

Abstract
N-heterocyclic carbenes (NHC) have been extensively studied as organocatalysts and ligands for transition metals, but the successful integration of NHCs and late transition metals in cooperative catalysis remains an underexplored area. We have developed a cooperative palladium-catalyzed allylation of NHC-activated aldehydes to access a variety of 3-allyl dihydrocoumarin derivatives. Kinetic experiments support a cooperative pathway for this transformation.
Introduction
The integration of two distinct catalytic pathways in a single flask is a powerful strategy in chemical synthesis.1 Through independent activation of separate nucleophilic and electrophilic species, this synergistic approach makes possible previously inaccessible transformations and can improve existing chemical reactions. In particular, the fusion of transition metal and organocatalysis concepts has become a major research endeavor over the last decade.1 Although there has been remarkable progress in this area, the combination of N-heterocyclic carbenes (NHCs)2 with late transition metals remains underexplored, and more importantly, quite counterintuitive given the strong propensity for NHCs to bind to transition metals with high affinity. While cooperative catalysis with NHCs and Lewis or Brønsted acids has been shown to increase reactivity and afford products with unprecedented levels of selectivity3, the incorporation of NHCs with late transition metals (TMs) presents a considerable challenge due to the potential for the formation of stable NHC-TM complexes (Figure 1),4 which do not possess the desired catalytic activity.
Fig. 1.

NHC/Transition metal cooperative catalysis.
Plan
Given the background above, we were motivated by the opportunity to harness the unconventional reactivity of NHCs with the assistance of transition metals to effect new chemical transformations. A highly desired objective is the addition of NHC-bound nucleophiles to TM-activated electrophiles. As proof of a concept to this general approach, the combination of Pdcatalyzed allylations5 with α,β-unsaturated aldehydes under carbene catalysis conditions could facilitate rapid access to synthetically valuable products, specifically dihydrocoumarin derivatives. A number of methods for accessing dihydrocoumarins have been developed6 as this structural motif is prevalent in natural products and biologically relevant small molecules.7 However, few methods exist to prepare 3-allylated dihydrocoumarins,8 and surprisingly the direct allylation of dihydrocoumarins is un-explored.9 It is clear that new catalytic methods to directly access this important molecular scaffold are necessary to facilitate structural diversification and efficient assembly of bioactive compounds. Herein we report the development of the novel cooperative NHC/TM strategy for the synthesis of allylated dihydrocoumarin derivatives through allylation and subsequent acylation of aldehyde 1a. The activation of the α,β-unsaturated aldehyde moiety within 1a through NHC catalysis and capture of a cationic Pd[π-allyl]Ln complex in a cooperative fashion facilitates access to dihydrocoumarins 2a or 3a through an enolate or homoenolate pathway, respectively.
Results and discussion
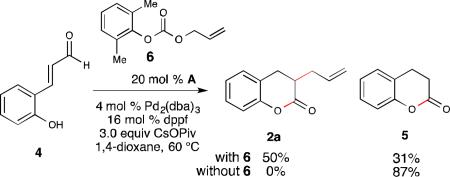
Initially, we investigated the intermolecular reaction of 4 with allyl carbonate 6 using our proposed cooperative NHC-Pd catalysis system (eq. 1). After extensive optimization, allylated dihydrocoumarin 2a could be obtained in 50% yield, but with dihydrocoumarin 5 as a competing side product in 31% yield. Further investigation showed that omitting 6 from the reaction mixture yielded the undesired dihydrocoumarin 5 in 87% yield. With the robustness of this competing pathway uncovered,6e an alternative strategy was deemed necessary to move forward with our reaction development.
 |
(1) |
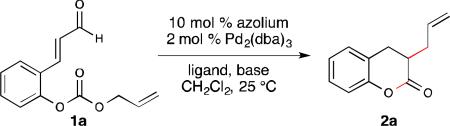
We hypothesized that the phenolic proton promotes the tautomerization of the NHC-enolate to the NHC-acyl adduct, which leads to the undesired dihydrocoumarin 5. To overcome this limititation, we envisoned using O-alloc aldehyde 1a to inhibit the formation of dihydrocoumarin 5. By masking the phenol with the allyl source, allylation of NHC-enolate intermediate would be the preferred pathway. With this hypothesis, we turned our focus toward utilizing aldehyde 1a in our cooperative catalysis system. Initial investigation with A (IMesCl) and DBU, followed by addition of a solution of Pd2(dba)3 and PPh3, provided enolate product 2a (14% yield by 1H NMR integration, Table 1, entry 1). The homoenolate adduct 3a (7% yield by 1H NMR integration, not shown) was also detected. In an effort to suppress formation of the homoenolate product (3a), a variety of bases were screened. Both Hünig's base and potassium carbonate gave solely the enolate product, with the latter resulting in a slightly higher yield (entry 2-3). Different NHC precatalysts were also evaluated, but only the NHC derived from A was found to be competent in this transformation (entry 4-6). Gratifyingly, the use of a large cone angle ligand, JohnPhos, (246°) led to an improved yield of 49% (entry 7).10
Table 1.
Optimization of Reaction Conditions.a
| ||||
|---|---|---|---|---|
| entry | azolium | ligand (mol %) | base (mol %) | yield (%)b |
| 1 | A | PPh3 (16) | DBU (20) | 14 |
| 2 | A | PPh3 (16) | i-Pr2NEt (20) | 24 |
| 3 | A | PPh3 (16) | K2CO3 (20) | 31 |
| 4 | B | PPh3 (16) | K2CO3 (20) | 5 |
| 5 | C | PPh3 (16) | K2CO3 (20) | 4 |
| 6 | D | PPh3 (16) | K2CO3 (20) | - |
| 7 | A | JohnPhos (16) | K2CO3 (20) | 49 |
| 8 | A | BINAP (8) | K2CO3 (20) | 19 |
| 9 | A | dppf (8) | K2CO3 (20) | 56 |
| 10c | A | dppf (8) | K2CO3 (10) | 61 |
Determined by 1H NMR integration (500 MHz, PhSiMe3 as an internal standard).
20 mol % azolium A.
We hypothesized that a bidentate ligand would coordinate strongly with palladium, thus minimizing Pd–NHC ligation. We initially explored the use of BINAP (natural bite angle11 = 92°), but were disappointed to obtain dihydrocoumarin 2a in a diminished 19% yield (entry 8). However, with dppf (natural bite angle = 99°) as the ligand, enolate product 2a was generated in 56% yield (entry 9). This result is consistent with observations that support the role of a larger natural bite angle in the heightened reactivity of the allyl moiety towards the enolate.12 There is a balance between angle and yield since DPE-Phos (natural bite angle = 131°) afford decreased yields (see ESI for details). Other Pd sources have been evaluated (such as [Pd(allyl)Cl]2 or Pd(OAc)2) with dppf as ligand, however reactions evaluated with these sources of Pd(0) afforded decreased yields of the desired product. Further evaluation of reaction solvents did not improve the yield of 2a, with polar solvents observed to depress reaction efficiency significantly (see ESI for details). Finally, upon modification of the stoichiometry of the azolium and base, the yield could be further improved to 61% (entry 10).
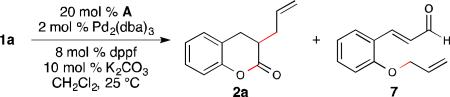
In control experiments, we observed that in the absence of NHC (A), aldehyde 7 was formed exclusively (Table 2, entry 2), while the omission of either dppf or Pd2(dba)3 from the reaction resulted in the recovery of the aldehyde starting material (Table 2, entry 3, 4). Surprisingly, when the reaction was conducted without an exogenous base, only a slight decrease in the yield of allylation product 2a was observed. Typically a base is required in NHC-catalyzed transformations to generate the active carbene, however, we hypothesize that in this case, the in situ generated phenoxide can serve this role competently (Table 2, entry 5).13
Table 2.
Evaluation of the Role of the Reaction Components.
| ||
|---|---|---|
| entry | variation of the standard conditions | resultsa |
| 1 | none | 61% yield 2a |
| 2 | no IMes (A) | 76% yield 7 |
| 3 | no Pd2(dba)3 | 69% recovered 1a |
| 4 | no ligand (dppf) | 62% recovered 1a |
| 5 | no base (K2CO3) | 56% yield 2a |
Yield determined by 1H NMR integration of the unpurified reaction.
The rapid production of aldehyde 7 under palladium catalysis conditions prompted us to investigate if 7 is a competent intermediate in the cooperative pathway. Thus, the exposure of 7 to the standard reaction conditions resulted in formation of dihydrocoumarin 2a, but in a significantly lower yield (31%). To further clarify if aldehyde 7 is a productive intermediate, the reaction was carried out under the standard conditions using GC-MS to monitor the presence of 1a, 7 and allylated adduct 2a (Fig. 2).14 This experiment indicated that the starting aldehyde was almost completely consumed after 1 hour, during which time aldehyde 7 accumulated and then was consumed. At prolonged reaction times (>100 min), the disappearance of aldehyde 7 corresponds approximately with the formation of desired product 2a, demonstrating that the allylation of the in situ generated phenoxide is faster than allylation of NHC-bound enolate.
Fig. 2.

Fate of aldehyde 7 during the reaction.
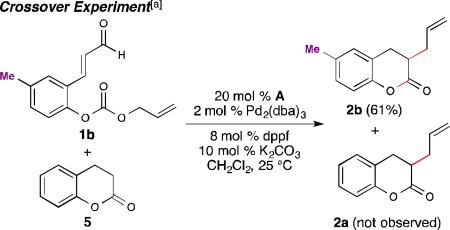
During our study, the formation of the undesired parent dihydrocoumarin 5 was detected in approximately 10% yield. This product could also serve as an intermediate en route to the desired product 2a through a serial process in which the NHC catalyzed unsubstituted coumarin formed first, followed by Pd-catalyzed allylation. Thus, dihydrocoumarin 5 was combined with aldehyde 1b under the standard reaction conditions (eq. 2). The exclusive formation of 2b and quantitative recovery of 5 discounts the serial pathway and fully supports a cooperative catalysis mechanism (vide infra).
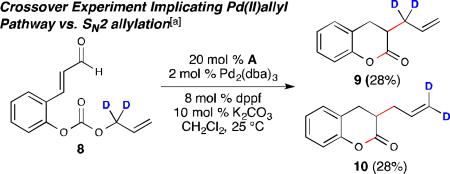
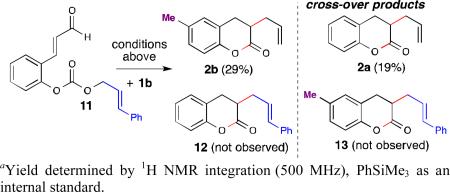
To support formation of the Pd[π-allyl]Ln complex, deuterated aldehyde 8 was subjected to the optimized reaction conditions. The resultant allylated products were obtained as a 1:1 mixture of regioisomers 9 and 10 (eq. 3). Moreover, when an equimolar amount of aldehyde 8 was mixed with 1b under the standard reaction conditions, cross-over products were observed (see ESI for details).15 These results, together with the information gained from the control experiments, support the initial formation of a Pd[π-allyl]Ln complex. In an effort to understand the kinetics of ion exchange within the reaction, aldehyde 11 was prepared and combined with 1b in a 1:1 ratio under the standard conditions (eq. 4). Only two products were obtained from this experiment: the non-cross-over product (2b) derived from aldehyde 1b in 29% yield and the crossover product (2a) from starting aldehyde 11 in 19% yield. The results from the cross-over experiment indicate that although ion exchange is rapid, the interaction between the phenoxide ion and Pd[π-allyl]Ln complex is important as the non-cross-over product is favored in the reaction.
 |
(2) |
 |
(3) |
 |
(4) |
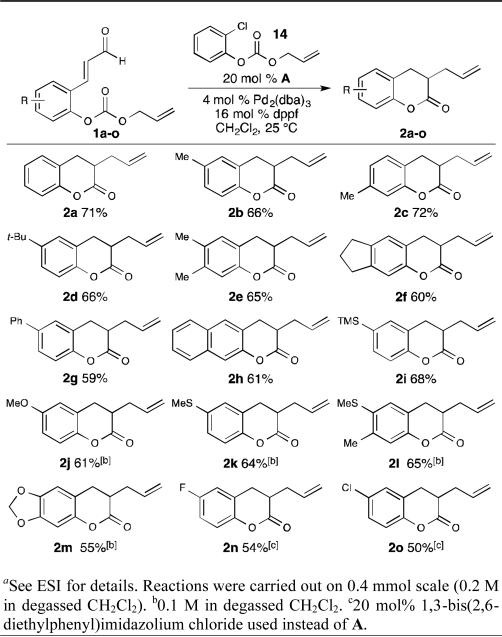
Based on the mechanistic investigations, we hypothesized that increasing the concentration of the allyl electrophile could improve the yield of the desired product (Table 3). Starting from carbonate 6, we systematically screened phenolic allyl carbonates arriving at carbonate 14 as the best allyl source additive. We hypothesize that the subtle electronic effect of the ortho-chloro substituent on the acidity of the phenol moiety plays a cruicial role in this result, but all experiments to probe this emperical observation have proven inconclusive to date. This modification, in combination with increasing the amount of palladium, afforded enolate allylation product 2a in an improved 71% isolated yield. With these optimized conditions, we explored the scope of this cooperative NHC/transition metal-catalyzed transformation. We found that the electronic character of the allylation precursor greatly affected the reaction outcome. We discovered that aldehydes with electron-donating groups provided the corresponding products in good yield (2a-2m). In certain cases, the yield of the allylated dihydrocoumarin could be increased by conducting the reaction at reduced concentration (2j-2m). We observed that aldehydes with electron-withdrawing groups on the phenol moiety provided lower yields with precatalyst A. One possible explanation is that weaker binding of the electron-deficient phenoxides to the Pd[π-allyl]Ln species leads to lower reactivity. For substrate aldehyde 1n, replacing the N-mesityl moieties of the imidazolium precatalyst with 2,6-diethylphenyl increased the yield of 2n slightly. This effect was also observed for chloro-substituted aldehyde 1o and was necessary to obtain 2o in moderate yield. This precatalyst effect was not observed for electron neutral aldehyde 1a. The substitution of the allyl moiety was also explored and found to be detrimental to reactivity. This limitation is similar to many reported transition metal-catalyzed allylation methods16 and is likely a result of unfavorable steric interactions. Investigations to understand this current restriction with our NHC/Pd platform are ongoing.
Table 3.
Substrate scope.a
|
Based on the mechanistic studies above, the current understanding of this allylation process is shown in Scheme 1. Since aldehyde 1a is rapidly consumed, we hypothesize that rate of palladium insertion is more facile than addition of the NHC to aldehyde 1a, resulting in formation of intermediate I or aldehyde 7 (formed Pd-allyl formation/allylation). Fortuitously, either aldehyde I or 7 can enter into the catalytic cycle. The NHC undergoes addition to either aldehyde I or 7 to generate extended Breslow intermediate IIA or IIB, respectively. At this point, NHC-bound homoenolate IIA can undergo either β-protonation to generate catalytic enol intermediate III, or allylation to arrive at an alternate catalytic enol to ultimately yield the undesired homoenolate product 3a. Alternatively, β-Protonation and palladium insertion of IIB can occur to afford the common enol intermediate III. The ionic interaction between the in situ generated phenoxide anion and cationic Pd[π-allyl]Ln species allows for pseudo-intramolecular allylation17 of the NHC-enol to generate acyl azolium IV, with concomitant regeneration of the Pd catalyst. Current data suggests that this ionic interaction is critical to achieve C-C bond formation over acylation of phenoxide V, which would instead furnish undesired dihydrocoumarin 5. Finally, intramolecular acylation of allylated acyl azolium IV affords the desired product 2a and regenerates the NHC catalyst.
Scheme 1.

Proposed Reaction Pathway.
Conclusions
In conclusion, a new cooperative NHC/TM catalysis approach has been developed. This proof of concept process involves the combination of an NHC-generated nucleophile with a TM-activated electrophile. Successful realization of this strategy required judicious choice of the reaction components to maintain the operability of the NHC and TM•ligand complex as separate, operative catalytic entities. Multiple control experiments provide strong evidence for the proposed cooperative catalysis pathway. These investigations also provided insight into the role of ion exchange in this transformation, prompting an increase in the concentration of the allyl electrophile and resulting in an improvement in efficiency. This system documents the feasibility of using NHCs and late TMs in a cooperative fashion and further exploration of this catalysis strategy involving N-heterocyclic carbenes and transition metals is underway.
Supplementary Material
Acknowledgements
This work has been supported by the National Institute of Health (R01-NIGMS). We thank Paul W. Siu (NU) for assistance with X-ray cyrstallography and Dr. Elizabeth O. McCusker for manuscript assistance.
Footnotes
† Electronic Supplementary Information (ESI) available: Experimental procedures and spectroscopic data for all new compounds. See DOI: 10.1039/b000000x/
Notes and references
- 1.a Jellerichs BG, Kong J-R, Krische MJ. J. Am. Chem. Soc. 2003;125:7758–7759. doi: 10.1021/ja0301469. [DOI] [PubMed] [Google Scholar]; b Lee JM, Na Y, Han H, Chang S. Chem. Soc. Rev. 2004;33:302–312. doi: 10.1039/b309033g. [DOI] [PubMed] [Google Scholar]; c Wasilke J-C, Obrey SJ, Baker RT, Bazan GC. Chem. Rev. 2005;105:1001–1020. doi: 10.1021/cr020018n. [DOI] [PubMed] [Google Scholar]; d Shao Z, Zhang H. Chem. Soc. Rev. 2009;38:2745–2755. doi: 10.1039/b901258n. [DOI] [PubMed] [Google Scholar]; e Allen AE, MacMillan DWC. Chem. Sci. 2012;3:633–658. doi: 10.1039/C2SC00907B. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Du Z, Shao Z. Chem. Soc. Rev. 2013;42:1337–1378. doi: 10.1039/c2cs35258c. [DOI] [PubMed] [Google Scholar]; g Tao Z-L, Zhang W-Q, Chen D-F, Adele A, Gong L-Z. J. Am. Chem. Soc. 2013;135:9255–9258. doi: 10.1021/ja402740q. [DOI] [PubMed] [Google Scholar]; h Ma G, Afewerki S, Deiana L, Palo-Nieto C, Liu L, Sun J, Ibrahem I, Córdova A. Angew. Chem. Int. Ed. 2013;52:6050–6054. doi: 10.1002/anie.201300559. [DOI] [PubMed] [Google Scholar]; i Tang W, Johnston S, Iggo JA, Berry NG, Phelan M, Lian L, Bacsa J, Xiao J. Angew. Chem. Int. Ed. 2013;52:1668–1672. doi: 10.1002/anie.201208774. [DOI] [PubMed] [Google Scholar]; j Hatano M, Horibe T, Ishihara K. Angew. Chem. Int. Ed. 2013;52:4549–4553. doi: 10.1002/anie.201300938. [DOI] [PubMed] [Google Scholar]; k Krautwald S, Sarlah D, Schafroth MA, Carreira EM. Science. 2013;340:1065–1068. doi: 10.1126/science.1237068. [DOI] [PubMed] [Google Scholar]
- 2.a Enders D, Niemeier O, Henseler A. Chem. Rev. 2007;107:5606–5655. doi: 10.1021/cr068372z. [DOI] [PubMed] [Google Scholar]; b Philips EM, Chan A, Scheidt KA. Aldrichimica Acata. 2009;42:55–66. [PMC free article] [PubMed] [Google Scholar]; c Nair V, Menon RS, Biju AT, Sinu CR, Paul RR, Jose A, Sreekumar V. Chem. Soc. Rev. 2011;40:5336–5346. doi: 10.1039/c1cs15139h. [DOI] [PubMed] [Google Scholar]; d Bugaut X, Glorius F. Chem. Soc. Rev. 2012;41:3511–3522. doi: 10.1039/c2cs15333e. [DOI] [PubMed] [Google Scholar]
- 3.a Zhao X, DiRocco DA, Rovis T. J. Am. Chem. Soc. 2011;133:12466–12469. doi: 10.1021/ja205714g. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Cohen DT, Scheidt KA. Chem. Sci. 2012;3:53–57. doi: 10.1039/C1SC00621E. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Dugal-Tessier J, O'Bryan EA, Schroeder TBH, Cohen DT, Scheidt KA. Angew. Chem. Int. Ed. 2012;51:4963–4967. doi: 10.1002/anie.201201643. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Mo J, Chen X, Chi YR. J. Am. Chem. Soc. 2012;134:8810–8813. doi: 10.1021/ja303618z. [DOI] [PubMed] [Google Scholar]; e ElSohly AM, Wespe DA, Poore TJ, Snyder SA. Angew. Chem. Int. Ed. 2013;52:5789–5794. doi: 10.1002/anie.201301849. [DOI] [PubMed] [Google Scholar]
- 4.a Nemoto T, Fukuda T, Hamada Y. Tetrahedron Lett. 2006;47:4365–4368. [Google Scholar]; b Lebeuf R. l., Hirano K, Glorius F. Org. Lett. 2008;10:4243–4246. doi: 10.1021/ol801644f. [DOI] [PubMed] [Google Scholar]; c Díez-González S, Marion N, Nolan SP. Chem. Rev. 2009;109:3612–3676. doi: 10.1021/cr900074m. [DOI] [PubMed] [Google Scholar]; d Chen Z, Yu X, Wu J. Chem. Comm. 2010;46:6356–6358. doi: 10.1039/c0cc01207f. [DOI] [PubMed] [Google Scholar]; e Rosa J. o. N., Reddy RS, Candeias NR, Cal PMSD, Gois PMP. Org. Lett. 2010;12:2686–2689. doi: 10.1021/ol100302e. [DOI] [PubMed] [Google Scholar]; f Adamo MFA, Bellini G, Suresh S. Tetrahedron. 2011;67:5784–5788. [Google Scholar]; g Reddy RS, Rosa JN, Veiros LF, Caddick S, Gois PMP. Org. Biomol. Chem. 2011;9:3126–3129. doi: 10.1039/c1ob05151b. [DOI] [PubMed] [Google Scholar]; h Nelson DJ, Nolan SP. Chem. Soc. Rev. 2013 doi: 10.1039/c3cs60146c. [DOI] [PubMed] [Google Scholar]; i Zhao J, Mück-Lichtenfeld C, Studer A. Adv. Synth. Catal. 2013;355:1098–1106. [Google Scholar]
- 5.a Trost BM, Crawley ML. Chem. Rev. 2003;103:2921–2944. doi: 10.1021/cr020027w. [DOI] [PubMed] [Google Scholar]; b Mohr JT, Stoltz BM. Chem. Asian J. 2007;2:1476–1491. doi: 10.1002/asia.200700183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.a Trost BM, Toste FD, Greenman K. J. Am. Chem. Soc. 2003;125:4518–4526. doi: 10.1021/ja0286573. [DOI] [PubMed] [Google Scholar]; b Matsuda T, Shigeno M, Murakami M. J. Am. Chem. Soc. 2007;129:12086–12087. doi: 10.1021/ja075141g. [DOI] [PubMed] [Google Scholar]; c Phillips EM, Wadamoto M, Roth HS, Ott AW, Scheidt KA. Org. Lett. 2008;11:105–108. doi: 10.1021/ol802448c. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Alden-Danforth E, Scerba MT, Lectka T. Org. Lett. 2008;10:4951–4953. doi: 10.1021/ol802029e. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Zeitler K, Rose CA. J. Org. Chem. 2009;74:1759–1762. doi: 10.1021/jo802285r. [DOI] [PubMed] [Google Scholar]; f Kim H, Yun J. Adv. Synth. Catal. 2010;352:1881–1885. [Google Scholar]; g Lu D, Li Y, Gong Y. J. Org. Chem. 2010;75:6900–6907. doi: 10.1021/jo101446d. [DOI] [PubMed] [Google Scholar]; h Park JO, Youn SW. Org. Lett. 2010;12:2258–2261. doi: 10.1021/ol100610v. [DOI] [PubMed] [Google Scholar]; i Hong B-C, Kotame P, Lee G-H. Org. Lett. 2011;13:5758–5761. doi: 10.1021/ol202331j. [DOI] [PubMed] [Google Scholar]; j Kuang Y, Liu X, Chang L, Wang M, Lin L, Feng X. Org. Lett. 2011;13:3814–3817. doi: 10.1021/ol201312y. [DOI] [PubMed] [Google Scholar]; k Jacobsen CB, Albrecht Ł, Udmark J, Jørgensen KA. Org. Lett. 2012;14:5526–5529. doi: 10.1021/ol302627u. [DOI] [PubMed] [Google Scholar]
- 7.Murray RDH, Mendez J, Brown SA. The Natural Coumarins: Occurrence, Chemistry And Biochemistry. Wiley; New York: 1982. [Google Scholar]
- 8.a Patra A, Misra SK. Magn. Rson. Chem. 1991;29:749–752. [Google Scholar]; b Moriarty RM, Epa WR, Prakash O. J. Chem. Res. (S) 1997:262–265. [Google Scholar]; c Murakata M, Jono T, Shoji T, Moriya A, Shirai Y. Tetrahedron Asymmetry. 2008:2479–2483. [Google Scholar]
-
9.Murakata M, Jono T, Mizuno Y, Hoshino O. J. Am. Chem. Soc. 1997;119:11713–11714. Using the reported method for alkylation of dihydrocoumarin reported by Hoshino et al., we observed complete consumption of starting material, but 2a could only be obtained in <20 % isolated yield after chromatography (multiple attempts).
- 10.a C. A. Tolman Chem. Rev. 1977;77:313–348. [Google Scholar]; b Rousseaux S, Davi M, Sofack-Kreutzer J, Pierre C, Kefalidis CE, Clot E, Fagnou K, Baudoin O. J. Am. Chem. Soc. 2010;132:10706–10716. doi: 10.1021/ja1048847. [DOI] [PubMed] [Google Scholar]; c Aranyos A, Old DW, Kiyomori A, Wolfe JP, Sadighi JP, Buclhwald SL. J. Am. Chem. Soc. 1999;121:4369–4378. [Google Scholar]
- 11.a Casey CP, Whiteker GT. Israel J. Chem. 1990;30:299–304. [Google Scholar]; b van Leeuwen PWNM, Kamer PCJ, Reek JNH. Pure Appl. Chem. 1999;71:1443–1452. [Google Scholar]
- 12.van Haaren RJ, Oevering H, Coussens BB, van Strijdonck GPF, Reek JNH, Kamer PCJ, van Leeuwen PWNM. Eur. J. Inorg. Chem. 1999;1999:1237–1241. [Google Scholar]
- 13.Estimated pKa of phenol in water is about 10 vs estimated pKa of NHC A is 20.8. For reference see: Kawanaka Y, Phillips EM, Scheidt KA. J. Am. Chem. Soc. 2009;131:18028–18029. doi: 10.1021/ja9094044.Higgins EM, Sherwood JA, Lindsay AG, Armstrong J, Massey RS, Alder RW, O'Donoghue AC. Chem. Comm. 2011;47:1559–1561. doi: 10.1039/c0cc03367g.Massey RS, Collett CJ, Lindsay AG, Smith AD, O'Donoghue AC. J. Am. Chem. Soc. 2012;134:20421–20432. doi: 10.1021/ja308420c.
- 14.Yield and conversion were determined by GC-MS of the unpurified reaction mixture with dodecane as the internal standard.
- 15.Trost BM, Xu J, Schmidt TJ. Am. Chem. Soc. 2009;131:18343–18357. doi: 10.1021/ja9053948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.a Behenna DC, Stoltz BM. J. Am. Chem. Soc. 2004;126:15044–15045. doi: 10.1021/ja044812x. [DOI] [PubMed] [Google Scholar]; b Trost BM, Xu J. J. Am. Chem. Soc. 2005;127:2846–2847. doi: 10.1021/ja043472c. [DOI] [PubMed] [Google Scholar]; c Trost BM, Bream RN, Xu J. Angew. Chem. Int. Ed. 2006;45:3109–3112. doi: 10.1002/anie.200504421. [DOI] [PubMed] [Google Scholar]; d Trost BM, Xu J, Reichle M. J. Am. Chem. Soc. 2007;129:282–283. doi: 10.1021/ja067342a. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Behenna DC, Liu Y, Yurino T, Kim J, White DE, Virgil SC, Stoltz BM. Nat. Chem. 2012;4:130–133. doi: 10.1038/nchem.1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.This process can be described as a pseudo-intramolecular allylation due to the presence of proposed intermediate III, which involves an ion pair. However, since ion exchange can occur, this is not a classic intramolecular allylation reaction. However, an alternative mechanism would be more inner-sphere with coordination of the NHC enolate to the pi-allyl Pd species. This assembly would provide a more intramolecular pathway. For a relevant detailed study of enolate/pi allyl Pd coordination, see: Keith JA, Behenna DC, Sherden N, Mohr JT, Ma S, Marinescu SC, Nielsen RJ, Oxgaard J, Stoltz BM, Goddard WA. J. Am. Chem. Soc. 2012;134:19050–19060. doi: 10.1021/ja306860n. and references cited therein.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.