) and GlcCer were not reduced (Fig.5).
Figure 3.
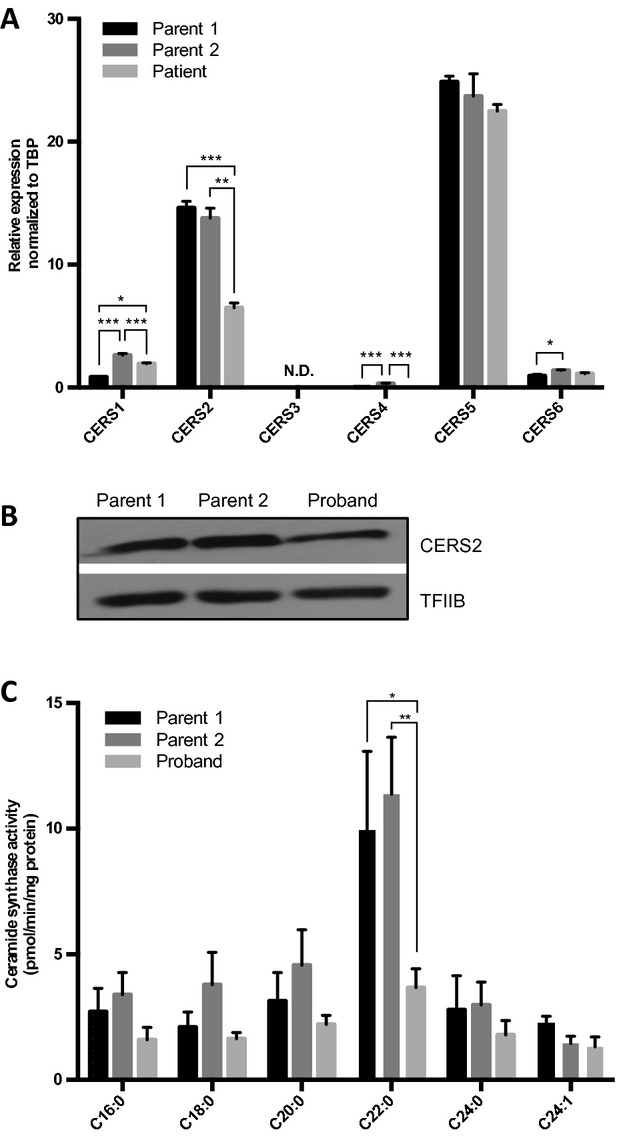
Heterozygous deletion of CERS2 reduces CERS2 expression, CERS2 protein level, and activity in fibroblasts. (A) Total RNA was isolated from cultured fibroblasts from the proband and controls and expression levels of CERS1-6 were determined by quantitative real-time PCR. Mean ± SD is shown, N = 3. Statistical analyses were performed using a multiple t-test with significance levels: *(P < 0.05), **(P < 0.01), and ***(P < 0.001). (B) Total cell extracts were prepared from cultured fibroblasts and CERS2 levels were determined by Western blotting. TFIIB served as loading control. (C) Ceramide synthase activities were determined in whole cell extracts using the indicated acyl-CoAs as substrates. Mean ± SEM is shown, N = 6–9. Statistical analyses were performed using multiple t-test with significance levels: *(P < 0.05) and **(P < 0.01).
Figure 4.

Ceramides and sphingomyelin species containing very long-chain fatty acids are reduced in proband fibroblasts. Lipid extracts prepared from cultured fibroblasts were analyzed using LC-MS. Ceramides containing long-chain fatty acids are shown in (A), while ceramides with very long-chain fatty acids are shown in (B). (C) Sphingomyelins containing long-chain fatty acids and (D) sphingomyelins containing very long-chain fatty acids from proband and parental control fibroblasts. Assuming that all ceramide and sphingomyelin species contain a C18-sphingoid base, sphingolipids were categorized into long-chain or very long-chain species. Levels of lipid species have been normalized to total cell number and internal standards. Three independent cultures of fibroblasts from the proband and controls were analyzed. Mean ± SD is shown, N = 3. Statistical analyses were performed using multiple t-test with significance levels: *(P < 0.05), **(P < 0.01), and ***(P < 0.001).
Figure 5.
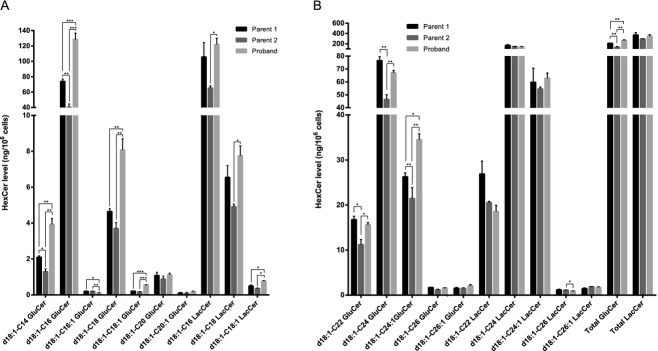
Lipid profile of glycosylceramide species in proband fibroblasts. Lipid extracts, prepared from cultured fibroblasts were analyzed using LC-MS. Glycosylceramide species containing long-chain fatty acids are shown in (A), while glycosylceramide species with very long-chain fatty acids are shown in (B). Lipids levels were normalized to total cell number and internal standards. Three independent cultures of fibroblasts from the proband and controls were analyzed. Mean ± SD is shown, N = 3. Statistical analyses were performed using multiple t-test with significance levels: *(P < 0.05) and **(P < 0.01).
SLs play important roles in the formation of membrane microdomains, which are lipid domains enriched in cholesterol and SLs. These domains orchestrate assembly of membrane proteins and ensure proper signal transduction (reviewed in22). Staining cells with cholera toxin B, a marker of membrane microdomains which binds to ganglioside GM1, revealed significantly reduced labeling in fibroblasts derived from the proband compared to both parental cell lines (Fig.6), indicating that membrane lipid composition, and possibly formation of microdomains, are altered when CERS2 activity is reduced.
Figure 6.
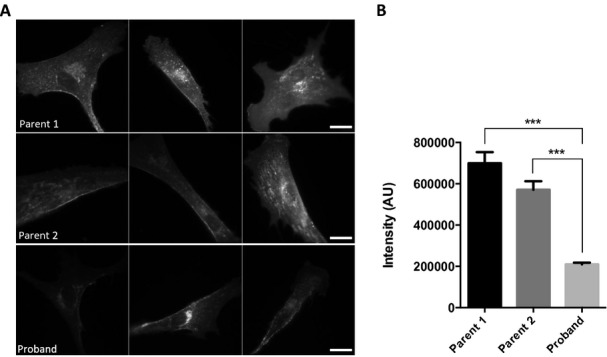
Cholera toxin B labeling of fibroblasts from proband and parental controls. (A) Fibroblasts from proband and controls were stained with Alexa488-labeled cholera toxin B for 20 min., and subsequently examined by fluorescence microscopy. (B) Images and fluorescence intensities were analyzed using ImageJ. Mean ± SEM is shown, N (parent 2) = 38 and N (proband) = 49. Statistical analyses were performed using unpaired two-tailed t-test with Welch's correction with significance level: ***(P = 0.0004).
Photosensitivity in CerS2 null mice
Finally, as the proband is photosensitive, we examined whether CerS2 null mice are sensitized to light, using freezing behavior, a well-known index of fear,23,24 under dim and bright illumination conditions. Although CerS2 heterozygosity in mice did not sensitize mice to light, CerS2 null mice were significantly more sensitive to light than CerS2+/+ mice (Fig.7).
Figure 7.
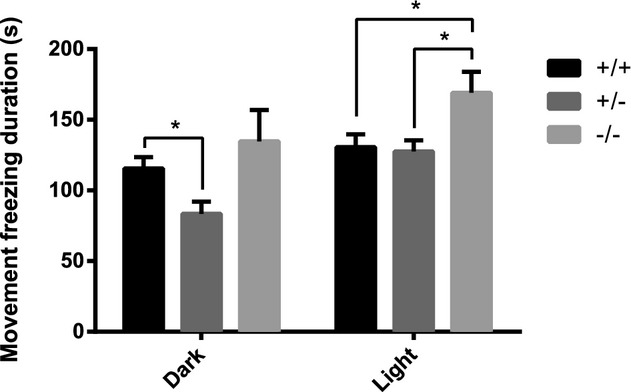
CERS2 KO mice exhibit increased sensitivity to light. Sensitivity to light was assessed by measuring freezing behavior, an index of fear, in a circular open field under two illumination conditions, dark (10 lux, 5 min) and light (120 lux, 5 min). ANOVA for GENE (KO/HT/WT) indicated no difference in freezing duration in the DARK (F(2,15) = 3.185; n.s.), but a statistically longer duration of freezing in CerS2 null mice than in both WT and heterozygote mice under light. N = 6. (ANOVA for GENE: F(2,15) = 4.42; P < 0.05; Dunnet post-hoc test: KO>het P < 0.05, KO>WT P < 0.05).
Discussion
In this study, we identified a de novo 27 kb heterozygous deletion of 1q21, including CERS2 and the distal part of SETDB1, in a male proband diagnosed with PME. In the brain, CERS2 is highly expressed in white matter tracts including the corpus callosum, striatum, and white matter of the cerebellum and brainstem, which is consistent with CERS2 expression in mature myelin-producing oligodendrocytes. Schwann cells of the peripheral nervous system display high CERS2 expression as well.10,4 SETDB1 is also expressed in the nervous system, and removal of Setdb1 in mice results in peri-implantation lethality.25 Studies of the forebrain of transgenic Setdb1 mice demonstrated that it targets ionotropic glutamatergic NMDA receptors.26 Recently, mutations and intragenic deletions of SETDB1 have been associated with Autism Spectrum Disorder (ASD) although all the detected SETDB1 variants were inherited from healthy parents.27 As our proband was not diagnosed with ASD, and heterozygous mutations and intragenetic deletions of SETDB1 have been identified in healthy individuals, we do not believe that the partial SETDB1 deletion causes the currently described phenotype.
Besides a recent genome-wide association study, which identified a CERS2 mutation to be associated with primary rhegmatogenous retinal detachment,28 CERS2 mutations causing human disorders have, until now, not been reported. This study suggests that CERS2 haploinsufficiency may cause PME. PMEs are a group of rare and devastating genetic disorders, which are often refractory to conventional treatment. Almost all PMEs reported so far have been associated with recessive inheritance. In the present report, we have confirmed that both parents have two functional CERS2 alleles, thus the CERS2 haploinsufficiency must have arisen due to a spontaneous deletion of one of the CERS2 alleles. Accordingly, fibroblasts isolated from the proband display reduced levels of CERS2 mRNA, protein, and activity, which cause reduced levels of very long-chain ceramides and SMs.
Little is known about how CERS2 deficiency might affect cellular functions in humans. In mice, total loss of CerS2 expression significantly diminishes levels of ceramides and more complex SLs such as C22-24 GalCer, which results in myelin instability and in degeneration of both white and gray matter in the cerebellum.12,13 This suggests that CERS2 activity is important in both neurons and formation of myelin sheath.12,13 CerS2 null mice also display astrogliosis and microglial activation in both white and gray matter, which may be related to the motor initiation difficulties and myoclonic jerks observed in these mice.13,29 EEG revealed abnormal fast rhythmic activity (>40 Hz) and, similar to that found in the proband, the null mice display no changes in EEG during myoclonic events.13 To this end, our present studies suggest that CerS2 null mice also are significantly more sensitive to light than wild-type mice (Fig.6), implying that the light sensitivity in the proband may be caused by impaired CERS2 function.
Disruption of CERS2 activity in mice not only causes neurological phenotypes but also results in impaired hepatic and lung functions.11,29–31 In the present proband, CERS2 haploinsufficiency does not appear to impair liver and kidney function (Table1). Although CERS2 is ubiquitously expressed, it is abundantly expressed in both liver and kidney, and found at significantly lower levels in the brain. We therefore propose that CERS2 haploinsufficiency in humans only has detrimental effects in tissues containing low levels of CERS2, such as the brain, while the remaining CERS2 activity in the liver and kidney may synthesize sufficient amounts of very long-chain SLs to sustain central cellular functions. Alternatively, compensatory mechanisms by other ceramide synthases may exist in vivo, which are not evident in isolated fibroblasts.
Ablation of CerS2 in mice results in a small and inconsistent reduction in total GM113 and in altered biophysical membrane properties.15 Consistent with this notion, we also found that CERS2 haploinsufficiency reduced cholera toxin B staining of the plasma membrane, suggesting that the altered SL composition can ultimately impair membrane microdomain formation and hence plasma membrane functions. Interestingly, such membrane microdomains have been shown to modulate neuronal excitability by controlling neurotransmitter receptor sensitivity and functions,32,33 which could contribute to the phenotypes displayed by the proband.
Collectively, we have identified a proband with PME coupled to altered synthesis and composition of very long-chain ceramides and SM. The proband has only one functional CERS2 allele, reduced CERS2 mRNA, protein, and activity levels, resulting in impaired synthesis of ceramides and SM containing very long acyl chains, which consequently can affect the formation of membrane microdomains. We foresee that this proband will provide a unique opportunity for addressing the functions of CERS2 and SLs in the development of PME, and lay the foundation for further exploration of the role of SL metabolism in human neurodegenerative disorders.
Acknowledgments
We thank Michael Witting, Andre Franke and Ingo Helbig from the POPGEN database at Christian-Albrechts-University (Kiel, Germany) for help with database analyses. We thank the patients and families for their essential help and support. The authors declare no conflicts of interest. This study was supported by The Lundbeck Foundation and The Danish Research Councils.
Author Contributions
M. B. M., A. S. B. O., D. N., O. B. D., L. H., A. H. F., and N. J. F. were all involved in designing and performing the described biochemical analyses. L. L. K. identified the family, analyzed CNV data, and designed and performed sequencing of CERS2. J. L. sequenced CERS2. H. H., A. S., N. T., and J. E. N. provided and examined the clinical information and discussed the functional effects of the CNV. J. V. provided clinical data and muscle tissue from the patient and parental controls. A. D. K. and M. M. T. performed the light sensitivity experiments in mice. M. B. M., A. S. B. O., A. H. F., R. S. M., and N. J. F. wrote the manuscript.
Conflict of Interest
None declared.
Funding Information
This study has been supported by The Lundbeck Foundation and The Danish Council for Independent Research, Natural Sciences.
References
- 1.Shahwan A, Farrell M, Delanty N. Progressive myoclonic epilepsies: a review of genetic and therapeutic aspects. Lancet Neurol. 2005;4:239–248. doi: 10.1016/S1474-4422(05)70043-0. [DOI] [PubMed] [Google Scholar]
- 2.Zupanc ML, Legros B. Progressive myoclonic epilepsy. Cerebellum. 2004;3:156–171. doi: 10.1080/14734220410035356. [DOI] [PubMed] [Google Scholar]
- 3.Satishchandra P, Sinha S. Progressive myoclonic epilepsy. Neurol India. 2010;58:514–522. doi: 10.4103/0028-3886.68660. [DOI] [PubMed] [Google Scholar]
- 4.Ben-David O, Futerman AH. The role of the ceramide acyl chain length in neurodegeneration: involvement of ceramide synthases. Neuromolecular Med. 2010;12:341–350. doi: 10.1007/s12017-010-8114-x. [DOI] [PubMed] [Google Scholar]
- 5.Baumann N, Pham-Dinh D. Biology of oligodendrocyte and myelin in the mammalian central nervous system. Physiol Rev. 2001;81:871–927. doi: 10.1152/physrev.2001.81.2.871. [DOI] [PubMed] [Google Scholar]
- 6.Sastry PS. Lipids of nervous tissue: composition and metabolism. Prog Lipid Res. 1985;24:69–176. doi: 10.1016/0163-7827(85)90011-6. [DOI] [PubMed] [Google Scholar]
- 7.Levy M, Futerman AH. Mammalian ceramide synthases. IUBMB Life. 2010;62:347–356. doi: 10.1002/iub.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mullen TD, Hannun YA, Obeid LM. Ceramide synthases at the centre of sphingolipid metabolism and biology. Biochem J. 2012;441:789–802. doi: 10.1042/BJ20111626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Laviad EL, Albee L, Pankova-Kholmyansky I, et al. Characterization of ceramide synthase 2: tissue distribution, substrate specificity, and inhibition by sphingosine 1-phosphate. J Biol Chem. 2008;283:5677–5684. doi: 10.1074/jbc.M707386200. [DOI] [PubMed] [Google Scholar]
- 10.Becker I, Wang-Eckhardt L, Yaghootfam A, et al. Differential expression of (dihydro)ceramide synthases in mouse brain: oligodendrocyte-specific expression of CerS2/Lass2. Histochem Cell Biol. 2008;129:233–241. doi: 10.1007/s00418-007-0344-0. [DOI] [PubMed] [Google Scholar]
- 11.Pewzner-Jung Y, Park H, Laviad EL, et al. A critical role for ceramide synthase 2 in liver homeostasis: I. alterations in lipid metabolic pathways. J Biol Chem. 2010;285:10902–10910. doi: 10.1074/jbc.M109.077594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Imgrund S, Hartmann D, Farwanah H, et al. Adult ceramide synthase 2 (CERS2)-deficient mice exhibit myelin sheath defects, cerebellar degeneration, and hepatocarcinomas. J Biol Chem. 2009;284:33549–33560. doi: 10.1074/jbc.M109.031971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ben-David O, Pewzner-Jung Y, Brenner O, et al. Encephalopathy caused by ablation of very long acyl chain ceramide synthesis may be largely due to reduced galactosylceramide levels. J Biol Chem. 2011;286:30022–30033. doi: 10.1074/jbc.M111.261206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zigdon H, Kogot-Levin A, Park JW, et al. Ablation of ceramide synthase 2 causes chronic oxidative stress due to disruption of the mitochondrial respiratory chain. J Biol Chem. 2013;288:4947–4956. doi: 10.1074/jbc.M112.402719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Silva LC, Ben David O, Pewzner-Jung Y, et al. Ablation of ceramide synthase 2 strongly affects biophysical properties of membranes. J Lipid Res. 2012;53:430–436. doi: 10.1194/jlr.M022715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yurlova L, Kahya N, Aggarwal S, et al. Self-segregation of myelin membrane lipids in model membranes. Biophys J. 2011;101:2713–2720. doi: 10.1016/j.bpj.2011.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Simpson MA, Cross H, Proukakis C, et al. Infantile-onset symptomatic epilepsy syndrome caused by a homozygous loss-of-function mutation of GM3 synthase. Nat Genet. 2004;36:1225–1229. doi: 10.1038/ng1460. [DOI] [PubMed] [Google Scholar]
- 18.Zhao L, Spassieva SD, Jucius TJ, et al. A deficiency of ceramide biosynthesis causes cerebellar purkinje cell neurodegeneration and lipofuscin accumulation. PLoS Genet. 2011;7:e1002063. doi: 10.1371/journal.pgen.1002063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lahiri S, Futerman AH. The metabolism and function of sphingolipids and glycosphingolipids. Cell Mol Life Sci. 2007;64:2270–2284. doi: 10.1007/s00018-007-7076-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Neess D, Bloksgaard M, Bek S, et al. Disruption of the acyl-CoA-binding protein gene delays hepatic adaptation to metabolic changes at weaning. J Biol Chem. 2011;286:3460–3472. doi: 10.1074/jbc.M110.161109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eckl KM, Tidhar R, Thiele H, et al. Impaired epidermal ceramide synthesis causes autosomal recessive congenital ichthyosis and reveals the importance of ceramide acyl chain length. J Invest Dermatol. 2013;133:2202–2211. doi: 10.1038/jid.2013.153. [DOI] [PubMed] [Google Scholar]
- 22.Zhang Y, Li X, Becker KA, Gulbins E. Ceramide-enriched membrane domains – structure and function. Biochim Biophys Acta. 2009;1788:178–183. doi: 10.1016/j.bbamem.2008.07.030. [DOI] [PubMed] [Google Scholar]
- 23.Blanchard DC, Griebel G, Blanchard RJ. The mouse defense test battery: pharmacological and behavioral assays for anxiety and panic. Eur J Pharmacol. 2003;463:97–116. doi: 10.1016/s0014-2999(03)01276-7. [DOI] [PubMed] [Google Scholar]
- 24.Crawley JN. Behavioral phenotyping of transgenic and knockout mice: experimental design and evaluation of general health, sensory functions, motor abilities, and specific behavioral tests. Brain Res. 1999;835:18–26. doi: 10.1016/s0006-8993(98)01258-x. [DOI] [PubMed] [Google Scholar]
- 25.Lohmann F, Loureiro J, Su H, et al. KMT1E mediated H3K9 methylation is required for the maintenance of embryonic stem cells by repressing trophectoderm differentiation. Stem Cells. 2010;28:201–212. doi: 10.1002/stem.278. [DOI] [PubMed] [Google Scholar]
- 26.Jiang Y, Jakovcevski M, Bharadwaj R, et al. Setdb1 histone methyltransferase regulates mood-related behaviors and expression of the NMDA receptor subunit NR2B. J Neurosci. 2010;30:7152–7167. doi: 10.1523/JNEUROSCI.1314-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cukier HN, Lee JM, Ma D, et al. The expanding role of MBD genes in autism: identification of a MECP2 duplication and novel alterations in MBD5, MBD6, and SETDB1. Autism Res. 2012;5:385–397. doi: 10.1002/aur.1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kirin M, Chandra A, Charteris DG, et al. Genome-wide association study identifies genetic risk underlying primary rhegmatogenous retinal detachment. Hum Mol Genet. 2013;22:3174–3185. doi: 10.1093/hmg/ddt169. [DOI] [PubMed] [Google Scholar]
- 29.Pewzner-Jung Y, Brenner O, Braun S, et al. A critical role for ceramide synthase 2 in liver homeostasis: II. insights into molecular changes leading to hepatopathy. J Biol Chem. 2010;285:10911–10923. doi: 10.1074/jbc.M109.077610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Park JW, Park WJ, Kuperman Y, et al. Ablation of very long acyl chain sphingolipids causes hepatic insulin resistance in mice due to altered detergent-resistant membranes. Hepatology. 2012;57:525–532. doi: 10.1002/hep.26015. [DOI] [PubMed] [Google Scholar]
- 31.Petrache I, Kamocki K, Poirier C, et al. Ceramide synthases expression and role of ceramide synthase-2 in the lung: insight from human lung cells and mouse models. PLoS One. 2013;8:e62968. doi: 10.1371/journal.pone.0062968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Allen JA, Halverson-Tamboli RA, Rasenick MM. Lipid raft microdomains and neurotransmitter signalling. Nat Rev Neurosci. 2007;8:128–140. doi: 10.1038/nrn2059. [DOI] [PubMed] [Google Scholar]
- 33.Zhu D, Xiong WC, Mei L. Lipid rafts serve as a signaling platform for nicotinic acetylcholine receptor clustering. J Neurosci. 2006;26:4841–4851. doi: 10.1523/JNEUROSCI.2807-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]