

Abstract
Streptococcus agalactiae (Group B streptococcus, GBS) is highly adapted to humans, where it is a normal constituent of the intestinal and vaginal flora. Yet, GBS has highly invasive potential and causes excessive inflammation, sepsis, and death at the beginning of life, in the elderly and in diabetic patients. Thus, GBS is a model pathobiont that thrives in the healthy host, but has not lost its potential virulence during coevolution with mankind. It remains incompletely understood how the innate immune system contains GBS in the natural niches, the intestinal and genital tracts, and which molecular events underlie breakdown of mucocutaneous resistance. Newborn infants between days 7 and 90 of life are at risk of a particularly striking sepsis manifestation (late-onset disease), where the transition from colonization to invasion and dissemination, and thus from health to severe sepsis is typically fulminant and not predictable. The great majority of late-onset sepsis cases are caused by one clone, GBS ST17, which expresses HvgA as a signature virulence factor and adhesin. In mice, HvgA promotes the crossing of both the mucosal and the blood–brain barrier. Expression levels of HvgA and other GBS virulence factors, such as pili and toxins, are regulated by the upstream two-component control system CovR/S. This in turn is modulated by acidic epithelial pH, high glucose levels, and during the passage through the mouse intestine. After invasion, GBS has the ability to subvert innate immunity by mechanisms like glycerinaldehyde-3-phosphate-dehydrogenase-dependent induction of IL-10 and β-protein binding to the inhibitory phagocyte receptors sialic acid binding immunoglobulin-like lectin 5 and 14. On the host side, sensing of GBS nucleic acids and lipopeptides by both Toll-like receptors and the inflammasome appears to be critical for host resistance against GBS. Yet, comprehensive models on the interplay between GBS and human immune cells at the colonizing site are just emerging.
Keywords: S. agalactiae, cellular innate immunity, intestinal microbiota, colonization, invasion, sepsis
Introduction
Streptococcus agalactiae, Group B Streptococcus (GBS), is a commensal of the human intestinal and vaginal tract in 15–30% of healthy adults, but remains one of the most important invasive pathogens in newborn infants and the elderly (1–4). Every 10th neonate acquires GBS vertically during passage through the birth canal or shortly thereafter. In most cases, GBS can be assumed to become a normal constituent of the child’s microbiome. In other cases, other colonizing bacteria that expand on the expense of GBS may replace it. These dynamic processes early in life are generally safe, as 99% of at least temporarily colonized infants will never develop invasive GBS disease (5–8). The achievement of establishing a microbiome including GBS can best be judged in light of GBS late-onset diseases (LOD) and meningitis: if GBS does not adapt, it may cause fulminant disease. Notably, crossing the intestinal barrier and the blood barrier seems to be mechanistically linked. GBS serotype III, a particularly frequent isolate in neonatal meningitis, has recently been found to exhibit specific neurotropism through expression of the adhesion factor HvgA (9, 10). HvgA efficiently supports bacterial adhesion and transfer through to the intestinal wall and later across the blood–brain barrier, specifically the vascular endothelium and the choroid plexus (9). In other words, protection of the neonatal brain from GBS starts in the gut. Long-term neurological impairment of variable degree, which affects about 35–50% of infants surviving meningitis, has become particularly important in the Western world, where improvements in intensive care have decreased lethality of GBS invasive disease below 10% (9, 11).
By definition, GBS is a normal constituent of the “intestinal microbiota,” which comprises numerous bacteria, fungi, and protozoa. In a normal adult, the microbiota comprises more than 10,000 species, adding up to 1500 g in biomass. The composition of the microbiota is unique to the host and can be viewed as a personal fingerprint that emerges in early infancy (12, 13). Members of the bacterial phyla Bacteroidetes, Firmicutes (e.g., Lactobacillus and Clostridum spp), Proteobacteria, Actinobacteria (e.g., Bifidobacterium spp.), Fusobacteria, and Spirochaetes are the most important constituents of the gastrointestinal flora (12, 14, 15). Notably, the microbial composition underlies temporal changes during the first year of life and differs between children born vaginally and by caesarian section (16–18). A modulating role of breast milk feeding in the composition of the neonatal microbiome, e.g., by maternal secretory IgA, antimicrobial peptides, lactoferrin, and sCD14, has been shown by several investigators (19–25). In twins, it appears that both genetic and environmental factors contribute to the composition of the intestinal microbiota (22, 26, 27).
Colonizers of the human intestine are generally considered as “symbionts” that stabilize the intestinal homeostasis by acidification of the intraluminal milieu, maintenance of the transepithelial resistance, prevention of pathogen adherence, and continuous immune stimulation (28–32). Pathobionts are potentially pathogenic colonizers that usually reside in the intestine in coexistence with the host, but can cause severe local or systemic disease. In newborn infants, the most important examples are E. coli, enterococci, and GBS. The Janus face of GBS is underlined by the fact that 10% of all neonates are at least temporarily colonized by GBS (1–4), yet only 1% of these develops invasive disease (5–8). Beginning at birth, GBS colonization rates continuously increase to 20–30% in adults (1–4). This indicates that GBS transmission occurs not only perinatally, but also horizontally later in life. Next to the exposure to various bacterial strains, external factors like stress, drugs, diet, gastrointestinal infections, and endogenous factors, like diabetes, alter the composition of the intestinal microbiota (21, 33). These individual life style and health factors may explain in part, why GBS is an important cause of soft-tissue and urinary tract infections, arthritis, and sepsis in patients >65 years and those with chronic diseases such as stroke (OR 3.5), diabetes (OR 3.0), kidney or liver (OR 9.7) failure, and cancer (breast cancer OR 4.0) (34). In healthy adults, GBS predominantly colonizes the outer mucus layer of the colon, yet may occasionally reside in the small intestine as well (35, 36). In pregnant women, GBS is a frequent cause of urinary and upper genital tract infections, intra-amniotic infections, and sepsis (37, 38). Whether GBS colonization usually, i.e., in infants as well as adults, starts in the intestinal tract and expands from there to other sites, or whether GBS subsets develop at independent colonization sites of the same human influenced by the site-specific microenvironment, has not yet been clarified. Accordingly, the origin of differences in serotype distribution between non-pregnant adults with invasive disease, where serotype V contributes to almost 30% of cases, and pregnant women or newborn infants, where serotypes III and Ia predominate, is not well understood (7, 39, 40).
The Pathogen Site: GBS Virulence Strategies to Progress from Colonization to Disease
What can we learn from neonatal GBS sepsis models with respect to factors regulating colonization and invasion? GBS sepsis in newborn infants can be divided into early-onset disease (EOD), which occurs within the first week of life by vertical GBS transmission during delivery, and LOD, which occurs through vertical or horizontal transfer and manifests between day 7 and 3 months of age (41). However, since more than 90% of all EOD cases manifest as sepsis, pneumonia, or meningitis within the first 24–48 h of life, some authors limit early-onset sepsis to the first 72 h after birth (40, 42). Peripartum antibiotic prophylaxis has markedly decreased the incidence and case fatality rate of EOD; the incidence or clinical course of LOD, however, has remained largely unaffected. The clinical picture of LOD typically manifests as hyperinflammatory syndrome with meningitis. As outlined above, GBS serotype III is greatly overrepresented in LOD, with clonotype ST17 contributing up to 90% of all clinical isolates found in meningitis (as compared to colonizing strains). This led to the phenotypic description of GBS III ST17 as hypervirulent clone (43–47). A recently published longitudinal study found that in LOD, GBS and other bacteria isolated from the blood of preterm infants genetically matched clones previously found in the patients’ stool (48). Accordingly, in many cases, LOD likely results from systemic spread of intestinal GBS, and not directly from vertical or horizontal transmission. Nevertheless, postnatal exposure with maternal GBS III ST17, e.g., via breast milk, appears to underlie some sepsis cases (49).
For the development of sepsis by GBS spreading from the intestine, the bacterium has to undertake three consecutive steps: (i) colonization of the colon and potentially the small intestine, (ii) translocation across the intestinal epithelium, and (iii) immune evasion preventing GBS clearance from the blood stream (summarized in Table 1).
Table 1.
GBS virulence factors and their role in transition from colonization to invasive disease.
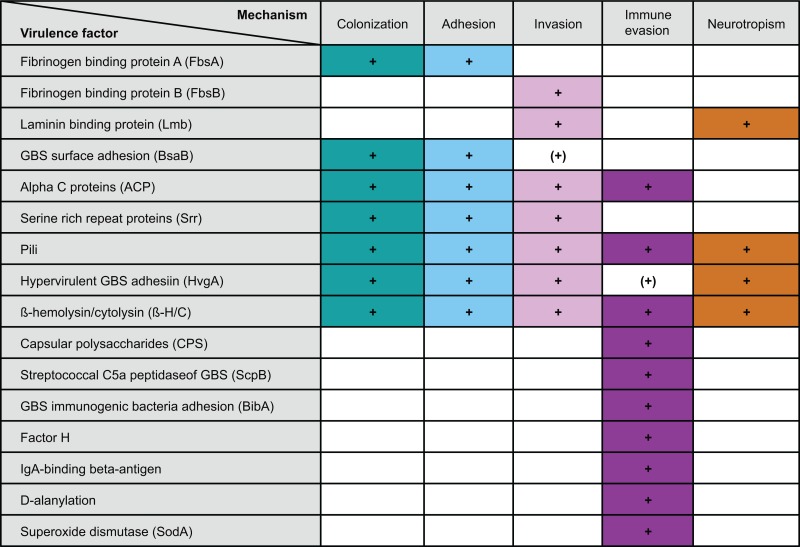 |
The first and pivotal step in GBS colonization is adhesion. Adhesion factors are expressed on the bacterial surface and allow GBS to bind to extracellular matrix proteins and epithelial cells of the colon and the genital tract resulting in biofilm formation (50, 51). Adhesion factors can additionally promote invasion, either by disruption of the epithelial cell layer or by modulation of the epithelial cytoskeleton and the junctional protein assembly, which in turn allows for paracellular translocation (52–55).
Two adhesion factors mediate attachment to the extracellular matrix. They are named according to their specific ligand: (i) fibrinogen-binding proteins and (ii) laminin-binding proteins (Lmb). While fibrinogen-binding protein A (FbsA) promotes adhesion, fibrinogen-binding-protein B (FbsB) mediates GBS invasion into host cells. FbsA and FbsB bind to both immobilized and soluble fibrinogen (56, 57). Bacterial attachment to extracellular matrix via Lmb seems important for GBS translocation across the intestinal epithelium and the blood–brain barrier (58, 59). Furthermore, the GBS surface adhesin BsaB binds to fibronectin (60). However, whether BsaB contributes to GBS invasion or mainly promotes colonization with GBS via its biofilm enhancing effects has not been fully resolved, yet.
Another group of GBS adhesion factors is characterized by the highly conserved LPxTG (Leu–Pro–X–Thr–Gly) motif at the C-terminus (61). GBS LPxTG is cleaved between Thr and Gly by the transpeptidase Sortase A, which covalently binds GBS to the cell wall and thus promotes both colonization and invasion (62). The first group of the LPxTG containing adhesion factors is the family of Alpha C proteins, which are encoded by the bca (group B, C protein alpha) gene and expressed on most strains of serotype Ia, Ib, and II (63). Alpha C proteins are further characterized by long tandem repeating elements, which allow antigenic variations, and a conserved N-terminal domain, encoding 185 amino acids (61, 64–67). GBS isolated from mothers who recently delivered a child with invasive GBS disease show increased expression of alpha C protein tandem repeats (“aa”). This is associated with increased susceptibility to opsonophagocytic antibody-mediated killing as compared to the GBS isolated previously from the respective newborn infant with sepsis (67). In line with these observations, low tandem repeat expression during infections seems to impair the specific antibody response and antibody-mediated killing (61, 64, 65, 68, 69). Furthermore, GBS alpha C proteins can promote invasion of human epithelial cells via α1β1-integrin binding (66).
The second group of LPxTG containing adhesion factors are serine-rich repeat (Srr) proteins, which interact with human keratin (Srr-1) and which enhance GBS virulence in mice (Srr-2) (70). Only recently, fibrinogen has been identified as Srr binding partner (71). Interestingly, while highly virulent serotype III strains express Srr-2 (70, 71), other GBS strains express Srr-1 (serotype Ia, Ib, Ic, II, and V).
Additional structures involved in adhesion are Pili, which were first described in GBS in 2005 (72). Pili promote colonization of epithelial cell surfaces, support biofilm formation, and facilitate translocation across the blood–brain barrier (51). They consist of a major shaft subunit, the backbone protein BP, which is critical for pilus assembly, and the two ancillary proteins AP1 and AP2 (73). Three pilus islands (PI1, PI2a, and PI2b) were identified. In mice, PI2a is essential for GBS virulence (74), and pilus island specific antibodies enhance opsonophagocytic killing and protect from sepsis (75, 76). Yet, development of pilus structures as vaccine candidates was hampered by variable pilus expression in GBS (75, 76).
Comparative expression analysis between clones with different clinical virulence (based on the disease phenotype in infants) led to the identification of the surface-anchored hypervirulent GBS adhesin (HvgA) as a specific virulence factor in GBS ST17 (9). Similar to pili, HvgA mediates both colonization and invasion in the intestine and confers meningeal tropism in neonatal mice (9, 10). Interestingly, GBS isolated from blood and cerebrospinal fluid during invasive disease express higher HvgA levels as compared to GBS cultured in vitro, indicating upregulation of HvgA expression during infection (9). Moreover, the ability of GBS ST17 to spread from the intestinal lumen is linked to the age of the mice, since 60–70% of preweaning mice (15–21 days old) succumb after enteral infection with hvgA-expressing GBS, whereas mice ≥4 weeks are protected (9). An experiment of nature, which we recently observed, suggests that yet to be identified changes in GBS virulence likely contribute to sepsis onset. An HvgA-positive strain of GBS, which was transmitted probably through breast milk from the mother, induced two episodes of sepsis each in twins, i.e., a total of four episodes, in a synchronous fashion. In all cases, sepsis started with soft-tissue infections of the lower oral cavity border, which implies a temporarily highly invasive and reproducible behavior of GBS (49).
Group B Streptococcus translocation across the epithelial barrier is further facilitated by the virulence factor β-hemolysin/cytolysin (β-H/C) (77–79). β-H/C induces cytolysis in eukaryotic cells and promotes bacterial invasion across epithelial and endothelial walls, including the blood–brain barrier. In mice, β-H/C induces placental inflammation and preterm birth, independently of bacterial ascension (80). β-H/C-deficient GBS show impaired virulence in various in vivo models including pneumonia, sepsis, and meningitis (77, 79, 81). However, at sublytic concentrations, β-H/C drives expression of the anti-inflammatory cytokine IL-10 and inhibits both IL-12 and NOS2 expression in GBS-infected macrophages (82). Thus expression levels of β-H/C appear to determine whether GBS stabilizes its niche to allow for colonization, or whether GBS becomes invasive. Additionally, the pore-forming toxin and co-hemolysin CAMP factor may contribute to GBS pathogenesis under certain circumstances (83, 84), but is dispensable for systemic virulence (83–86).
Distinct types of capsular polysaccharides (CPS), which underlie the serotyping system, allow for immune evasion, since α2 → 3 linked sialic acid modifications use molecular mimicry with host sugar epitopes. Direct binding to immunoglobulin-like lectins (Siglecs) on leukocytes inhibits complement C3 activation on the bacterial surface (87–89). Complement inactivation is further supported by ScpB (Streptococcal C5a peptidase of GBS)-mediated proteolytic C5a inactivation (90) and reduced complement binding through the inhibitory factors BibA (GBS immunogenic bacterial adhesin) (91) and factor H (92). Immunoglobulin binding, an essential precondition for opsonophagocytosis, is hampered by the IgA-binding beta-antigen of the c protein complex, which recognizes the Fc region of human immunoglobulin A (93). GBS evades phagolysosomal processing by neutralizing reactive oxygen species via BibA, β-H/C, superoxide dismutase (SodA), and additional unknown factors (77, 78, 91, 94–96). Similarly, binding of intestinal and circulating antimicrobial peptides can be suppressed by intrinsic GBS mechanisms such as d-alanylation of lipteichoic acid, which is catalyzed by the dlt operon. Down-modulation of d-alanylation decreases the negative surface charge, which in turn is important for cationic binding of antimicrobial peptides such as colistin (97).
Regulation of GBS Virulence
As outlined above, the human intestine is the natural niche for GBS. In other words, GBS thrives in healthy hosts. Accordingly, it is for the benefit of both GBS and the host if GBS is kept in a colonization state and does not exhibit its aggressive traits. Thus, expression of virulence factors in GBS must be tightly regulated. Two-component control systems, which typically consist of a membrane-linked histidine kinase sensor and a cytoplasmatic transcriptional element, are common in bacteria and more than 4000 regulatory systems have been described (98, 99). In GBS, CovS/CovR (Control of virulence Sensor/Regulator) tightly regulates the expression of pili, BsaB, hvgA, cyIE, which is involved in β-H/C expression, and many other genes (60, 100, 101). CovS acts as a pH sensor. In an acidic milieu, such as the vagina, CovS remains in an autophosphorylated state and activates the regulatory domain CovR by transphosphorylation at the aspartate residue D53. Subsequently, the phosphorylated form of CovR acts as a repressor. An increase in pH reduces CovS phosphorylation and induces the expression of β-H/C (102). In addition to pH, high glucose levels, peripheral insulin resistance, and passage through the intestine modulate CovR/CovS in mice (103–105). Inactivation of the CovR system increases GBS adherence to epithelial cells (106) by upregulation of PI1 expression (104). In line with this, GBS mutant in either CovS or CovR show increased hemolysis and approximately 80-fold upregulated HvgA (9).
Next to CovS, the serine/threonine kinase Stk1 acts as a sensor for environmental stimuli. Stk1 activation leads to phosphorylation of CovR at threonine 65. GBS with Stk1 mutations show reduced expression of β-H/C; susceptibility to opsonophagocytic killing and oxidative stress is increased (107, 108). Thus, while CovR phosphorylation at the aspartate residue D53 by CovS leads to protein activation associated with reduced virulence, Stk1-mediated phosphorylation at the threonine residue T63 increases the expression of β-H/C (108). Abx1, an additional partner within the CovS/CovR system, displays complex counterbalancing activity (107–109). As transmembrane protein Abx1 holds direct proximity to CovS, which it partially antagonizes. Both functional loss and overexpression of Abx1 hampers GBS virulence expression, and Abx1 expression itself is critically dependent on functional β-H/C formation (109). This system “fine tunes” GBS and may explain, at least in part, the loss of virulence of hyperhemolytic mutants due to CovR inactivation or Abx1 overexpression.
In summary, the CovS/CovR system inhabits a central role in the control of GBS virulence. It is tightly regulated, and specific environmental factors allow for subtle adaptation of the bacterial phenotype to the site of colonization/infection.
The Host Site: Toll-Like Receptors and the Inflammasome Mediate Host Resistance and Fatal Inflammation
Neonatal mice are exquisitely sensitive for GBS. Less than 100 bacteria injected s.c. constitute the lethal dose 90%. Under these conditions, disease is at least partially immune mediated, since the inflammatory cytokine TNF alone accounts for approximately half of the deaths (110). An important ligand receptor interaction resulting in TNF formation is that between diacylated lipoproteins and Toll-like receptors (TLR)2/6 heterodimers (111). Deletion of the diacyl transferase Lgt from GBS results in the same change in GBS sepsis phenotype as that observed in conventional TLR2 knock-out mice. The TLR-dependent recognition of bacterial lipopeptides can be assumed to primarily occur on the cell surface (112), although endosomal recognition of lipopeptides has recently been demonstrated (113). Next to lipoproteins, nucleic acids from GBS potently activate inflammatory genes in phagocytes. At least three distinct signaling pathways engaged by GBS nucleic acids have been identified. First, intracellular recognition of GBS DNA by one or several currently unknown cytosolic receptors results in the formation of type I interferons in a TBK1 and IRF3-dependent fashion (114) (IFN pathway). Second, two pathways involve recognition of GBS RNA. The first engages an UNC-93B- and MyD88-dependent pathway (115, 116) (TLR pathway), which has been shown to utilize TLR7 in dendritic cells (117), whereas the cognate TLR in macrophages is still elusive (116). The second pathway involves cytosolic sensing of RNA and β-hemolysin through the intracellular NLRP3 inflammasome (NOD-like receptor family, pyrin domain containing 3), which mediates IL-1β maturation in macrophages and dentritic cells (118, 119). Accordingly, in a mouse GBS sepsis model, NLRP3-deficiency is associated with significantly increased lethality (118, 119). It appears that NLRP3 and GBS RNA closely interact in the cytosol, and disruption of the GBS bearing phagosomes allows NLRP3 and GBS RNA to get into close contact (118). Discrimination of bacterial RNA from human ribosomal and transfer RNA appears to depend at least partially on the RNA methylation status (120). In S. aureus, methylated ribosomal RNA is recognized by TLR13, a TLR that currently lacks a human homolog. Accordingly, S. aureus strains that have acquired the methylase erm, conferring erythromycin resistance, have lost their TLR13-activating potential (121).
In summary, sensing of GBS lipoproteins and nucleic acids mediates activation of macrophages and dendritic cells and contributes to resistance against GBS and disease progression during sepsis. Yet, the contribution of these systems to containing GBS at mucosal sites is currently unclear.
IL-10 and Its Role in GBS Colonization and Disease
Very recently, the glycolytic enzyme glycerinaldehyde-3-phosphate-dehydrogenase (GAPDH) from GBS was shown to subvert immunity by inducing IL-10 (103, 122, 123). This adds to the kaleidoscope of functions of an enzyme, which is largely known for its role in bacterial energy generation (124–127) (summarized in Figure 1). GBS GAPDH is expressed as a surface molecule and in a soluble form (126, 128, 129). In mice, it directly activates B- and, to a lower extent, T-lymphocytes and induces the maturation of anti-GAPDH Ig-secreting plasma cells (130). Both endogenous and systemically administered GAPDH specific antibodies significantly reduce intestinal GBS colonization and protect from invasion of GAPDH-expression GBS strains. On the other hand, administration of recombinant GAPDH induces systemic release of IL-10 in mice and increases susceptibility to invasive GBS disease and bacterial invasion of per se non-invasive strains (82, 122, 130, 131). In line with these observations, IL-10-deficient mice are protected from invasive GBS disease and show improved survival in various sepsis models (122, 131–133). These effects are further consistent with the observation that reduced IL-10 activity increases neutrophil recruitment and bacterial killing (122, 130, 131). However, in earlier investigations, IL-10 given before infection protected neonatal mice from GBS sepsis and correlated with TNF suppression and improved survival (134). This effect was strictly dependent on the time of IL-10 application, since simultaneous or post-infectious administration failed to mediate protective effects. In contrast, in LPS-TLR4 induced septic shock models IL-10 improved survival even when given simultaneously with or early after LPS administration (133, 135, 136). Monocytes, macrophages, and T- and B-lymphocytes are all sources of IL-10 (Figure 1). IL-10 induction in macrophages depends on JNK signaling, which in turn is critical for the outcome in GBS sepsis (137–140). Expression of IL-10R (IL-10 receptor) and TGFBR1/TGFBR2 (TGF-beta receptors 1 and 2) and the presence of their specific ligands are pivotal to preserve immune homeostasis (141–144). In vitro, IL-10 reduces NF-kB signaling and the expression of TLRs, MyD88, and TIR in resident intestinal macrophages (iM) and circulating monocytes (145). In neonatal mice, the inflammatory program of iM is shifted toward IL-10 as the signature effector molecule via TGF-beta as a signaling intermediate (146). Patients with defects in the IL-10 or IL-10R gene develop severe, early-onset inflammatory bowel disease. Thus IL-10 is critical for intestinal inflammation control (147–149). Since, mice with a targeted deletion of STAT3 in macrophages and neutrophils (LysMcre/Stat3flox/−) develop spontaneous enterocolitis, it seems that tissue macrophages constitute the major source of IL-10 (150). Macrophage expression of IL-10 is dependent on TLR-dependent sensing of the microbiota and signal transduction by the adapter protein MyD88 (151). The multitude of functions of IL-10, its site-specific functions and the dynamics in its inducible effects all contribute to its highly complex role in stabilizing the interface between GBS and host.
Figure 1.

GBS transition to invasive disease. GBS lipoproteins, GAPDH, and nucleic acids majorly contribute to intestinal immune activation inducing pro-inflammatory responses, neutrophil recruitment, cellular proliferation, maturation and, finally, microbial clearance. Early induction of specific antibody release by B plasma cells and IL-10 secretion by both B-lymphocytes and phagocytes act as a negative feedback loop to counter-regulate hyperinflammation. Uncontrolled increase of IL-10, in contrast, hampers neutrophil recruitment and bacterial elimination. At the same time, T-lymphocytes can directly interact with GBS ligands or receive pro-inflammatory signals via monocytic cytokines. While Th1 and Th17 cells promote infection control, activation of regulatory T cells prevents hyperinflammation and supports post-infectious healing.
GBS in (Transient) Immunodeficiency
The so-called human model, where patients with monogenetic variations provide clues for the role of cell specific immunity, has been powerful in better understanding host–microbe interactions leading to coexistence and disease. An intriguing example is MyD88/IRAK-4-deficiency with its specific susceptibility to staphylococci, streptococci, and Pseudomonas spp. However, the search for monogenetic immunodeficiency disorders underlying susceptibility to invasive GBS infections has only been partially successful so far. One patient with very late-onset GBS sepsis suffering from IRAK-4 deficiency has been reported, supporting that cellular innate immunity and the TLR system are important for resistance against GBS (152–154). It can be expected that whole exome or genome analysis in families with GBS sepsis will provide further information in this regard in the future.
Next to the genetic absence of specific factors in cellular innate immunity, transient changes in innate immunity may underlie susceptibility to invasive GBS disease in newborn infants. Neonatal sepsis is characterized by excessive inflammation, since high levels of pro-inflammatory cytokines can be measured in the serum of newborn infants suffering from invasive GBS disease. In vitro, PBMC from healthy neonates rapidly respond to GBS with the formation of large amounts of IL-6, TNF, IL-8, and IL-1β (155–157). Since insulin suppresses the cytokine formation in response to GBS, peripheral insulin resistance present in newborn infants and particularly during sepsis may promote the inflammatory process (158). Excessive stimulation of immune cells may be further enhanced by a reduction of antimicrobial phagocyte properties, which are markedly impaired in neonates and may allow for pathogen persistence and failure to contract the immune response (159–161). GBS persistence likely results from reduced G-actin polarization and L-selectin expression in newborn, especially preterm infants, which results in impaired neutrophil adhesion and migration (162, 163). Moreover, low expression of opsonizing complement components, immunoglobulins, reactive oxygen species, the integrin CD11b/CD18, and Fc-gamma receptors II and III (164–166) and a predominant polarization of T-lymphocytes toward Th2 and Th17 may overall interfere with timely and rigorous clearance of invasive GBS (161, 167–173). How can the high levels of IL-10 in neonatal GBS infections, as observed in mice (174–177) and humans (178–180), be explained in view of the generally highly inflammatory state? Although conclusive evidence on this matter is lacking, it is tempting to speculate that the negative impact of IL-10 on neutrophil function, which allows for pathogen expansion, overrides the direct anti-inflammatory properties of IL-10 on the mononuclear phagocyte system. In this scenario, the net result in neonatal GBS sepsis is inflammatory despite high IL-10 levels.
Conclusion
Group B Streptococcus comprises several regulatory systems that respond to the microenvironment and, by steering adhesion and virulence factors, allow for colonization of mucosal niches in the genital and (lower) intestinal tract. Colonization is further propagated by the GBS-intrinsic ability to manipulate local cellular immunity. Yet, under only partially understood conditions GBS looses its colonizing trait and invades the host. Then, immune mechanisms that usually stabilize the natural GBS niche may lead to detrimental immunopathology. Rather subtle changes on the single immune cell level in newborn infants appear to facilitate the escalation from a beneficial site-specific response to sepsis and meningitis. Better understanding of the dynamic expression of virulence traits in GBS, and of the cellular immunology that shapes the GBS niche, will hopefully pave the way for preventing livelong disabilities inflicted by a normal component of the microbiota.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- 1.Matsubara K, Yamamoto G. Invasive group B streptococcal infections in a tertiary care hospital between 1998 and 2007 in Japan. Int J Infect Dis (2009) 13:679–84. 10.1016/j.ijid.2008.10.007 [DOI] [PubMed] [Google Scholar]
- 2.Stoll BJ, Hansen NI, Bell EF, Shankaran S, Laptook AR, Walsh MC, et al. Neonatal outcomes of extremely preterm infants from the NICHD neonatal research network. Pediatrics (2010) 126:443–56. 10.1542/peds.2009-2959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stoll BJ, Hansen NI, Sánchez PJ, Faix RG, Poindexter BB, Van Meurs KP, et al. Early onset neonatal sepsis: the burden of group B streptococcal and E. coli disease continues. Pediatrics (2011) 127:817–26. 10.1542/peds.2010-2217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Skoff TH, Farley MM, Petit S, Craig AS, Schaffner W, Gershman K, et al. Increasing burden of invasive group B streptococcal disease in nonpregnant adults, 1990-2007. Clin Infect Dis (2009) 49(1):85–92. 10.1086/599369 [DOI] [PubMed] [Google Scholar]
- 5.Trijbels-Smeulders MA, Kimpen JL, Kollée LA, Bakkers J, Melchers W, Spanjaard L, et al. Serotypes, genotypes, and antibiotic susceptibility profiles of group B streptococci causing neonatal sepsis and meningitis before and after introduction of antibiotic prophylaxis. Pediatr Infect Dis J (2006) 25:945–8. 10.1097/01.inf.0000237821.65559.08 [DOI] [PubMed] [Google Scholar]
- 6.Trijbels-Smeulders MA, Kollée LA, Adriaanse AH, Kimpen JL, Gerards LJ. Neonatal group B streptococcal infection: incidence and strategies for prevention in Europe. Pediatr Infect Dis J (2004) 23:172–3. 10.1097/01.inf.0000111212.94913.5f [DOI] [PubMed] [Google Scholar]
- 7.Kunze M, Ziegler A, Fluegge K, Hentschel R, Proempeler H, Berner R. Colonization, serotypes and transmission rates of group B streptococci in pregnant women and their infants born at a single University Center in Germany. J Perinat Med (2011) 39:417–22. 10.1515/jpm.2011.037 [DOI] [PubMed] [Google Scholar]
- 8.Fluegge K, Siedler A, Heinrich B, Schulte-Moenting J, Moennig MJ, Bartels DB, et al. Incidence and clinical presentation of invasive neonatal group B streptococcal infections in Germany. Pediatrics (2006) 117:e1139–45. 10.1542/peds.2005-2481 [DOI] [PubMed] [Google Scholar]
- 9.Tazi A, Disson O, Bellais S, Bouaboud A, Dmytruk N, Dramsi S, et al. The surface protein HvgA mediates group B Streptococcus hypervirulence and meningeal tropism in neonates. J Exp Med (2010) 207(11):2313–22. 10.1084/jem.20092594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tazi A, Bellais S, Tardieux I, Dramsi S, Trieu-Cuot P, Poyart C. Group B Streptococcus surface proteins as major determinants for meningeal tropism. Curr Opin Microbiol (2012) 15(1):44–9. 10.1016/j.mib.2011.12.002 [DOI] [PubMed] [Google Scholar]
- 11.Lewin EB, Amstey MS. Natural history of group B Streptococcus colonization and its therapy during pregnancy. Am J Obstet Gynecol (1981) 139(5):512–5. [DOI] [PubMed] [Google Scholar]
- 12.Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, et al. Diversity of the human intestinal microbial flora. Science (2005) 308(5728):1635–8. 10.1126/science.1110591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zoetendal EG, Akkermans AD, De Vos WM. Temperature gradient gel electrophoresis analysis of 16S rRNA from human fecal samples reveals stable and host-specific communities of active bacteria. Appl Environ Microbiol (1998) 64(10):3854–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Garrett WS, Gordon JI, Glimcher LH. Homeostasis and inflammation in the intestine. Cell (2010) 140(6):859–70. 10.1016/j.cell.2010.01.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Masanta WO, Heimesaat MM, Bereswill S, Tareen AM, Lugert R, Groß U, et al. Modification of intestinal microbiota and its consequences for innate immune response in the pathogenesis of campylobacteriosis. Clin Dev Immunol (2013) 2013:526860. 10.1155/2013/526860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Neut C, Bezirtzoglou E, Romond C, Beerens H, Delcroix M, Noel AM. Bacterial colonization of the large intestine in newborns delivered by cesarean section. Zentralbl Bakteriol Mikrobiol Hyg A (1987) 266(3–4):330–7. [DOI] [PubMed] [Google Scholar]
- 17.Bennet R, Nord CE. Development of the faecal anaerobic microflora after caesarean section and treatment with antibiotics in newborn infants. Infection (1987) 15(5):332–6. 10.1007/BF01647733 [DOI] [PubMed] [Google Scholar]
- 18.Penders J, Thijs C, Vink C, Stelma FF, Snijders B, Kummeling I, et al. Factors influencing the composition of the intestinal microbiota in early infancy. Pediatrics (2006) 118(2):511–21. 10.1542/peds.2005-2824 [DOI] [PubMed] [Google Scholar]
- 19.Guaraldi F, Salvatori G. Effect of breast and formula feeding on gut microbiota shaping in newborns. Front Cell Infect Microbiol (2012) 2:94. 10.3389/fcimb.2012.00094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rogier EW, Frantz AL, Bruno ME, Wedlund L, Cohen DA, Stromberg AJ, et al. Secretory antibodies in breast milk promote long-term intestinal homeostasis by regulating the gut microbiota and host gene expression. Proc Natl Acad Sci U S A (2014) 111(8):3074–9. 10.1073/pnas.1315792111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Palmer C, Bik EM, DiGiulio DB, Relman DA, Brown PO. Development of the human infant intestinal microbiota. PLoS Biol (2007) 5(7):e177. 10.1371/journal.pbio.0050177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Civardi E, Garofoli F, Mazzucchelli I, Angelini M, Manzoni P, Stronati M. Enteral nutrition and infections: the role of human milk. Early Hum Dev (2014) 90(Suppl 1):S57–9. 10.1016/S0378-3782(14)70019-2 [DOI] [PubMed] [Google Scholar]
- 23.Wold AE, Adlerberth I. Breast feeding and the intestinal microflora of the infant – implications for protection against infectious diseases. Adv Exp Med Biol (2000) 478:77–93. 10.1007/0-306-46830-1_7 [DOI] [PubMed] [Google Scholar]
- 24.Jakaitis BM, Denning PW. Human breast milk and the gastrointestinal innate immune system. Clin Perinatol (2014) 41(2):423–35. 10.1016/j.clp.2014.02.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cederlund A, Kai-Larsen Y, Printz G, Yoshio H, Alvelius G, Lagercrantz H, et al. Lactose in human breast milk an inducer of innate immunity with implications for a role in intestinal homeostasis. PLoS One (2013) 8(1):e53876. 10.1371/journal.pone.0053876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stewart CJ, Marrs EC, Nelson A, Lanyon C, Perry JD, Embleton ND, et al. Development of the preterm gut microbiome in twins at risk of necrotising enterocolitis and sepsis. PLoS One (2013) 8(8):e73465. 10.1371/journal.pone.0073465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Azad MB, Konya T, Maughan H, Guttman DS, Field CJ, Chari RS, et al. Gut microbiota of healthy Canadian infants: profiles by mode of delivery and infant diet at 4 months. CMAJ (2013) 185(5):385–94. 10.1503/cmaj.121189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stappenbeck TS, Hooper LV, Gordon JI. Developmental regulation of intestinal angiogenesis by indigenous microbes via Paneth cells. Proc Natl Acad Sci U S A (2002) 99(24):15451–5. 10.1073/pnas.202604299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu J, Gordon JI. Honor thy symbionts. Proc Natl Acad Sci U S A (2003) 100(18):10452–9. 10.1073/pnas.1734063100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hooper LV, Midtvedt T, Gordon JI. How host-microbial interactions shape the nutrient environment of the mammalian intestine. Annu Rev Nutr (2002) 22:283–307. 10.1146/annurev.nutr.22.011602.092259 [DOI] [PubMed] [Google Scholar]
- 31.MacDonald TT, Gordon JN. Bacterial regulation of intestinal immune responses. Gastroenterol Clin North Am (2005) 34(3):401–12, vii–viii. 10.1016/j.gtc.2005.05.012 [DOI] [PubMed] [Google Scholar]
- 32.Fukuda S, Toh H, Hase K, Oshima K, Nakanishi Y, Yoshimura K, et al. Bifidobacteria can protect from enteropathogenic infection through production of acetate. Nature (2011) 469(7331):543–7. 10.1038/nature09646 [DOI] [PubMed] [Google Scholar]
- 33.Maynard CL, Elson CO, Hatton RD, Weaver CT. Reciprocal interactions of the intestinal microbiota and immune system. Nature (2012) 489(7415):231–41. 10.1038/nature11551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jackson LA, Hilsdon R, Farley MM, Harrison LH, Reingold AL, Plikaytis BD, et al. Risk factors for group B streptococcal disease in adults. Ann Intern Med (1995) 123(6):415–20. 10.7326/0003-4819-123-6-199509150-00003 [DOI] [PubMed] [Google Scholar]
- 35.Anthony BF, Carter JA, Eisenstadt R, Rimer DG. Isolation of group B streptococci from the proximal small intestine of adults. J Infect Dis (1983) 147(4):776. 10.1093/infdis/147.4.776 [DOI] [PubMed] [Google Scholar]
- 36.Kneafsey PD, Kelly JK, Church DL, Rapp EF, Lafreniere R. Phlegmonous duodenitis complicating multiple myeloma: a successfully treated case. Am J Gastroenterol (1987) 82(12):1322–5. [PubMed] [Google Scholar]
- 37.Krohn MA, Hillier SL, Baker CJ. Maternal peripartum complications associated with vaginal group B streptococci colonization. J Infect Dis (1999) 179(6):1410–5. 10.1086/314756 [DOI] [PubMed] [Google Scholar]
- 38.Regan JA, Klebanoff MA, Nugent RP, Eschenbach DA, Blackwelder WC, Lou Y, et al. Colonization with group B streptococci in pregnancy and adverse outcome. VIP Study Group. Am J Obstet Gynecol (1996) 174(4):1354–60. 10.1016/S0002-9378(96)70684-1 [DOI] [PubMed] [Google Scholar]
- 39.Lamy M-C, Dramsi S, Billoët A, Réglier-Poupet H, Tazi A, Raymond J, et al. Rapid detection of the “highly virulent” group B Streptococcus ST-17 clone. Microbes Infect (2006) 8(7):1714–22. 10.1016/j.micinf.2006.02.008 [DOI] [PubMed] [Google Scholar]
- 40.Phares CR, Lynfield R, Farley MM, Mohle-Boetani J, Harrison LH, Petit S, et al. Epidemiology of invasive group B streptococcal disease in the United States, 1999-2005. JAMA (2008) 299(17):2056–65. 10.1001/jama.299.17.2056 [DOI] [PubMed] [Google Scholar]
- 41.Baker CJ, Barrett FF. Transmission of group B streptococci among parturient women and their neonates. J Pediatr (1973) 83(6):919–25. 10.1016/S0022-3476(73)80524-4 [DOI] [PubMed] [Google Scholar]
- 42.Haque KN. Definitions of bloodstream infection in the newborn. Pediatr Crit Care Med (2005) 6(3 Suppl):S45–9. 10.1097/01.PCC.0000161946.73305.0A [DOI] [PubMed] [Google Scholar]
- 43.Poyart C, Réglier-Poupet H, Tazi A, Billoët A, Dmytruk N, Bidet P, et al. Invasive group B streptococcal infections in infants, France. Emerg Infect Dis (2008) 14(10):1647–9. 10.3201/eid1410.080185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brochet M, Couvé E, Zouine M, Vallaeys T, Rusniok C, Lamy M-C, et al. Genomic diversity and evolution within the species Streptococcus agalactiae. Microbes Infect (2006) 8(5):1227–43. 10.1016/j.micinf.2005.11.010 [DOI] [PubMed] [Google Scholar]
- 45.Musser JM, Mattingly SJ, Quentin R, Goudeau A, Selander RK. Identification of a high-virulence clone of type III Streptococcus agalactiae (group B Streptococcus) causing invasive neonatal disease. Proc Natl Acad Sci U S A (1989) 86(12):4731–5. 10.1073/pnas.86.12.4731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jones N, Bohnsack JF, Takahashi S, Oliver KA, Chan M-S, Kunst F, et al. Multilocus sequence typing system for group B Streptococcus. J Clin Microbiol (2003) 41(6):2530–6. 10.1128/JCM.41.6.2530-2536.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Manning SD, Springman AC, Lehotzky E, Lewis MA, Whittam TS, Davies HD. Multilocus sequence types associated with neonatal group B streptococcal sepsis and meningitis in Canada. J Clin Microbiol (2009) 47(4):1143–8. 10.1128/JCM.01424-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Carl MA, Ndao IM, Springman AC, Manning SD, Johnson JR, Johnston BD, et al. Sepsis from the gut: the enteric habitat of bacteria that cause late-onset neonatal bloodstream infections. Clin Infect Dis (2014) 58(9):1211–8. 10.1093/cid/ciu084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Elling AR, Hufnagel M, de Zoysa A, Lander F, Zumstein K, Krueger M, et al. Synchronous recurrence of group B streptococcal late-onset sepsis in twins. Pediatrics (2014) 133(5):1–5. 10.1542/peds.2013-0426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Borges S, Silva J, Teixeira P. Survival and biofilm formation by group B streptococci in simulated vaginal fluid at different pHs. Antonie Van Leeuwenhoek (2012) 101(3):677–82. 10.1007/s10482-011-9666-y [DOI] [PubMed] [Google Scholar]
- 51.Konto-Ghiorghi Y, Mairey E, Mallet A, Duménil G, Caliot E, Trieu-Cuot P, et al. Dual role for pilus in adherence to epithelial cells and biofilm formation in Streptococcus agalactiae. PLoS Pathog (2009) 5(5):e1000422. 10.1371/journal.ppat.1000422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nizet V, Gibson RL, Chi EY, Framson PE, Hulse M, Rubens CE. Group B streptococcal beta-hemolysin expression is associated with injury of lung epithelial cells. Infect Immun (1996) 64(9):3818–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gibson RL, Nizet V, Rubens CE. Group B streptococcal beta-hemolysin promotes injury of lung microvascular endothelial cells. Pediatr Res (1999) 45(5 Pt 1):626–34. 10.1203/00006450-199905010-00003 [DOI] [PubMed] [Google Scholar]
- 54.Duménil G, Nassif X. Extracellular bacterial pathogens and small GTPases of the Rho family: an unexpected combination. Curr Top Microbiol Immunol (2005) 291:11–28. 10.1007/3-540-27511-8_2 [DOI] [PubMed] [Google Scholar]
- 55.Soriani M, Santi I, Taddei A, Rappuoli R, Grandi G, Telford JL. Group B Streptococcus crosses human epithelial cells by a paracellular route. J Infect Dis (2006) 193(2):241–50. 10.1086/498982 [DOI] [PubMed] [Google Scholar]
- 56.Jacobsson K. A novel family of fibrinogen-binding proteins in Streptococcus agalactiae. Vet Microbiol (2003) 96(1):103–13. 10.1016/S0378-1135(03)00206-2 [DOI] [PubMed] [Google Scholar]
- 57.Schubert A, Zakikhany K, Schreiner M, Frank R, Spellerberg B, Eikmanns BJ, et al. A fibrinogen receptor from group B Streptococcus interacts with fibrinogen by repetitive units with novel ligand binding sites. Mol Microbiol (2002) 46(2):557–69. 10.1046/j.1365-2958.2002.03177.x [DOI] [PubMed] [Google Scholar]
- 58.Spellerberg B, Rozdzinski E, Martin S, Weber-Heynemann J, Schnitzler N, Lütticken R, et al. Lmb, a protein with similarities to the LraI adhesin family, mediates attachment of Streptococcus agalactiae to human laminin. Infect Immun (1999) 67(2):871–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tenenbaum T, Spellerberg B, Adam R, Vogel M, Kim KS, Schroten H. Streptococcus agalactiae invasion of human brain microvascular endothelial cells is promoted by the laminin-binding protein Lmb. Microbes Infect (2007) 9(6):714–20. 10.1016/j.micinf.2007.02.015 [DOI] [PubMed] [Google Scholar]
- 60.Jiang S, Wessels MR. BsaB, a novel adherence factor of group B Streptococcus. Infect Immun (2014) 82(3):1007–16. 10.1128/IAI.01014-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Michel JL, Madoff LC, Olson K, Kling DE, Kasper DL, Ausubel FM. Large, identical, tandem repeating units in the C protein alpha antigen gene, bca, of group B streptococci. Proc Natl Acad Sci U S A (1992) 89(21):10060–4. 10.1073/pnas.89.21.10060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lalioui L, Pellegrini E, Dramsi S, Baptista M, Bourgeois N, Doucet-Populaire F, et al. The SrtA sortase of Streptococcus agalactiae is required for cell wall anchoring of proteins containing the LPXTG motif, for adhesion to epithelial cells, and for colonization of the mouse intestine. Infect Immun (2005) 73(6):3342–50. 10.1128/IAI.73.6.3342-3350.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Madoff LC, Hori S, Michel JL, Baker CJ, Kasper DL. Phenotypic diversity in the alpha C protein of group B streptococci. Infect Immun (1991) 59(8):2638–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gravekamp C, Kasper DL, Michel JL, Kling DE, Carey V, Madoff LC. Immunogenicity and protective efficacy of the alpha C protein of group B streptococci are inversely related to the number of repeats. Infect Immun (1997) 65(12):5216–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pannaraj PS, Kelly JK, Rench MA, Madoff LC, Edwards MS, Baker CJ. Alpha C protein-specific immunity in humans with group B streptococcal colonization and invasive disease. Vaccine (2008) 26(4):502–8. 10.1016/j.vaccine.2007.11.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bolduc GR, Madoff LC. The group B streptococcal alpha C protein binds alpha1beta1-integrin through a novel KTD motif that promotes internalization of GBS within human epithelial cells. Microbiology (2007) 153(Pt 12):4039–49. 10.1099/mic.0.2007/009134-0 [DOI] [PubMed] [Google Scholar]
- 67.Madoff LC, Michel JL, Gong EW, Kling DE, Kasper DL. Group B streptococci escape host immunity by deletion of tandem repeat elements of the alpha C protein. Proc Natl Acad Sci U S A (1996) 93(9):4131–6. 10.1073/pnas.93.9.4131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rubens CE, Wessels MR, Heggen LM, Kasper DL. Transposon mutagenesis of type III group B Streptococcus: correlation of capsule expression with virulence. Proc Natl Acad Sci U S A (1987) 84(20):7208–12. 10.1073/pnas.84.20.7208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Madoff LC, Michel JL, Gong EW, Rodewald AK, Kasper DL. Protection of neonatal mice from group B streptococcal infection by maternal immunization with beta C protein. Infect Immun (1992) 60(12):4989–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Seifert KN, Adderson EE, Whiting AA, Bohnsack JF, Crowley PJ, Brady LJ. A unique serine-rich repeat protein (Srr-2) and novel surface antigen (epsilon) associated with a virulent lineage of serotype III Streptococcus agalactiae. Microbiology (2006) 152(4):1029–40. 10.1099/mic.0.28516-0 [DOI] [PubMed] [Google Scholar]
- 71.Seo HS, Minasov G, Seepersaud R, Doran KS, Dubrovska I, Shuvalova L, et al. Characterization of fibrinogen binding by glycoproteins Srr1 and Srr2 of Streptococcus agalactiae. J Biol Chem (2013) 288(50):35982–96. 10.1074/jbc.M113.513358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lauer P, Rinaudo CD, Soriani M, Margarit I, Maione D, Rosini R, et al. Genome analysis reveals pili in group B Streptococcus. Science (2005) 309(5731):105. 10.1126/science.1111563 [DOI] [PubMed] [Google Scholar]
- 73.Rosini R, Rinaudo CD, Soriani M, Lauer P, Mora M, Maione D, et al. Identification of novel genomic islands coding for antigenic pilus-like structures in Streptococcus agalactiae. Mol Microbiol (2006) 61(1):126–41. 10.1111/j.1365-2958.2006.05225.x [DOI] [PubMed] [Google Scholar]
- 74.Papasergi S, Brega S, Mistou M-Y, Firon A, Oxaran V, Dover R, et al. The GBS PI-2a pilus is required for virulence in mice neonates. PLoS One (2011) 6(4):e18747. 10.1371/journal.pone.0018747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nuccitelli A, Cozzi R, Gourlay LJ, Donnarumma D, Necchi F, Norais N. Structure-based approach to rationally design a chimeric protein for an effective vaccine against group B Streptococcus infections. Proc Natl Acad Sci U S A (2011) 108(25):10278–83. 10.1073/pnas.1106590108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Margarit I, Rinaudo CD, Galeotti CL, Maione D, Ghezzo C, Buttazzoni E, et al. Preventing bacterial infections with pilus-based vaccines: the group B Streptococcus paradigm. J Infect Dis (2009) 199(1):108–15. 10.1086/595564 [DOI] [PubMed] [Google Scholar]
- 77.Doran KS, Liu GY, Nizet V. Group B streptococcal beta-hemolysin/cytolysin activates neutrophil signaling pathways in brain endothelium and contributes to development of meningitis. J Clin Invest (2003) 112(5):736–44. 10.1172/JCI200317335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Liu GY, Doran KS, Lawrence T, Turkson N, Puliti M, Tissi L, et al. Sword and shield: linked group B streptococcal beta-hemolysin/cytolysin and carotenoid pigment function to subvert host phagocyte defense. Proc Natl Acad Sci U S A (2004) 101(40):14491–6. 10.1073/pnas.0406143101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Doran KS, Chang JC, Benoit VM, Eckmann L, Nizet V. Group B streptococcal beta-hemolysin/cytolysin promotes invasion of human lung epithelial cells and the release of interleukin-8. J Infect Dis (2002) 185(2):196–203. 10.1086/338475 [DOI] [PubMed] [Google Scholar]
- 80.Randis TM, Gelber SE, Hooven TA, Abellar RG, Akabas LH, Lewis EL, et al. Group B Streptococcus β-hemolysin/cytolysin breaches maternal-fetal barriers to cause preterm birth and intrauterine fetal demise in vivo. J Infect Dis (2014) 210(2):265–73. 10.1093/infdis/jiu067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hensler ME, Liu GY, Sobczak S, Benirschke K, Nizet V, Heldt GP. Virulence role of group B Streptococcus beta-hemolysin/cytolysin in a neonatal rabbit model of early-onset pulmonary infection. J Infect Dis (2005) 191(8):1287–91. 10.1086/428946 [DOI] [PubMed] [Google Scholar]
- 82.Bebien M, Hensler ME, Davanture S, Hsu L-C, Karin M, Park JM, et al. The pore-forming toxin β hemolysin/cytolysin triggers p38 MAPK-dependent IL-10 production in macrophages and inhibits innate immunity. PLoS Pathog (2012) 8(7):e1002812. 10.1371/journal.ppat.1002812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lang S, Palmer M. Characterization of Streptococcus agalactiae CAMP factor as a pore-forming toxin. J Biol Chem (2003) 278(40):38167–73. 10.1074/jbc.M303544200 [DOI] [PubMed] [Google Scholar]
- 84.Jürgens D, Sterzik B, Fehrenbach FJ. Unspecific binding of group B streptococcal cocytolysin (CAMP factor) to immunoglobulins and its possible role in pathogenicity. J Exp Med (1987) 165(3):720–32. 10.1084/jem.165.3.720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hensler ME, Quach D, Hsieh C-J, Doran KS, Nizet V. CAMP factor is not essential for systemic virulence of group B Streptococcus. Microb Pathog (2008) 44(1):84–8. 10.1016/j.micpath.2007.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Podbielski A, Blankenstein O, Lütticken R. Molecular characterization of the cfb gene encoding group B streptococcal CAMP-factor. Med Microbiol Immunol (1994) 183(5):239–56. 10.1007/BF00198458 [DOI] [PubMed] [Google Scholar]
- 87.Chang Y-C, Olson J, Louie A, Crocker PR, Varki A, Nizet V. Role of macrophage sialoadhesin in host defense against the sialylated pathogen group B Streptococcus. J Mol Med (Berl) (2014) 92(9):951–9. 10.1007/s00109-014-1157-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pezzicoli A, Ruggiero P, Amerighi F, Telford JL, Soriani M. Exogenous sialic acid transport contributes to group B Streptococcus infection of mucosal surfaces. J Infect Dis (2012) 206(6):924–31. 10.1093/infdis/jis451 [DOI] [PubMed] [Google Scholar]
- 89.Chang Y-C, Olson J, Beasley FC, Tung C, Zhang J, Crocker PR, et al. Group B Streptococcus engages an inhibitory Siglec through sialic acid mimicry to blunt innate immune and inflammatory responses in vivo. PLoS Pathog (2014) 10(1):e1003846. 10.1371/journal.ppat.1003846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bohnsack JF, Takahashi S, Hammitt L, Miller DV, Aly AA, Adderson EE. Genetic polymorphisms of group B Streptococcus scpB alter functional activity of a cell-associated peptidase that inactivates C5a. Infect Immun (2000) 68(9):5018–25. 10.1128/IAI.68.9.5018-5025.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Santi I, Scarselli M, Mariani M, Pezzicoli A, Masignani V, Taddei A, et al. BibA: a novel immunogenic bacterial adhesin contributing to group B Streptococcus survival in human blood. Mol Microbiol (2007) 63(3):754–67. 10.1111/j.1365-2958.2006.05555.x [DOI] [PubMed] [Google Scholar]
- 92.Jarva H, Hellwage J, Jokiranta TS, Lehtinen MJ, Zipfel PF, Meri S. The group B streptococcal beta and pneumococcal Hic proteins are structurally related immune evasion molecules that bind the complement inhibitor factor H in an analogous fashion. J Immunol (2004) 172(5):3111–8. 10.4049/jimmunol.172.5.3111 [DOI] [PubMed] [Google Scholar]
- 93.Jerlström PG, Chhatwal GS, Timmis KN. The IgA-binding beta antigen of the c protein complex of group B streptococci: sequence determination of its gene and detection of two binding regions. Mol Microbiol (1991) 5(4):843–9. 10.1111/j.1365-2958.1991.tb00757.x [DOI] [PubMed] [Google Scholar]
- 94.Poyart C, Pellegrini E, Gaillot O, Boumaila C, Baptista M, Trieu-Cuot P. Contribution of Mn-cofactored superoxide dismutase (SodA) to the virulence of Streptococcus agalactiae. Infect Immun (2001) 69(8):5098–106. 10.1128/IAI.69.8.5098-5106.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Cornacchione P, Scaringi L, Fettucciari K, Rosati E, Sabatini R, Orefici G, et al. Group B streptococci persist inside macrophages. Immunology (1998) 93(1):86–95. 10.1046/j.1365-2567.1998.00402.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Teixeira CF, Azevedo NL, Carvalho TM, Fuentes J, Nagao PE. Cytochemical study of Streptococcus agalactiae and macrophage interaction. Microsc Res Tech (2001) 54(4):254–9. 10.1002/jemt.1137 [DOI] [PubMed] [Google Scholar]
- 97.Poyart C, Lamy MC, Boumaila C, Fiedler F, Trieu-Cuot P. Regulation of d-alanyl-lipoteichoic acid biosynthesis in Streptococcus agalactiae involves a novel two-component regulatory system. J Bacteriol (2001) 183(21):6324–34. 10.1128/JB.183.21.6324-6334.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ulrich LE, Koonin EV, Zhulin IB. One-component systems dominate signal transduction in prokaryotes. Trends Microbiol (2005) 13(2):52–6. 10.1016/j.tim.2004.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Beier D, Gross R. Regulation of bacterial virulence by two-component systems. Curr Opin Microbiol (2006) 9(2):143–52. 10.1016/j.mib.2006.01.005 [DOI] [PubMed] [Google Scholar]
- 100.Lembo A, Gurney MA, Burnside K, Banerjee A, de los Reyes M, Connelly JE, et al. Regulation of CovR expression in group B Streptococcus impacts blood-brain barrier penetration. Mol Microbiol (2010) 77(2):431–43. 10.1111/j.1365-2958.2010.07215.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Jiang S, Cieslewicz MJ, Kasper DL, Wessels MR. Regulation of virulence by a two-component system in group B Streptococcus. J Bacteriol (2005) 187(3):1105–13. 10.1128/JB.187.3.1105-1113.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Santi I, Grifantini R, Jiang S-M, Brettoni C, Grandi G, Wessels MR, et al. CsrRS regulates group B Streptococcus virulence gene expression in response to environmental pH: a new perspective on vaccine development. J Bacteriol (2009) 191(17):5387–97. 10.1128/JB.00370-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Oliveira L, Madureira P, Andrade EB, Bouaboud A, Morello E, Ferreira P, et al. Group B Streptococcus GAPDH is released upon cell lysis, associates with bacterial surface, and induces apoptosis in murine macrophages. PLoS One (2012) 7(1):e29963. 10.1371/journal.pone.0029963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Jiang S, Park SE, Yadav P, Paoletti LC, Wessels MR. Regulation and function of pilus island 1 in group B Streptococcus. J Bacteriol (2012) 194(10):2479–90. 10.1128/JB.00202-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Patras KA, Wang N-Y, Fletcher EM, Cavaco CK, Jimenez A, Garg M, et al. Group B Streptococcus CovR regulation modulates host immune signalling pathways to promote vaginal colonization. Cell Microbiol (2013) 15(7):1154–67. 10.1111/cmi.12105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lamy M-C, Zouine M, Fert J, Vergassola M, Couve E, Pellegrini E, et al. CovS/CovR of group B Streptococcus: a two-component global regulatory system involved in virulence. Mol Microbiol (2004) 54(5):1250–68. 10.1111/j.1365-2958.2004.04365.x [DOI] [PubMed] [Google Scholar]
- 107.Rajagopal L, Vo A, Silvestroni A, Rubens CE. Regulation of cytotoxin expression by converging eukaryotic-type and two-component signalling mechanisms in Streptococcus agalactiae. Mol Microbiol (2006) 62(4):941–57. 10.1111/j.1365-2958.2006.05431.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lin W-J, Walthers D, Connelly JE, Burnside K, Jewell KA, Kenney LJ, et al. Threonine phosphorylation prevents promoter DNA binding of the group B Streptococcus response regulator CovR. Mol Microbiol (2009) 71(6):1477–95. 10.1111/j.1365-2958.2009.06616.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Firon A, Tazi A, Da Cunha V, Brinster S, Sauvage E, Dramsi S, et al. The Abi-domain protein Abx1 interacts with the CovS histidine kinase to control virulence gene expression in group B Streptococcus. PLoS Pathog (2013) 9(2):e1003179. 10.1371/journal.ppat.1003179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Mancuso G, Midiri A, Beninati C, Biondo C, Galbo R, Akira S, et al. Dual role of TLR2 and myeloid differentiation factor 88 in a mouse model of invasive group B streptococcal disease. J Immunol (2004) 172:6324–9. 10.4049/jimmunol.172.10.6324 [DOI] [PubMed] [Google Scholar]
- 111.Henneke P, Dramsi S, Mancuso G, Chraibi K, Pellegrini E, Theilacker C, et al. Lipoproteins are critical TLR2 activating toxins in group B streptococcal sepsis. J Immunol (2008) 180(9):6149–58. 10.4049/jimmunol.180.9.6149 [DOI] [PubMed] [Google Scholar]
- 112.Takeda K, Takeuchi O, Akira S. Recognition of lipopeptides by toll-like receptors. J Endotoxin Res (2002) 8(6):459–63. 10.1177/09680519020080060101 [DOI] [PubMed] [Google Scholar]
- 113.Motoi Y, Shibata T, Takahashi K, Kanno A, Murakami Y, Li X, et al. Lipopeptides are signaled by toll-like receptor 1, 2 and 6 in endolysosomes. Int Immunol (2014) 26(10):563–73. 10.1093/intimm/dxu054 [DOI] [PubMed] [Google Scholar]
- 114.Charrel-Dennis M, Latz E, Halmen KA, Trieu-Cuot P, Fitzgerald KA, Kasper DL, et al. TLR-independent type I interferon induction in response to an extracellular bacterial pathogen via intracellular recognition of its DNA. Cell Host Microbe (2008) 4(6):543–54. 10.1016/j.chom.2008.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Deshmukh SD, Müller S, Hese K, Rauch KS, Wennekamp J, Takeuchi O, et al. NO is a macrophage autonomous modifier of the cytokine response to streptococcal single-stranded RNA. J Immunol (2012) 188(2):774–80. 10.4049/jimmunol.1101383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Deshmukh SD, Kremer B, Freudenberg M, Bauer S, Golenbock DT, Henneke P. Macrophages recognize streptococci through bacterial single-stranded RNA. EMBO Rep (2011) 12(1):71–6. 10.1038/embor.2010.189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Mancuso G, Gambuzza M, Midiri A, Biondo C, Papasergi S, Akira S, et al. Bacterial recognition by TLR7 in the lysosomes of conventional dendritic cells. Nat Immunol (2009) 10(6):587–94. 10.1038/ni.1733 [DOI] [PubMed] [Google Scholar]
- 118.Gupta R, Ghosh S, Monks B, Deoliveira R, Tzeng T, Kalantari P, et al. RNA and β-hemolysin of group B Streptococcus induce IL-1β by activating NLRP3 inflammasomes in mouse macrophages. J Biol Chem (2014) 289(20):13701–5. 10.1074/jbc.C114.548982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Costa A, Gupta R, Signorino G, Malara A, Cardile F, Biondo C, et al. Activation of the NLRP3 inflammasome by group B streptococci. J Immunol (2012) 188(4):1953–60. 10.4049/jimmunol.1102543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Jöckel S, Nees G, Sommer R, Zhao Y, Cherkasov D, Hori H, et al. The 2′-O-methylation status of a single guanosine controls transfer RNA-mediated toll-like receptor 7 activation or inhibition. J Exp Med (2012) 209(2):235–41. 10.1084/jem.20111075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Hochrein H, Kirschning CJ. Bacteria evade immune recognition via TLR13 and binding of their 23S rRNA by MLS antibiotics by the same mechanisms. Oncoimmunology (2013) 2(3):e23141. 10.4161/onci.23141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Madureira P, Andrade EB, Gama B, Oliveira L, Moreira S, Correia-Neves M, et al. Inhibition of IL-10 production by maternal antibodies against group B Streptococcus GAPDH confers immunity to offspring by favoring neutrophil recruitment. PLoS Pathog (2011) 7(11):e1002363. 10.1371/journal.ppat.1002363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Seifert KN, McArthur WP, Bleiweis AS, Brady LJ. Characterization of group B streptococcal glyceraldehyde-3-phosphate dehydrogenase: surface localization, enzymatic activity, and protein-protein interactions. Can J Microbiol (2003) 49(5):350–6. 10.1139/w03-042 [DOI] [PubMed] [Google Scholar]
- 124.Kim J-W, Dang CV. Multifaceted roles of glycolytic enzymes. Trends Biochem Sci (2005) 30(3):142–50. 10.1016/j.tibs.2005.01.005 [DOI] [PubMed] [Google Scholar]
- 125.Pancholi V, Fischetti VA. A major surface protein on group A streptococci is a glyceraldehyde-3-phosphate-dehydrogenase with multiple binding activity. J Exp Med (1992) 176(2):415–26. 10.1084/jem.176.2.415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Bergmann S, Rohde M, Hammerschmidt S. Glyceraldehyde-3-phosphate dehydrogenase of Streptococcus pneumoniae is a surface-displayed plasminogen-binding protein. Infect Immun (2004) 72(4):2416–9. 10.1128/IAI.72.4.2416-2419.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Ling E, Feldman G, Portnoi M, Dagan R, Overweg K, Mulholland F, et al. Glycolytic enzymes associated with the cell surface of Streptococcus pneumoniae are antigenic in humans and elicit protective immune responses in the mouse. Clin Exp Immunol (2004) 138(2):290–8. 10.1111/j.1365-2249.2004.02628.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Fluegge K, Schweier O, Schiltz E, Batsford S, Berner R. Identification and immunoreactivity of proteins released from Streptococcus agalactiae. Eur J Clin Microbiol Infect Dis (2004) 23(11):818–24. 10.1007/s10096-004-1229-y [DOI] [PubMed] [Google Scholar]
- 129.Alvarez RA, Blaylock MW, Baseman JB. Surface localized glyceraldehyde-3-phosphate dehydrogenase of Mycoplasma genitalium binds mucin. Mol Microbiol (2003) 48(5):1417–25. 10.1046/j.1365-2958.2003.03518.x [DOI] [PubMed] [Google Scholar]
- 130.Madureira P, Baptista M, Vieira M, Magalhaes V, Camelo A, Oliveira L, et al. Streptococcus agalactiae GAPDH is a virulence-associated immunomodulatory protein. J Immunol (2007) 178(3):1379–87. 10.4049/jimmunol.178.3.1379 [DOI] [PubMed] [Google Scholar]
- 131.Andrade EB, Alves J, Madureira P, Oliveira L, Ribeiro A, Cordeiro-da-Silva A, et al. TLR2-induced IL-10 production impairs neutrophil recruitment to infected tissues during neonatal bacterial sepsis. J Immunol (2013) 191(9):4759–68. 10.4049/jimmunol.1301752 [DOI] [PubMed] [Google Scholar]
- 132.Kamanaka M, Kim ST, Wan YY, Sutterwala FS, Lara-Tejero M, Galán JE, et al. Expression of interleukin-10 in intestinal lymphocytes detected by an interleukin-10 reporter knockin tiger mouse. Immunity (2006) 25(6):941–52. 10.1016/j.immuni.2006.09.013 [DOI] [PubMed] [Google Scholar]
- 133.Gérard C, Bruyns C, Marchant A, Abramowicz D, Vandenabeele P, Delvaux A, et al. Interleukin 10 reduces the release of tumor necrosis factor and prevents lethality in experimental endotoxemia. J Exp Med (1993) 177(2):547–50. 10.1084/jem.177.2.547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Cusumano V, Genovese F, Mancuso G, Carbone M, Fera MT, Teti G. Interleukin-10 protects neonatal mice from lethal group B streptococcal infection. Infect Immun (1996) 64(7):2850–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Smith SR, Terminelli C, Kenworthy-Bott L, Calzetta A, Donkin J. The cooperative effects of TNF-alpha and IFN-gamma are determining factors in the ability of IL-10 to protect mice from lethal endotoxemia. J Leukoc Biol (1994) 55(6):711–8. [DOI] [PubMed] [Google Scholar]
- 136.Howard M, Muchamuel T, Andrade S, Menon S. Interleukin 10 protects mice from lethal endotoxemia. J Exp Med (1993) 177(4):1205–8. 10.1084/jem.177.4.1205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Sing A, Rost D, Tvardovskaia N, Roggenkamp A, Wiedemann A, Kirschning CJ, et al. Yersinia V-antigen exploits toll-like receptor 2 and CD14 for interleukin 10-mediated immunosuppression. J Exp Med (2002) 196(8):1017–24. 10.1084/jem.20020908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Jang S, Uematsu S, Akira S, Salgame P. IL-6 and IL-10 induction from dendritic cells in response to Mycobacterium tuberculosis is predominantly dependent on TLR2-mediated recognition. J Immunol (2004) 173(5):3392–7. 10.4049/jimmunol.173.5.3392 [DOI] [PubMed] [Google Scholar]
- 139.Dillon S, Agrawal A, Van Dyke T, Landreth G, McCauley L, Koh A, et al. A toll-like receptor 2 ligand stimulates Th2 responses in vivo, via induction of extracellular signal-regulated kinase mitogen-activated protein kinase and c-Fos in dendritic cells. J Immunol (2004) 172(8):4733–43. 10.4049/jimmunol.172.8.4733 [DOI] [PubMed] [Google Scholar]
- 140.Kenzel S, Mancuso G, Malley R, Teti G, Golenbock DT, Henneke P. c-Jun kinase is a critical signaling molecule in a neonatal model of group B streptococcal sepsis. J Immunol (2006) 176:3181–8. 10.4049/jimmunol.176.5.3181 [DOI] [PubMed] [Google Scholar]
- 141.Moore KW, De Waal Malefyt R, Coffman RL, O’Garra A. Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol (2001) 19:683–765. 10.1146/annurev.immunol.19.1.683 [DOI] [PubMed] [Google Scholar]
- 142.Couper KN, Blount DG, Riley EM. IL-10: the master regulator of immunity to infection. J Immunol (2008) 180(9):5771–7. 10.4049/jimmunol.180.9.5771 [DOI] [PubMed] [Google Scholar]
- 143.Smythies LE, Maheshwari A, Clements R, Eckhoff D, Novak L, Vu HL, et al. Mucosal IL-8 and TGF-β recruit blood monocytes: evidence for cross-talk between the lamina propria stroma and myeloid cells. J Leukoc Biol (2006) 80(September):492–9. 10.1189/jlb.1005566 [DOI] [PubMed] [Google Scholar]
- 144.Varol C, Zigmond E, Jung S. Securing the immune tightrope: mononuclear phagocytes in the intestinal lamina propria. Nat Rev Immunol (2010) 10(6):415–26. 10.1038/nri2778 [DOI] [PubMed] [Google Scholar]
- 145.Smythies LE, Shen R, Bimczok D, Novak L, Clements RH, Eckhoff DE, et al. Inflammation anergy in human intestinal macrophages is due to Smad-induced IkappaBalpha expression and NF-kappaB inactivation. J Biol Chem (2010) 285(25):19593–604. 10.1074/jbc.M109.069955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Maheshwari A, Kelly DR, Nicola T, Jain SK, Murphy-Ullrich J, Athar M, et al. TGF-β2 suppresses macrophage cytokine production and mucosal inflammatory responses in the developing intestine. Gastroenterology (2012) 140(1):242–53. 10.1053/j.gastro.2010.09.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Begue B, Verdier J, Rieux-Laucat F, Goulet O, Morali A, Canioni D, et al. Defective IL10 signaling defining a subgroup of patients with inflammatory bowel disease. Am J Gastroenterol (2011) 106(8):1544–55. 10.1038/ajg.2011.112 [DOI] [PubMed] [Google Scholar]
- 148.Gertz EM, Ph D, Schäffer AA, Noyan F, Perro M, Sc M, et al. Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. N Engl J Med (2009) 361(21):2033–45. 10.1056/NEJMoa0907206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Glocker E, Frede N, Perro M, Sebire N, Elawad M, Shah N, et al. Case report infant colitis – it’s in the genes. Lancet (2010) 376:1272. 10.1016/S0140-6736(10)61008-2 [DOI] [PubMed] [Google Scholar]
- 150.Reindl W, Weiss S, Lehr H-A, Förster I. Essential crosstalk between myeloid and lymphoid cells for development of chronic colitis in myeloid-specific signal transducer and activator of transcription 3-deficient mice. Immunology (2007) 120(1):19–27. 10.1111/j.1365-2567.2006.02473.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Hoshi N, Schenten D, Nish SA, Walther Z, Gagliani N, Flavell RA, et al. MyD88 signalling in colonic mononuclear phagocytes drives colitis in IL-10-deficient mice. Nat Commun (2012) 3:1120. 10.1038/ncomms2113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Krause JC, Ghandil P, Chrabieh M, Casanova J, Picard C, Puel A. Very late-onset group B Streptococcus meningitis, sepsis, and systemic shigellosis due to interleukin-1 receptor-associated kinase-4 deficiency. Clin Infect Dis (2009) 49:1393–6. 10.1086/630206 [DOI] [PubMed] [Google Scholar]
- 153.Picard C, von Bernuth H, Ghandil P, Chrabieh M, Levy O, Arkwright PD, et al. Clinical features and outcome of patients with IRAK-4 and MyD88 deficiency. Medicine (Baltimore) (2010) 89(6):403–25. 10.1097/MD.0b013e3181fd8ec3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Picard C, Casanova J, Puel A. Infectious diseases in patients with IRAK-4, MyD88, NEMO, or IκBα deficiency. Clin Microbiol Rev (2011) 24(3):490–7. 10.1128/CMR.00001-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Berner R, Welter P, Brandis M. Cytokine expression of cord and adult blood mononuclear cells in response to Streptococcus agalactiae. Pediatr Res (2002) 51:304–9. 10.1203/00006450-200203000-00007 [DOI] [PubMed] [Google Scholar]
- 156.Levy O, Jean-Jacques RM, Cywes C, Sisson RB, Zarember KA, Godowski PJ, et al. Critical role of the complement system in group B Streptococcus-induced tumor necrosis factor alpha release. Infect Immun (2003) 71(11):6344–53. 10.1128/IAI.71.11.6344-6353.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Williams PA, Bohnsack JF, Augustine NH, Drummond WK, Rubens CE, Hill HR. Production of tumor necrosis factor by human cells in vitro and in vivo, induced by group B streptococci. J Pediatr (1993) 123(2):292–300. 10.1016/S0022-3476(05)81706-8 [DOI] [PubMed] [Google Scholar]
- 158.Kenzel S, Mergen M, von Süßkind-Schwendi J, Wennekamp J, Deshmukh SD, Haeffner M, et al. Insulin modulates the inflammatory granulocyte response to streptococci via phosphatidylinositol 3-kinase. J Immunol (2012) 189(9):4582–91. 10.4049/jimmunol.1200205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Wennekamp J, Henneke P. Induction and termination of inflammatory signaling in group B streptococcal sepsis. Immunol Rev (2008) 225:114–27. 10.1111/j.1600-065X.2008.00673.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Pong A, Bradley JS. Bacterial meningitis and the newborn infant. Infect Dis Clin North Am (1999) 13(3):711–33, viii. 10.1016/S0891-5520(05)70102-1 [DOI] [PubMed] [Google Scholar]
- 161.Filias A, Theodorou GL, Mouzopoulou S, Varvarigou AA, Mantagos S, Karakantza M. Phagocytic ability of neutrophils and monocytes in neonates. BMC Pediatr (2011) 11:29. 10.1186/1471-2431-11-29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Harris MC, Shalit M, Southwick FS. Diminished actin polymerization by neutrophils from newborn infants. Pediatr Res (1993) 33(1):27–31. 10.1203/00006450-199301000-00006 [DOI] [PubMed] [Google Scholar]
- 163.Sacchi F, Augustine NH, Coello MM, Morris EZ, Hill HR. Abnormality in actin polymerization associated with defective chemotaxis in neutrophils from neonates. Int Arch Allergy Appl Immunol (1987) 84(1):32–9. 10.1159/000234395 [DOI] [PubMed] [Google Scholar]
- 164.Drossou V, Kanakoudi F, Diamanti E, Tzimouli V, Konstantinidis T, Germenis A, et al. Concentrations of main serum opsonins in early infancy. Arch Dis Child Fetal Neonatal Ed (1995) 72(3):F172–5. 10.1136/fn.72.3.F172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Abughali N, Berger M, Tosi MF. Deficient total cell content of CR3 (CD11b) in neonatal neutrophils. Blood (1994) 83(4):1086–92. [PubMed] [Google Scholar]
- 166.Nupponen I, Pesonen E, Andersson S, Mäkelä A, Turunen R, Kautiainen H, et al. Neutrophil activation in preterm infants who have respiratory distress syndrome. Pediatrics (2002) 110(1 Pt 1):36–41. 10.1542/peds.110.1.36 [DOI] [PubMed] [Google Scholar]
- 167.Källman J, Schollin J, Schalèn C, Erlandsson A, Kihlström E. Impaired phagocytosis and opsonisation towards group B streptococci in preterm neonates. Arch Dis Child Fetal Neonatal Ed (1998) 78(1):F46–50. 10.1136/fn.78.1.F46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Ballinger MN, Peters-Golden M, Moore BB. Impaired neonatal macrophage phagocytosis is not explained by overproduction of prostaglandin E2. Respir Res (2011) 12(1):155. 10.1186/1465-9921-12-155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Rose S, Lichtenheld M, Foote MR, Adkins B. Murine neonatal CD4+ cells are poised for rapid Th2 effector-like function. J Immunol (2007) 178(5):2667–78. 10.4049/jimmunol.178.5.2667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Zaghouani H, Hoeman CM, Adkins B. Neonatal immunity: faulty T-helpers and the shortcomings of dendritic cells. Trends Immunol (2009) 30(12):585–91. 10.1016/j.it.2009.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Netea MG, Quintin J, van der Meer JW. Trained immunity: a memory for innate host defense. Cell Host Microbe (2011) 9(5):355–61. 10.1016/j.chom.2011.04.006 [DOI] [PubMed] [Google Scholar]
- 172.Aksoy E, Albarani V, Nguyen M, Laes J-F, Ruelle J-L, De Wit D, et al. Interferon regulatory factor 3-dependent responses to lipopolysaccharide are selectively blunted in cord blood cells. Blood (2007) 109(7):2887–93. 10.1182/blood-2006-06-027862 [DOI] [PubMed] [Google Scholar]
- 173.De Wit D, Tonon S, Olislagers V, Goriely S, Boutriaux M, Goldman M, et al. Impaired responses to toll-like receptor 4 and toll-like receptor 3 ligands in human cord blood. J Autoimmun (2003) 21(3):277–81. 10.1016/j.jaut.2003.08.003 [DOI] [PubMed] [Google Scholar]
- 174.Zhang X, Deriaud E, Jiao X, Braun D, Leclerc C, Lo-Man R. Type I interferons protect neonates from acute inflammation through interleukin 10-producing B cells. J Exp Med (2007) 204(5):1107–18. 10.1084/jem.20062013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Chelvarajan RL, Collins SM, Doubinskaia IE, Goes S, Van Willigen J, Flanagan D, et al. Defective macrophage function in neonates and its impact on unresponsiveness of neonates to polysaccharide antigens. J Leukoc Biol (2004) 75(6):982–94. 10.1189/jlb.0403179 [DOI] [PubMed] [Google Scholar]
- 176.Genovese F, Mancuso G, Cuzzola M, Biondo C, Beninati C, Delfino D, et al. Role of IL-10 in a neonatal mouse listeriosis model. J Immunol (1999) 163(5):2777–82. [PubMed] [Google Scholar]
- 177.Sun C-M, Deriaud E, Leclerc C, Lo-Man R. Upon TLR9 signaling, CD5+ B cells control the IL-12-dependent Th1-priming capacity of neonatal DCs. Immunity (2005) 22(4):467–77. 10.1016/j.immuni.2005.02.008 [DOI] [PubMed] [Google Scholar]
- 178.Kollmann TR, Crabtree J, Rein-Weston A, Blimkie D, Thommai F, Wang XY, et al. Neonatal innate TLR-mediated responses are distinct from those of adults. J Immunol (2009) 183(11):7150–60. 10.4049/jimmunol.0901481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Wynn JL, Cvijanovich NZ, Allen GL, Thomas NJ, Freishtat RJ, Anas N, et al. The influence of developmental age on the early transcriptomic response of children with septic shock. Mol Med (2011) 17(11–12):1146–56. 10.2119/molmed.2011.00169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Belderbos ME, van Bleek GM, Levy O, Blanken MO, Houben ML, Schuijff L, et al. Skewed pattern of toll-like receptor 4-mediated cytokine production in human neonatal blood: low LPS-induced IL-12p70 and high IL-10 persist throughout the first month of life. Clin Immunol (2009) 133(2):228–37. 10.1016/j.clim.2009.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]