

Abstract
Objective
Amyotrophic lateral sclerosis (ALS) and myasthenia gravis (MG) are caused, respectively, by motor neuron degeneration and neuromuscular junction (NMJ) dysfunction. The membrane protein LRP4 is crucial in the development and function of motor neurons and NMJs and LRP4 autoantibodies have been recently detected in some MG patients. Because of the critical role in motor neuron function we searched for LRP4 antibodies in ALS patients.
Methods
We developed a cell-based assay and a radioimmunoassay and with these we studied the sera from 104 ALS patients.
Results
LRP4 autoantibodies were detected in sera from 24/104 (23.4%) ALS patients from Greece (12/51) and Italy (12/53), but only in 5/138 (3.6%) sera from patients with other neurological diseases and 0/40 sera from healthy controls. The presence of LRP4 autoantibodies in five of six tested patients was persistent for at least 10 months. Cerebrospinal fluid samples from six of seven tested LRP4 antibody-seropositive ALS patients were also positive. No autoantibodies to other MG autoantigens (AChR and MuSK) were detected in ALS patients. No differences in clinical pattern were seen between ALS patients with or without LRP4 antibodies.
Conclusions
We infer that LRP4 autoantibodies are involved in patients with neurological manifestations affecting LRP4-containing tissues and are found more frequently in ALS patients than MG patients. LRP4 antibodies may have a direct pathogenic activity in ALS by participating in the denervation process.
Introduction
Amyotrophic lateral sclerosis (ALS), a heterogeneous neurodegenerative disease affecting motor neurons of the motor cortex and spinal anterior horn, has a mean survival of 3–5 years1 and exists as a sporadic and a familial form. The pathogenesis of sporadic ALS (˜90% of all ALS cases) remains largely obscure, explaining the absence of effective treatments. ALS can be viewed as a “phenotypic tank” in which groups of pathogenically heterogeneous patients coexist while activation of the immune system during the neurodegeneration process has been observed.2–5 Identifying specific biomarkers might allow subgrouping of ALS patients, early diagnosis, and effective intervention.6
In ALS, upper motor neuron dysfunction causes spasticity, whereas lower motor neuron dysfunction leads to muscle wasting, weakness, and fasciculation. Electromyographic (EMG) changes are strongly supportive for ALS diagnosis.7 Although suggested long ago,8 the extent of neuromuscular junction (NMJ) dysfunction in ALS is not known.
LRP4 is located at the postsynaptic membrane of the NMJ9 and on motor neurons in the brain10 and spinal cord.11 Upon binding to agrin, muscle LRP4 induces activation of MuSK, resulting in acetylcholine receptor (AChR) clustering, necessary for proper NMJ function.9 Recent data have shown that LRP4 expression in both motor neurons and muscle is critical for the presynaptic differentiation and survival of motor neuronal axons.11,12
Due to the critical function of LRP4, anti-LRP4 autoantibodies could cause NMJ-related diseases. Myasthenia gravis (MG), mainly characterized by autoantibodies to AChR or MuSK,13 has recently been associated also with LRP4 autoantibodies.14–16 LRP4 autoantibodies inhibit agrin-mediated AChR cluster formation and are probably pathogenic in these patients. They could also play a role in ALS pathogenesis by inhibiting the binding of muscle LRP4 to proteins on motor axons and inhibiting the presynaptic differentiation of the motor axons,12 leading to premature withdrawal of motor nerve terminals, an early step in ALS.17 In addition, animal LRP4 antibodies have been shown to reduce viability of neurons in cell culture and to impair synaptic structure.18
We detected a high and persistent frequency of LRP4 autoantibodies in the sera and cerebrospinal fluid (CSF) of two cohorts of ALS patients suggesting these antibodies may play a role in the pathogenic process underlying muscle denervation.
Material and Methods
Patients and collection of serum and CSF samples
Serum and CSF samples were collected from two independent cohorts of sporadic ALS patients followed by the University Departments of Neurology in Athens (51 sera and 14 CSF) and Milan (53 sera and 10 CSF). Overall, 164 control sera and 54 control CSFs were also used (see Results). Samples were collected and used after approval by the internal Review Boards of the involved institutions and with written informed consent from patients or relatives. The diagnosis of ALS was based on the revised El Escorial criteria1 while other diseases were excluded.
Assays for the detection of LRP4, AChR and MuSK autoantibodies
LRP4 autoantibodies were detected blindly by a cell-based assay (CBA) involving immunofluorescence with HEK293 cells transfected with human LRP4 fused to green fluorescent protein (GFP), using enhanced green fluorescent protein (EGFP) transfected cells as negative controls (modified from Pevzner et al.15; see Supplementary Methods). Anti-LRP4 antibodies were also tested by a radioimmunoprecipitation assay (RIPA) with a polypeptide fragment of LRP4 (amino acids 21–737; see Supplementary Methods). AChR and MuSK autoantibodies were detected by the classical radioimmunoassays for these antibodies (Supplementary Methods) whereas antibodies to AChR clusters were detected by a CBA with HEK293 cells transfected with human muscle AChR subunits and rapsin as described by Leite et al.19
Results
LRP4 antibodies in sera from ALS patients
Sera from 104 Greek and Italian sporadic ALS patients and 164 controls were screened (at 1/100 dilution) for LRP4 antibodies using a CBA with human LRP4 (Fig.1). LRP4 antibodies were detected in 23.1% of the ALS sera (23.5% of the Greek patients and 22.6% of the Italian patients; Table1). Similarly, all sera were tested for binding to the N-terminal polypeptide fragment of LRP4 (amino acids 21–737; 42% of the extracellular domain of LRP4) by a RIPA. Eight of the 24 CBA-positive sera were found also positive by the RIPA; In addition four sera found “ambiguous” by the CBA assay (but declared as CBA-negative) were RIPA-positive (Fig. S1).
Figure 1.
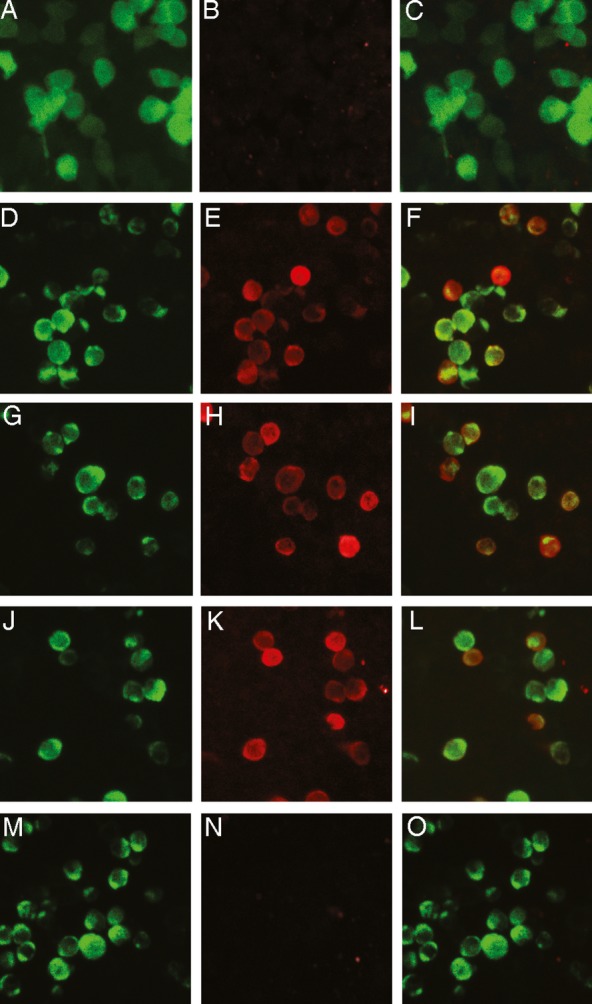
Detection of LRP4 antibodies in sera of ALS patients using a cell-based assay (CBA). Immunofluorescence study of the binding of antibodies to HEK293 cells transfected with pCMV6-LRP4-tGFP or pEGFP. Left column: GFP fluorescence; middle column: bound antibody staining; right column: merged images Rows 1 and 2: Commercial rabbit LRP4 antibody (1:750 dilution) was incubated with pEGFP-transfected cells (row 1; A–C) or pCMV6-LRP4-tGFP-transfected cells (row 2; D–F) and bound antibodies visualized using Alexa 568-conjugated anti-rabbit IgG antibodies. Rows 3–5: LRP4-GFP-transfected cells were incubated with a 1:100 dilution of serum from two ALS patients (rows 3–4; G–L) or a healthy control (row 5, M–O) and bound antibodies visualized with Alexa 568-conjugated anti-human IgG antibodies. Only the commercial antibody and patients' sera bound on expressed LRP4 as visualized with red staining (E, H, K) and the merged images (F, I, L).
Table 1.
Detection of autoantibodies to LRP4 and other NMJ antigens in the serum and CSF from ALS patients and controls1
| Patients | No of samples | Anti-LRP4 | Anti-AChR | Anti-AChR clusters | Anti-MuSK |
|---|---|---|---|---|---|
| Greek sporadic ALS | 51 | 12 (23.5%) | 0 | 0 | 0 |
| Italian sporadic ALS | 53 | 12 (22.6%) | 0 | 0 | 02 |
| Familial ALS | 4 | 0 | 0 | 0 | 0 |
| MG (anti-AChR positive) | 40 | 1 (2.5%) | 40 | 22/22 | 0 |
| MS | 84 | 4 (4.8%) | NT | NT | NT |
| OND | 10 | 0 | NT | NT | NT |
| Healthy controls | 40 | 0 | NT | NT | NT |
| CSF from anti-LRP4 positive ALS patients | 7 | 6 | NT | NT | NT |
| CSF from anti-LRP4 negative ALS patients | 17 | 0 | NT | NT | NT |
| CSF from patients with MS or OND | 44 | 13 (2.3%) | NT | NT | NT |
MG, myasthenia gravis; MS, multiple sclerosis; OND, other neurological diseases.
Serum samples from patients in two ALS cohorts (Greek and Italian) and patients with familial ALS, MG (myasthenia gravis) with AChR antibodies, MS, and other neurological disease (OND) and healthy controls were tested in the cell-based assay at a 1/100 dilution for the presence of LRP4 or AChR cluster antibodies and in the radioimmunoprecipitation assay for the presence of AChR and MuSK antibodies. CSF samples from patients with ALS, MS, and OND were also tested, undiluted, for the presence of LRP4 antibodies.
Two sera were found ambiguous for MuSK antibodies (titer 0.02 nmol/L). Both were LRP4-negative.
This patient was one of the four LRP4-seropositive MS patients.
Sera from 6 LRP4-ALS patients were also collected ˜10 months later (Fig. S2); one was found negative (with stable clinical condition), in two the antibody titer was doubled (both with marked worsening of their clinical condition), whereas in three the antibody titer remained stable (with marginal worsening of their clinical condition).
We then compared the anti-LRP4 titers between anti-LRP4 positive ALS and MG sera. Serial dilutions of 21/24 LRP4-positive ALS sera were compared with those of 11 randomly chosen positive MG sera. We found that both ALS and MG groups contained low and high titer sera (Fig.2) In fact, the average titer of the ALS sera (endpoint dilutions 1/895 + 563) was considerably higher than that of the MG sera (1/632 + 361). Yet, we could not notice any correlation between disease severity and serum titer.
Figure 2.
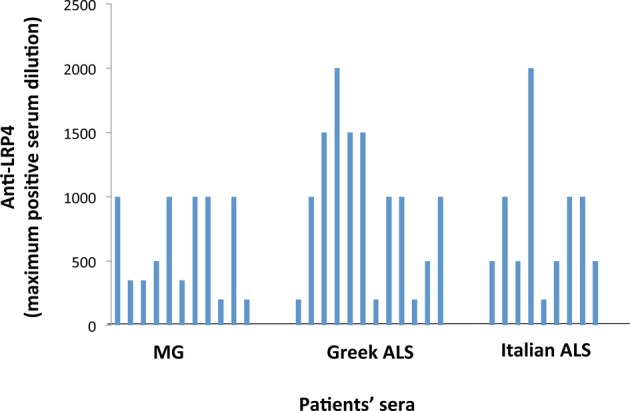
Comparison of the anti-LRP4 titers between ALS and MG anti-LRP4 positive sera. Twenty-one out of the 24 positive ALS sera were tested and compared with 11 randomly chosen LRP4-positive MG sera. Sera were tested by the CBA at dilutions 1/100, 1/200, 1/400, 1/500, 1/1000, 1/2000 and 1/4000. Ambiguous result in a certain dilution is presented as an intermediate titer (e.g., ambiguous result at 1/2000 is presented as titer 1/1500)
LRP4 antibodies were not detected in sera from 40 healthy individuals, four patients with familiar ALS, and 10 patients with other neurological diseases (OND), and were detected in only 4/84 (4.8%) multiple sclerosis (MS) patients and 1/40 (2.5%) AChR antibody-positive MG patients. None of the tested ALS sera had detectable antibodies to AChR, AChR clusters or MuSK (Table1).
LRP4 antibodies in the CSF of seropositive ALS patients
CSF samples from 68 ALS, MS, and OND patients were then analysed for LRP4 antibodies. CSF samples from 6/7 tested LRP4-ALS patients and one LRP4-seropositive MS patient were anti-LRP4-positive, while none of the 60 CSF from the LRP4-seronegative patients was positive (Table1). Comparison of serum and CSF from the same patient at the same IgG concentration (performed for three patients) showed that a roughly similar amount of IgG was required for a positive LRP4 antibody result (Fig. S3).
IgG subclass characterization of ALS anti-LRP4 antibodies
We then examined the IgG subclass of the LRP4 antibodies in sera from 17 LRP4-ALS and 13 LRP4-MG patients (Table S1). All contained LRP4 antibodies of the predominant complement activating IgG1 subclass. However, whereas most (11/13, 85%) LRP4-MG sera also contained IgG2 anti-LRP4 antibodies, only 4/17 (24%) ALS sera contained IgG2 anti-LRP4 antibodies used. Fifty percent of the LRP4-ALS and LRP4-MG sera contained IgG3 autoantibodies, while none contained detectable IgG4 autoantibodies (Table S1).
Clinical features of ALS patients positive or negative for LRP4 antibodies
We then compared the characteristics of the anti-LRP4-positive and anti-LRP4-negative ALS patients in each cohort (Table2). In both cohorts, LRP4 antibodies were detected both in the predominant upper and lower motor neuron disease variants; no significant differences in terms of clinical presentation were observed between the anti-LRP4-positive and anti-LRP4-negative groups. In the Greek cohort, LRP4 antibodies were more common in patients with a bulbar onset than in those with a spinal onset. In both cohorts, there were no significant differences between anti-LRP4-positive and anti-LRP4-negative patients in any other tested parameter.
Table 2.
Clinical data for the ALS patients used in the study
| Greek cohort |
Italian cohort |
Both cohorts |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Total | Anti-LRP4 antibody-positive | Anti-LRP4 antibody-negative | Total | Anti-LRP4 antibody-positive | Anti-LRP4 antibody-negative | Total | Anti-LRP4 antibody-positive | Anti-LRP4 antibody-negative | |
| Total no. of patients | 51 | 12 | 39 | 53 | 12 | 41 | 104 | 24 | 80 |
| % anti-LRP4 positive | 23.5 | 22.6 | 23.1 | ||||||
| Mean age at onset (years) | 60.8 | 67.4 | 58.6 | 53.8 | 54.6 | 53.6 | 57.2 | 61.0 | 56.0 |
| Mean disease duration (months) | 35.6 | 19.5 | 41.0 | 36.9 | 35.5 | 37.3 | 36.3 | 27.5 | 39.1 |
| ALSFRS-R scale | 36.6 | 42.1 | 34.4 | 35.1 | 34.0 | 35.4 | 35.8 | 38.1 | 34.9 |
| F/M ratio | 0.70 | 0.50 | 0.77 | 1.21 | 1.40 | 1.16 | 0.93 | 0.85 | 0.95 |
| Upper motor (predominant) | 20/3% (64.5%) | 6/10 (60.0%) | 14/21 (66.7%) | 3/39 (7.7%) | 1/9 (11.1%) | 2/30 (6.7%) | 23/70 (32.9%) | 7/19 (36.8%) | 16/51 (31.3%) |
| Lower motor (predominant) | 11/31 (35.5%) | 4/10 (40.0%) | 7/21 (33.3%) | 36/39 (92.3%) | 8/9 (88.9%) | 28/30 (93.3%) | 47/70 (69.3%) | 12/19 (66.7%) | 35/51 (70.4%) |
| Bulbar (onset) | 17/38 (44.7%) | 8/10 (80.0%) | 9/28 (32.1%) | 12/53 (22.6%) | 2/12 (16.7%) | 10/41 (24.4%) | 29/91 (31.9%) | 10/22 (45.5%) | 19/69 (27.5%) |
| Spinal (onset) | 21/38 (55.3%) | 2/10 (20.0%) | 19/28 (67.9%) | 41/53 (77.4%) | 10/12 (83.3%) | 31/41 (75.6%) | 62/91 (68.9%) | 12/22 (54.5%) | 50/72 (72.5%) |
| EMG | 7 in 1 seg | 2 in 1 seg | 5 in 1 seg | 7 in 1 seg | 2 in 1 seg | 5 in 1 seg | |||
| 13 in 2 seg | 8 in 2 seg | 5 in 2 seg | 4 in 2 seg | 11 in 3 seg | 4 in 2 seg | 17 in 2 seg | 8 in 2 seg | 9 in 2 seg | |
| 18 in 3 seg | 2 in 3 seg | 16 in 3 seg | 42 in 3 seg | 31 in 3 seg | 60 in 3 seg | 13 in 3 seg | 47 in 3 seg | ||
| 1 in 4 seg | 1 in 4 seg | 1 in 4 seg | 1 in 4 seg | ||||||
| El Escorial classification1 | 7 defin. | 7 defin. | 7 defin. | 7 defin. | |||||
| 30 prob. | 9 prob. | 21 prob. | 36 prob. | 10 prob. | 26 prob. | 66 prob. | 19 prob. | 47 prob. | |
| 7 pos. | 3 pos. | 4 pos. | 11 pos. | 1 pos. | 10 pos. | 18 pos. | 4 pos. | 14 pos. | |
ALSFRS-R, revised ALS functional rating scale; defin, definite; EMG, electromyography (data refer to the number of segments with EMG findings); pos, possible; prob. probable; seg, segments.
By the end of the study all possible ALS patients had become either probable or definite. The diagnosis of probable ALS, supported by laboratory data, based on revised El Escorial criteria, is practically equivalent to definite ALS.
Discussion
LRP4 antibodies are present in some MG patients and have been shown to inhibit AChR clustering in cell culture,14,16 strongly suggesting they may play a pathogenic role in LRP4-MG. Interestingly, we found that LRP4 autoantibodies were also present in 23.1% of sera from ALS patients and in 6/7 CSF samples from LRP4 antibody-seropositive ALS patients; in contrast, no antibodies to AChR, AChR clusters or MuSK were detected. Furthermore, LRP4 antibodies were present in 5/6 patients for ˜10 months after their first detection. Interestingly, changes in their autoantibody titer roughly correlated with changes in their clinical condition (stable condition upon decrease in antibody titer, considerable worsening upon increase in antibody titer). This interesting indication needs further evaluation in a larger LRP4-ALS population. The ratio of LRP4 antibodies to total IgG was roughly similar in the serum and CSF, suggesting that LRP4 antibodies are transported into the CSF through the blood-brain barrier, although a parallel moderate intrathecal LRP4 antibody production is not unlikely. LRP4 autoantibodies were almost absent in patients with MS, OND, and familial ALS.
The titers of LRP4 antibodies in ALS patient sera varied considerably between patients and were comparable with those of the MG patients and somewhat higher than their titer values (about 50% higher) (Fig.2). As the titers overlapped between the two diseases, we conclude that the anti-LRP4 titer does not determine the clinical outcome of the disease. Interestingly, on the basis of the incidence of ALS (˜1/100,000) and anti-AChR/anti-MuSK seronegative MG (˜1/300,000), we calculated that the incidence of LRP4-ALS may be three to four times higher than that of LRP4-MG. Yet, the LRP4-ALS patients did not cluster into any specific phenotype (Table2).
We compared the IgG subclasses of LRP4 antibodies in ALS and MG patients. In contrast to MuSK antibodies, which are of the non-complement binding IgG4 subclass, we did not detect IgG4 LRP4 autoantibodies whereas all LRP4-ALS sera contained mostly IgG1 LRP4 autoantibodies (Table S1). Interestingly, IgG2 LRP4 antibodies were found in 85% of the LRP4-MG sera, but only in 24% of LRP4-ALS sera. IgG1 antibodies can activate complement much more efficiently than IgG2 and this might be of pathogenic significance; in fact, recent data support a significant role of complement in ALS.2,4
Although LRP4-MG is much more frequent in women (F/M ratio ˜3/1; Zisimopoulou et al., unpubl. obs.), the gender ratio in the ALS-LRP4 patients was ˜1.00. This difference between LRP4-MG and LRP4-ALS, as well as the difference between the two groups in the occurrence of IgG2 autoantibodies, suggests different immune mechanisms for the production of LRP4 antibodies that could result in the production of antibodies with different functional characteristics, possibly responsible for the very different pathologies.
The identification of anti-LRP4 autoantibodies in several ALS patients offers a potentially useful biomarker for diagnostic purposes, but much work is needed to investigate the pathogenic potential of these antibodies. Shen et al.20 have very recently shown that mice and rabbits immunized with LRP4 exhibit MG symptoms. Elaborate animal studies are needed by active immunization with LRP4 and LRP4 fragments and by passive transfer of antibodies from immunized animals, MG and ALS LRP4-patients, injected both in the body and in the brain of the animals. Such kind of studies should prove the ALS-pathogenic potential of the LRP4 antibodies, if any, and identify any functional differences between MG and ALS LRP4-antibodies. The earliest changes in motor neurons of ALS patients seem to be at the nerve terminal ends.21 This may justify a possible pathogenic role of the anti-LRP4 antibodies, at least during the first few steps of ALS induction. Nevertheless, even if LRP4-antibodies play a pathogenic role in ALS, clearly they are not necessary for ALS induction in the majority of patients, as 77% of our ALS patients did not have detectable anti-LRP4 antibodies. This is not surprising as many studies have shown that ALS is an heterogeneous spectrum of diseases rather than a single disease with a single etiology.1
As LRP4 antibodies were also detected in the CSF (Table2), they might have an effect on both neuronal and muscle LRP4. Since animal LRP4 antibodies reduce the neuronal viability in cell culture,18 LRP4 autoantibodies with access to motor neurons could potentially cause motor neuron degeneration.
Although an autoimmune component in ALS has been suggested for many years,22–24 a critical role of autoimmunity in ALS is not widely accepted, partially due to the absence of a convincing effect of immunotherapy.25–27 Nevertheless, the timing of immunotherapy may be a critical issue in this context, as it is generally applied at advanced stages of ALS when tissue damage has become irreversible. In addition, a potential biomarker such as anti-LRP4, may help to identify a group of ALS patients in which immunotherapies may be applicable.
In conclusion, we have shown that LRP4 antibodies are not only present in MG patients and may be more broadly related to damage to LRP4 expressing tissues, such as motor neurons and the NMJ. Although their pathogenic role is still hypothetical, a direct involvement of the immune system in the neurodegenerative process of denervation in ALS may be envisaged due to the critical role of LRP4 in both NMJ and motor neurons, opening up new therapeutic approaches. Yet, even if LRP4 autoantibodies are simply secondary to unknown pathogenic mechanisms, they might serve as useful biomarkers for a subgroup of ALS patients.
Acknowledgments
This study was supported by the European Commission FP7 REGPOT 2010-1 NeuroSign grant and the Greek NSRF “Thalis” Autoimmunity grant. We thank L. Oros and K. Lazaridis for valuable discussions. The plasmids for the AChR subunits and for rapsyn were a gift from D. Beeson, Oxford University.
Conflict of Interest
S. Tzartos runs a diagnostics laboratory (Tzartos Neurodiagnostics) in Athens. R. Mantegazza is the recipient of the Annual Research Funding of the Italian Ministry of Health (RC2010/LR3 and RC2011/LR3) and a grant from Regione Lombardia (8598/Ricerca Indipendente 2009). All other authors declare no conflict of interest.
Funding Information
This study was supported by the European Commission FP7 REGPOT 2010-1 NeuroSign grant and the Greek NSRF “Thalis” Autoimmunity grant.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Supplementary Methods.
Figure S1. Measurement of anti-LRP4 antibodies in the sera of ALS patients and healthy controls (HC) by RIPA. LRP4 fragment, (amino acids 21–737; 42% of the LRP4 extracellular domain) was 125I-labeled and used in the RIPA. The two horizontal lines at about 2500 and 3000 cpm denote the cutoff values for ambiguous (average of controls plus 3SD) and positive (average of controls plus 4SD) result, respectively.
Figure S2. Persistence of LRP4 autoantibodies in the sera of LRP4-ALS patients. Sera from 6 LRP4-ALS patients were collected ˜10 months after the initial test. Original (first) and new (second) sera were then tested simultaneously at various dilutions (1/100–1/1600). The symbols show the maximum dilution of each serum found positive for LRP4 antibodies. Each line represents one patient and joins the titer of the first and second serum. It is shown that one patient became negative, in two the antibody titer was doubled, whereas in three the antibody titer remained stable.
Figure S3. Comparison of the LRP4 antibody content of serum and CSF from the same seropositive ALS patient. The serum and CSF samples from three LRP4 antibody-positive ALS patients were tested at similar IgG concentrations in the CBA for LRP4 antibodies. For patients 1 and 2 (and in another two patients tested), serum and CSF IgG were of roughly similar potency; in patient 3, CSF IgG were about five times less potent than serum IgG.
Table S1. IgG subclass characteristics of LRP4 antibodies from ALS and MG patients.
References
- 1.Turner MR, Hardiman O, Benatar M, et al. Controversies and priorities in amyotrophic lateral sclerosis. Lancet Neurol. 2013;12:310–322. doi: 10.1016/S1474-4422(13)70036-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hovden H, Frederiksen JL, Pedersen SW. Immune system alterations in amyotrophic lateral sclerosis. Acta Neurol Scand. 2013 doi: 10.1111/ane.12125. doi: 10.1111/ane.12125. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 3.Rentzos M, Rombos A, Nikolaou C, et al. Interleukin-17 and interleukin-23 are elevated in serum and cerebrospinal fluid of patients with ALS: a reflection of Th17 cells activation? Acta Neurol Scand. 2010;122:425–429. doi: 10.1111/j.1600-0404.2010.01333.x. [DOI] [PubMed] [Google Scholar]
- 4.Sta M, Sylva-Steenland RM, Casula M, et al. Innate and adaptive immunity in amyotrophic lateral sclerosis: evidence of complement activation. Neurobiol Dis. 2011;42:211–220. doi: 10.1016/j.nbd.2011.01.002. [DOI] [PubMed] [Google Scholar]
- 5.Pagani MR, Gonzalez LE, Uchitel OD. Autoimmunity in amyotrophic lateral sclerosis: past and present. Neurol Res Int. 2011;2011:497080. doi: 10.1155/2011/497080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ludolph AC. Motor neuron disease: urgently needed-biomarkers for amyotrophic lateral sclerosis. Nat Rev Neurol. 2011;7:13–14. doi: 10.1038/nrneurol.2010.196. [DOI] [PubMed] [Google Scholar]
- 7.Joyce NC, Carter GT. Electrodiagnosis in persons with amyotrophic lateral sclerosis. PM R. 2013 doi: 10.1016/j.pmrj.2013.03.020. doi: 10.1016/j.pmrj.2013.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mulder DW, Lambert EH, Eaton LM. Myasthenic syndrome in patients with amyotrophic lateral sclerosis. Neurology. 1959;9:627–631. doi: 10.1212/wnl.9.10.627. [DOI] [PubMed] [Google Scholar]
- 9.Weatherbee SD, Anderson KV, Niswander LA. LDL-receptor-related protein 4 is crucial for formation of the neuromuscular junction. Development. 2006;133:4993–5000. doi: 10.1242/dev.02696. [DOI] [PubMed] [Google Scholar]
- 10.Tian QB, Suzuki T, Yamauchi T, et al. Interaction of LDL receptor-related protein 4 (LRP4) with postsynaptic scaffold proteins via its C-terminal PDZ domain-binding motif, and its regulation by Ca/calmodulin-dependent protein kinase II. Eur J Neurosci. 2006;23:2864–2876. doi: 10.1111/j.1460-9568.2006.04846.x. [DOI] [PubMed] [Google Scholar]
- 11.Wu H, Lu Y, Shen C, et al. Distinct roles of muscle and motoneuron LRP4 in neuromuscular junction formation. Neuron. 2012;75:94–107. doi: 10.1016/j.neuron.2012.04.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yumoto N, Kim N, Burden SJ. Lrp4 is a retrograde signal for presynaptic differentiation at neuromuscular synapses. Nature. 2012;489:438–442. doi: 10.1038/nature11348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leite MI, Waters P, Vincent A. Diagnostic use of autoantibodies in myasthenia gravis. Autoimmunity. 2010;43:371–379. doi: 10.3109/08916930903541208. [DOI] [PubMed] [Google Scholar]
- 14.Higuchi O, Hamuro J, Motomura M, et al. Autoantibodies to low-density lipoprotein receptor-related protein 4 in myasthenia gravis. Ann Neurol. 2011;69:418–422. doi: 10.1002/ana.22312. [DOI] [PubMed] [Google Scholar]
- 15.Pevzner A, Schoser B, Peters K, et al. Anti-LRP4 autoantibodies in AChR- and MuSK-antibody-negative myasthenia gravis. J Neurol. 2012;259:427–435. doi: 10.1007/s00415-011-6194-7. [DOI] [PubMed] [Google Scholar]
- 16.Zhang B, Tzartos JS, Belimezi M, et al. Autoantibodies to lipoprotein-related protein 4 in patients with double-seronegative myasthenia gravis. Arch Neurol. 2012;69:445–451. doi: 10.1001/archneurol.2011.2393. [DOI] [PubMed] [Google Scholar]
- 17.Pun S, Santos AF, Saxena S, et al. Selective vulnerability and pruning of phasic motoneuron axons in motoneuron disease alleviated by CNTF. Nat Neurosci. 2006;9:408–419. doi: 10.1038/nn1653. [DOI] [PubMed] [Google Scholar]
- 18.Lu Y, Tian QB, Endo S, et al. A role for LRP4 in neuronal cell viability is related to apoE-binding. Brain Res. 2007;26:19–28. doi: 10.1016/j.brainres.2007.08.035. [DOI] [PubMed] [Google Scholar]
- 19.Leite MI, Jacob S, Viegas S, et al. IgG1 antibodies to acetylcholine receptors in ‘seronegative’ myasthenia gravis. Brain. 2008;131:1940–1952. doi: 10.1093/brain/awn092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shen C, Lu Y, Zhang B, et al. Antibodies against low-density lipoprotein receptor-related protein 4 induce myasthenia gravis. J Clin Invest. 2013;123:5190–5202. doi: 10.1172/JCI66039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fischer LR, Glass JD. Axonal degeneration in motor neuron disease. Neurodegener Dis. 2007;4:431–442. doi: 10.1159/000107704. [DOI] [PubMed] [Google Scholar]
- 22.Wolfgram F. Blind studies on the effect of amyotrophic lateral sclerosis sera on motor neurons in vitro. UCLA Forum Med Sci. 1976;(19):145–149. [PubMed] [Google Scholar]
- 23.Roisen FJ, Bartfeld H, Donnenfeld H, et al. Neuron specific in vitro cytotoxicity of sera from patients with amyotrophic lateral sclerosis. Muscle Nerve. 1982;5:48–53. doi: 10.1002/mus.880050109. [DOI] [PubMed] [Google Scholar]
- 24.Donnenfeld H, Kascsak RJ, Bartfeld H. Deposits of IgG and C3 in the spinal cord and motor cortex of ALS patients. J Neuroimmunol. 1984;6:51–57. doi: 10.1016/0165-5728(84)90042-0. [DOI] [PubMed] [Google Scholar]
- 25.Olarte MR, Schoenfeldt RS, McKiernan G, et al. Plasmapheresis in amyotrophic lateral sclerosis. Ann Neurol. 1980;8:644–645. doi: 10.1002/ana.410080625. [DOI] [PubMed] [Google Scholar]
- 26.Brown RH, Jr, Hauser SL, Harrington H, et al. Failure of immunosuppression with a ten- to 14-day course of high-dose intravenous cyclophosphamide to alter the progression of amyotrophic lateral sclerosis. Arch Neurol. 1986;43:383–384. doi: 10.1001/archneur.1986.00520040063021. [DOI] [PubMed] [Google Scholar]
- 27.Drachman DB, Chaudhry V, Cornblath D, et al. Trial of immunosuppression in amyotrophic lateral sclerosis using total lymphoid irradiation. Ann Neurol. 1994;35:142–150. doi: 10.1002/ana.410350205. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Methods.
Figure S1. Measurement of anti-LRP4 antibodies in the sera of ALS patients and healthy controls (HC) by RIPA. LRP4 fragment, (amino acids 21–737; 42% of the LRP4 extracellular domain) was 125I-labeled and used in the RIPA. The two horizontal lines at about 2500 and 3000 cpm denote the cutoff values for ambiguous (average of controls plus 3SD) and positive (average of controls plus 4SD) result, respectively.
Figure S2. Persistence of LRP4 autoantibodies in the sera of LRP4-ALS patients. Sera from 6 LRP4-ALS patients were collected ˜10 months after the initial test. Original (first) and new (second) sera were then tested simultaneously at various dilutions (1/100–1/1600). The symbols show the maximum dilution of each serum found positive for LRP4 antibodies. Each line represents one patient and joins the titer of the first and second serum. It is shown that one patient became negative, in two the antibody titer was doubled, whereas in three the antibody titer remained stable.
Figure S3. Comparison of the LRP4 antibody content of serum and CSF from the same seropositive ALS patient. The serum and CSF samples from three LRP4 antibody-positive ALS patients were tested at similar IgG concentrations in the CBA for LRP4 antibodies. For patients 1 and 2 (and in another two patients tested), serum and CSF IgG were of roughly similar potency; in patient 3, CSF IgG were about five times less potent than serum IgG.
Table S1. IgG subclass characteristics of LRP4 antibodies from ALS and MG patients.