

Abstract
There is substantial controversy regarding the causative role of amyloid β (Aβ) deposition in Alzheimer's disease (AD). The cerebrovasculature plays an important role in the elimination of Aβ from the brain and hypertension is a well-known risk factor for AD. In spontaneously hypertensive stroke-prone rats (SHRSP), an animal model of chronic arterial hypertension, cerebral small vessel disease (CSVD) leads to age-dependent parenchymal Aβ accumulation similar to that observed in AD. These data approve the neuropathological link between CSVD and AD, confirm the challenge that parenchymal Aβ deposition is a specific marker for AD and disclose the meaning of SHRSP as valid experimental model to investigate the association between hypertension, CSVD, and Aβ plaques.
Introduction
Accumulation of amyloid β (Aβ) in the brain parenchyma is one of the pathological hallmarks of Alzheimer's disease (AD). There is substantial controversy regarding the causative role of amyloid deposition in AD.1 In the majority of sporadic AD cases, Aβ accumulation is thought to result from the failure of Aβ clearance from the brain.2 The cerebrovasculature plays an important role in the elimination of Aβ from the brain, including receptor-mediated transport across the blood–brain barrier and perivascular drainage along vascular basement membranes and hypertension is a well-known risk factor for AD and Aβ deposition.3–5 We used the spontaneously hypertensive stroke-prone rats (SHRSP), an animal model of chronic arterial hypertension, to assess the role of cerebral small vessel disease (CSVD) in Aβ accumulation.6 Here we show that arterial hypertension and chronic small vessel disease lead to an age-dependent parenchymal Aβ accumulation similar to that observed in AD. These findings approve the neuropathological link between cerebrovascular diseases and AD and indicate that Aβ accumulation is a nonspecific marker of tissue damage that can be elicited by hypertension, small vessel disease and AD or rather by the combined effects of all.
Efficient clearance of Aβ requires healthy endothelial cell function and strong arterial pulsatility. Factors that affect vascular health, including age, hypercholesterolemia, and diabetes, are also risk factors for AD and have been shown to influence Aβ deposition in the brain.7 Hypertension is associated with CSVD and is a risk factor for vascular dementia (VaD).8 CSVD pathology is commonly observed in AD brains and systolic hypertension has been shown to be associated with greater rate of cognitive decline in AD patients.9 However, the link between hypertension, CSVD, and Aβ deposition remains undefined.
Experimental models of AD rely on the expression of rare human familial mutations and often develop Aβ pathology due to an overproduction of Aβ peptides, rather than a failure of endogenous clearance mechanisms.10 Furthermore, histopathologic findings characteristic for CSVD are missing in most AD models and inbred-provoked uniformity of animals does not adequately address the heterogeneity of human disease.11
In contrast to the broader assortment of experimental AD models, there are several available CSVD models generated by either (1) the induction of local and global hypoperfusion, (2) transgenic modifications leading to vasoconstrictions of cerebral arteries and small vessel wall damage, and by (3) provoked chronic hypertension.12 Hypoperfusion models lack adequate reproducibility of human CSVD as they result in (1) scattered infarcts in distinct vascular territories and (2) a pathophysiological state found in patients with advanced disease of larger cerebral arteries (e.g., atherosclerosis). Such changes are not necessarily associated with small vessel wall changes and do not include hypertension, one of the main risk factors for CSVD.
SHRSP, an animal model of chronic arterial hypertension, develop a broad spectrum of CSVD histopathology, including early microvascular dysfunction with subsequent blood–brain barrier breakdown.6 This results in degenerative small vessel wall changes with perivascular rupture and oozing bleeds and in many cases, reactive small vessel thrombosis with associated converging small infarcts all similar to that observed in hypertensive humans.13,14
However, whether SHRSP develop Aβ deposition resulting from their risk profile of chronic hypertension and CSVD has not been reported. To address this, we investigated Aβ pathology in the brains of 88 male SHRSP and 44 age-matched male Wistar rats using conventional histology including hematoxylin eosin (H&E) and Congo red staining and by immunohistochemistry using various anti-Aβ antibodies.
Material and Methods
Animals
All animal procedures were approved by the local Ethical Committee in accordance with the animal protection guidelines and European Communities Council guidelines (42502-2-1148 DZNE, 2-943 FAN). Male SHRSP (SHRSP, 12–18 weeks n = 18, 20–31 weeks n = 27, 32–36 weeks n = 20, 40–44 weeks n = 23) and 44 Wistar controls (12–18 weeks n = 8, 20–31 weeks n = 4, 32–36 weeks n = 12, 40–44 weeks n = 20) were obtained from Charles River Laboratories International Inc. (Wilmington, MA). Tg2576 mice were obtained from Dr. Wolfgang Härtig at the University of Leipzig (Leipzig, Germany) and FAD mice were obtained from Jackson Laboratory, Bar Harbor, ME.
Human tissue
Brain tissue sections from control and cases of AD were provided by Newcastle Brain Tissue resource (https://nbtr.ncl.ac.uk/) and ethical approval was granted in 2008 by the Newcastle and North Tyneside 1 Research Ethics Committee under the National Research Ethics Service (08/H0906/136).
Histology and immunocytochemistry
Rats and mice were transcardially perfused with phosphate-buffered saline (PBS) followed by 4% paraformaldehyde (PFA), brains were removed and sectioned (30 μm thickness). Rodent brain tissues were processed for hematoxylin eosin (H&E) and Congo red staining. For Aβ immunocytochemistry, brain sections were pretreated with either citrate buffer (65°C, 30 min) or formic acid (70–98%, 3–5 min), blocked with 10% donkey serum or 15% goat serum (Sigma, St. Louis, MO), and incubated overnight at 4°C with STL-FITC (Axxora, 1:500; Loerrach, Germany), anti-rodent-Aβ (Covance, 1:500; Dedham, MA) or anti-human-Aβ (Covance, clone 4G8, 1:500, DakoCytomation (Glostrup, Denmark), clone 6F3D, 1:500, Millipore (Billerica, MA), anti-Aβ 1–17 1:500). For fluorescent immunolabeling, sections were developed using Cy3-donkey-anti-rabbit-IgG (Dianova, 1:500; Hamburg, Germany) or Alexa Fluor 594-anti-mouse-IgG (Invitrogen, 1:200; Carlsbad, CA), and rat sections were stained with 4′,6-diamidino-2-phenylindole (DAPI) (20 min, room temperature). Enzyme-linked immunocytochemistry was developed using biotinylated anti-mouse-IgG (Vector Labs, 1:500; Burlingame, CA) with nickel-enhanced diaminobenzidine as chromogen. To verify staining specificity, tissues were incubated with Cy5-donkey-anti-rat-IgG (Dianova, 1:500) to detect IgG or with anti-human-Aβ (clone 4G8) that was preabsorbed with human Aβ40 (Cambridge Biosciences, Cambridge, U.K., 25 μmol/L).
Results and Discussion
Sixteen out of 88 SHRSP (18%) showed deposition of parenchymal Aβ plaques while cortical regions were affected in 69%, basal ganglia in 44%, hippocampus formation in 31% and thalamus and corpus callosum, respectively, in 6% of the Aβ positive animals (Figs.1, 2, S1). Aβ positive deposits did not colocalize with IgG or DAPI (Figs.1F, H, and I, 2B, F, G, and J), suggesting that the staining was not due to leakage of the blood–brain barrier or nonspecific cellular labeling. These deposits were morphologically comparable to parenchymal Aβ accumulations detectable in transgenic mouse models of AD and human AD (Fig.3) and could be demonstrated specifically using various anti-Aβ antibodies simultaneously applied to AD mice (Figs.1C, 2C and D, 3A and D–F). Age of the SHRSP had a significant effect on Aβ deposition with a nearly threefold increase in affected animals from about 9% in the younger groups (age 12–31 weeks, four of 45 SHRSP were affected by parenchymal Aβ plaques) to about 28% in the aged cohort (age 32–44 weeks, 12 of 43 SHRSP were affected by parenchymal Aβ plaques); P < 0.05, univariate analysis of variance. Thus, 12 (75%) out of the 16 SHRSP affected by parenchymal Aβ deposits were 32 weeks or even older with the highest prevalence of parenchymal Aβ positivity in the oldest age group (40–44 weeks, 9 (56%) out of all 16 SHRSP with Aβ plaques). Single deposition surrounded small vessel walls (Fig. S1A).
Figure 1.
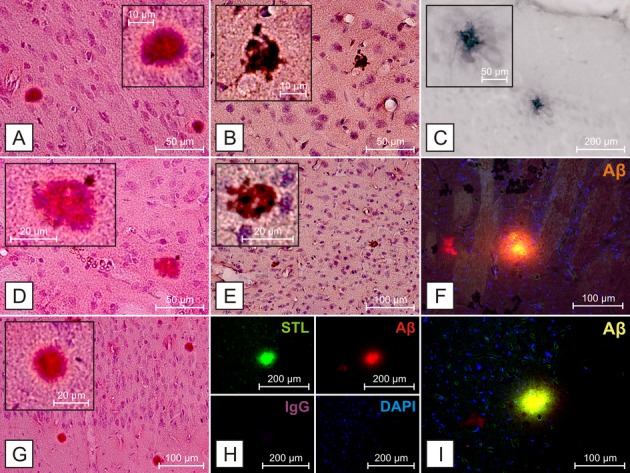
Aβ deposition in SHRSP 304, 26 weeks. Aβ deposition in cortical regions (A–C, E, G–I) and the basal ganglia (D, F) detectable using H&E (A, D, G) and Congo red (B, E) staining, as well as antibodies against Aβ (C) anti-human-Aβ clone 4G8, (F, H, I) anti-rodent-Aβ. Aβ-positive deposits do not colocalize with IgG (H) or DAPI (F, H, and I), suggesting that the staining is not due to leakage of the blood–brain barrier or nonspecific cellular labeling. Aβ deposits do colocalize with STL, as previously described19 (H). STL – Solanum tuberosum lectin, endothelial marker,20 DAPI – 4′,6-diamidino-2-phenylindole, for DNA detection.
Figure 2.
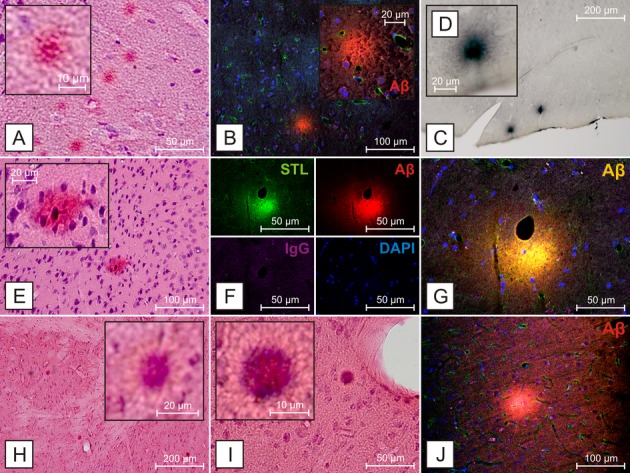
Aβ deposition in SHRSP of different age groups. Hippocampal (A), cortical (B–D, E, J) and striatal (F–I) Aβ deposition in SHRSP of different age groups. Again, Aβ-positive deposits do not colocalize with IgG (F) or DAPI (B, F, G, and J), suggesting that the staining is not due to leakage of the blood–brain barrier or nonspecific cellular labeling. (A–D) SHRSP 331, 40 weeks, (E–G), SHRSP 309, 44 weeks, (H–J), SHRSP 308, 28 weeks, (A, E, H, I) H&E staining, (B, F, G, J) anti-rodent-Aβ, (C and D) anti-human-Aβ 1–17. STL – Solanum tuberosum lectin, endothelial marker,20 DAPI – 4′,6-diamidino-2-phenylindole, for DNA detection.
Figure 3.
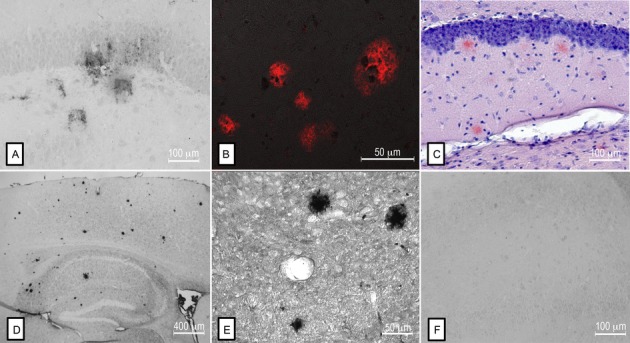
Comparison of Aβ deposition between SHRSP, Tg mouse models of AD, and human AD brains. Aβ-positive deposits in the hippocampus of a SHRSP (A) resemble those found in human AD brains (B). Similarly, plaques identified by Congo red staining (C) and by anti-Aβ immunohistochemistry (D, E) in mice overexpressing the Swedish human APP mutation, have a similar morphology to those identified in the SHRSP. Preabsorption of anti-Aβ (clone 4G8) with human Aβ40 attenuated the detection of Aβ staining in SHRSP (F).
Age-matched controls exhibited no parenchymal Aβ deposition (data not shown) and no staining was observed following preabsorption of the anti-Aβ antibody with purified Aβ peptides, confirming the specificity of the staining (Fig.3F).
Within the growing burden of dementia in the elderly AD is the most common and VaD a frequent subtype of dementia, respectively. Age and hypertension are among common risk factors for AD, VaD, and CSVD. Aging and hypertension are associated with morphological and functional changes in the cerebral vasculature, including increased arterial stiffness and tortuosity, abnormal inclusions, splitting, duplication and thickening of the basement membrane, and microvascular dysfunction, as found in chronic hypertensive SHRSP.15–17
Healthy maintenance of the neurovascular unit is important for transendothelial transport of Aβ from the brain parenchyma. Furthermore, the efficiency of perivascular drainage of Aβ correlated with the amplitude of arterial pulsations.18 Our present findings that endogenous parenchymal Aβ deposits in the brains of SHRSP, but not in aged-matched controls, suggests that hypertension accelerates the age-dependent failure of mechanisms that remove Aβ from the brain. These data provide additional support for a link between cerebrovascular health, AD, and parenchymal Aβ accumulation, challenge the notion that Aβ parenchymal deposition is a specific marker for AD and disclose the meaning of SHRSP as valid experimental model to investigate the association between hypertension, CSVD, and Aβ plaques.
Limitations and perspectives
Major limitations of this study include the lack of a broader method spectrum to prove parenchymal amyloid accumulations in SHRSP and, the absent ability of the used antibodies to distinguish between different forms of Aβ. However, promising data concerning elevated expressions of preliminary forms of fibrillar Aβ plaques in already young SHRSP are under progress.
Our ongoing work now focusses on the detailed understanding of the link between hypertension, age and a possible (1) failure of enzymatic amyloid degradation, (2) diminution of Aβ clearance across the BBB, and (3) along vascular basement membranes. Additionally, a closer look at the contribution of further vascular risk factors to that pathological interplay between small cerebrovascular damage, age, and parenchymal Aβ load is planned in SHRSP.
Acknowledgments
We thank Kathrin Baldauf, German Center for Neurodegenerative Diseases (DZNE), Magdeburg, Germany, for providing brain slices of FAD mice. This study was supported financially by a DZNE intersite project on vascular dementia. The funding body was not involved in the study design, the data collection, analysis, and interpretation. Human tissue for this study was provided by the Newcastle Brain Tissue Resource which is funded in part by a grant from the UK Medical Research Council (G0400074) and by Brains for Dementia research, a joint venture between Alzheimer's Society and Alzheimer's Research UK.
Author Contributions
C. B., C. H., C. G., R. C., and S. S. directed the study, designed experiments, analyzed data, and drafted the manuscript; J. A. performed neuropathological assessment of human tissue; S. V., J. A., and R. T. K. contributed to the manuscript writing and data formulation; H. J. H. and K. R. funded major parts of the study and contributed to manuscript writing. All authors read and approved the final manuscript.
Conflict of Interest
None declared.
Funding Information
This study was supported financially by a DZNE intersite project on vascular dementia. The funding body was not involved in the study design, the data collection, analysis, and interpretation. Human tissue for this study was provided by the Newcastle Brain Tissue Resource, which is funded in part by a grant from the UK Medical Research Council (G0400074) and by Brains for Dementia research, a joint venture between Alzheimer's Society and Alzheimer's Research UK.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Cortical Aβ deposition in aged SHRSP detected by anti-rodent-Aβ. (A) SHRSP 331, 40 weeks, (B, C) SHRSP K3R3, 44 weeks, (D) SHRSP K4R1, 44 weeks, (E) SHRSP 333, 40 weeks, (F) SHRSP K4R4, 44 weeks.
References
- 1.Gilbert BJ. The role of amyloid beta in the pathogenesis of Alzheimer's disease. J Clin Pathol. 2013;66:362–366. doi: 10.1136/jclinpath-2013-201515. [DOI] [PubMed] [Google Scholar]
- 2.Mawuenyega KG, Sigurdson W, Ovod V, et al. Decreased clearance of CNS beta-amyloid in Alzheimer's disease. Science. 2010;330:1774. doi: 10.1126/science.1197623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bell RD, Zlokovic BV. Neurovascular mechanisms and blood-brain barrier disorder in Alzheimer's disease. Acta Neuropathol. 2009;118:103–113. doi: 10.1007/s00401-009-0522-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carare RO, Hawkes CA, Jeffrey M, et al. Cerebral amyloid angiopathy, prion angiopathy, CADASIL and the spectrum of Protein Elimination-Failure Angiopathies (PEFA) in neurodegenerative disease with a focus on therapy. Neuropathol Appl Neurobiol. 2013;39:593–611. doi: 10.1111/nan.12042. [DOI] [PubMed] [Google Scholar]
- 5.Rodrigue KM, Rieck JR, Kennedy KM, et al. Risk factors for beta-amyloid deposition in healthy aging: vascular and genetic effects. JAMA Neurol. 2013;70:600–606. doi: 10.1001/jamaneurol.2013.1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schreiber S, Bueche CZ, Garz C, et al. Blood brain barrier breakdown as the starting point of cerebral small vessel disease? – New insights from a rat model. Exp Transl Stroke Med. 2013;5:4. doi: 10.1186/2040-7378-5-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Qiu C, Xu W, Fratiglioni L. Vascular and psychosocial factors in Alzheimer's disease: epidemiological evidence toward intervention. J Alzheimers Dis. 2010;20:689–697. doi: 10.3233/JAD-2010-091663. [DOI] [PubMed] [Google Scholar]
- 8.Sharp SI, Aarsland D, Day S, et al. Hypertension is a potential risk factor for vascular dementia: systematic review. Int J Geriatr Psychiatry. 2011;26:661–669. doi: 10.1002/gps.2572. [DOI] [PubMed] [Google Scholar]
- 9.Mielke MM, Rosenberg PB, Tschanz J, et al. Vascular factors predict rate of progression in Alzheimer disease. Neurology. 2007;69:1850–1858. doi: 10.1212/01.wnl.0000279520.59792.fe. [DOI] [PubMed] [Google Scholar]
- 10.Anand A, Banik A, Thakur K, et al. The animal models of dementia and Alzheimer's disease for pre-clinical testing and clinical translation. Curr Alzheimer Res. 2012;9:1010–1029. doi: 10.2174/156720512803569055. [DOI] [PubMed] [Google Scholar]
- 11.LaFerla FM, Green KN. Animal models of Alzheimer disease. Cold Spring Harb Perspect Med. 2012;2:1–13. doi: 10.1101/cshperspect.a006320. . Pii: a006320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jiwa NS, Garrard P, Hainsworth AH. Experimental models of vascular dementia and vascular cognitive impairment: a systematic review. J Neurochem. 2010;115:814–828. doi: 10.1111/j.1471-4159.2010.06958.x. [DOI] [PubMed] [Google Scholar]
- 13.Braun H, Schreiber S. Microbleeds in cerebral small vessel disease. Lancet Neurol. 2013;12:735–736. doi: 10.1016/S1474-4422(13)70148-0. [DOI] [PubMed] [Google Scholar]
- 14.Schreiber S, Bueche CZ, Garz C, et al. The pathologic cascade of cerebrovascular lesions in SHRSP: is erythrocyte accumulation an early phase? J Cereb Blood Flow Metab. 2012;32:278–290. doi: 10.1038/jcbfm.2011.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hunter JM, Kwan J, Malek-Ahmadi M, et al. Morphological and pathological evolution of the brain microcirculation in aging and Alzheimer's disease. PLoS One. 2012;7:e36893. doi: 10.1371/journal.pone.0036893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kalaria RN. Small vessel disease and Alzheimer's dementia: pathological considerations. Cerebrovasc Dis. 2002;13(Suppl. 2):48–52. doi: 10.1159/000049150. [DOI] [PubMed] [Google Scholar]
- 17.Wisniewski HM, Wegiel J, Wang KC, et al. Ultrastructural studies of the cells forming amyloid in the cortical vessel wall in Alzheimer's disease. Acta Neuropathol. 1992;84:117–127. doi: 10.1007/BF00311383. [DOI] [PubMed] [Google Scholar]
- 18.Schley D, Carare-Nnadi R, Please CP, et al. Mechanisms to explain the reverse perivascular transport of solutes out of the brain. J Theor Biol. 2006;238:962–974. doi: 10.1016/j.jtbi.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 19.Hartig W, Reichenbach A, Voigt C, et al. Triple fluorescence labelling of neuronal, glial and vascular markers revealing pathological alterations in various animal models. J Chem Neuroanat. 2009;37:128–138. doi: 10.1016/j.jchemneu.2008.10.003. [DOI] [PubMed] [Google Scholar]
- 20.Michalski D, Grosche J, Pelz J, et al. A novel quantification of blood-brain barrier damage and histochemical typing after embolic stroke in rats. Brain Res. 2010;1359:186–200. doi: 10.1016/j.brainres.2010.08.045. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Cortical Aβ deposition in aged SHRSP detected by anti-rodent-Aβ. (A) SHRSP 331, 40 weeks, (B, C) SHRSP K3R3, 44 weeks, (D) SHRSP K4R1, 44 weeks, (E) SHRSP 333, 40 weeks, (F) SHRSP K4R4, 44 weeks.