

Abstract
Nuclear factor (NF)-κB plays a major role in the pathogenesis of B-cell neoplasms. A broad array of mostly extracellular stimuli has been reported to activate NF-κB, to various degrees, in chronic lymphocytic leukemia (CLL) cells. Because CLL cells harbor high levels of unphosphorylated (U) signal transducer and activator of transcription (STAT)-3 protein and U-STAT3 was reported to activate NF-κB, we sought to determine whether U-STAT3 activates NF-κB in CLL. Using the electrophoretic mobility shift assay (EMSA) we studied peripheral blood low-density cells from 15 patients with CLL and found that CLL cell nuclear extracts from all the samples bound to an NF-κB DNA probe, suggesting that NF-κB is constitutively activated in CLL. Immunoprecipitation studies showed that STAT3 bound NF-κB p65, and confocal microscopy studies detected U-STAT3/NF-κB complexes in the nuclei of CLL cells, thereby confirming these findings. Furthermore, infection of CLL cells with retroviral STAT3-shRNA attenuated the binding of NF-κB to DNA, as assessed by EMSA, and downregulated mRNA levels of NF-κB-regulated genes, as assessed by quantitative polymerase chain reaction. Taken together, our data suggest that U-STAT3 binds to the NF-κB p50/p65 dimers and that the U-STAT3/NF-κB complexes bind to DNA and activate NF-κB-regulated genes in CLL cells.
Introduction
Chronic lymphocytic leukemia (CLL) is the most common adult leukemia in the Western Hemisphere. CLL is characterized by a dynamic imbalance between the proliferation and apoptosis of neoplastic B-lymphocytes co-expressing CD5 and CD19 antigens, leading to the accumulation of these cells in the peripheral blood, bone marrow, and lymphatic tissues (1). Several mechanisms have been reported to provide CLL cells with a survival advantage. One such mechanism involves the activation of nuclear factor (NF)-κB (2-8). NF-κB plays an important role in the survival and proliferation of normal and neoplastic B cells. In CLL, NF-κB has been found to be activated, to a variable degree, regardless of disease stage or treatment status (2-8), suggesting that NF-κB might be a target for therapy in CLL (9-11).
The NF-κB family of transcription factors consists of p50, p52, p65 (RelA), c-Rel, and RelB, which share an N-terminal Rel homology domain responsible for DNA binding and homo- and heterodimerization. NF-κB dimers bind to κB binding sites within the promoters/enhancers of NF-κB target genes and regulate transcription. Typically, NF-κB dimers are associated with one of three IκB proteins, IκBα, IκBβ, or IκBε, or the precursor proteins p100 and p105, which maintain NF-κB dimers in the cytoplasm in an inactive state. The activation of NF-κB can be mediated by either the canonical pathway or the alternative pathway. The canonical pathway is mainly activated by extracellular factors such as ligands, whose interaction with their corresponding cellular receptors results in activation of the β subunit of the IκB kinase (IKK) complex (IKKβ) that induces the phosphorylation and degradation of the NF-κB inhibitor IκBα. NF-κB activation in response to antigen receptor ligation is mediated by the IKK enzymatic subunits, IKKα, IKKβ, and IKKγ (NEMO). Following IκBα degradation, NF-κB heterodimers (composed of p50, p65, and/or c-Rel) translocate to the nucleus and bind to DNA. Activation of the alternative NF-κB pathway results from the transformation of NF-κB2/p100 to p52, which is triggered by the phosphorylation of NF-κB2/p100 by the α-subunit of the IKK complex (IKKα). This allows for nuclear translocation of p52, along with RelB, and induction of NF-κB-regulated genes (12).
NF-κB is activated in CLL by several different mechanisms: interaction with stroma cells (13), activation of the tumor necrosis factor receptor (TNFR) family members (14-17), activation of the cell surface receptor CD40 by its ligand CD154 (4, 18, 19), activation of the B-cell receptor (20), activation of Notch signaling (21), induction of nitric oxide synthase (22), deregulation of the caspase-recruitment domain (CARD) membrane-associated guanylate kinase protein 1(23), deregulation of the T cell leukemia/lymphoma-1 (TCL1) oncogene (24), induction of glycogen synthase kinase-3β (GSK-3β) (25), and modulation of epigenetic regulators (26).
In 2005, Yang et al. described another, previously unknown, mechanism of NF-κB activation (27). They found that unphosphorylated (U) signal transducer and activator of transcription (STAT)-3 binds to the NF-κB dimers p65/p50 in competition with IκB. The U-STAT3/NF-κB complex translocates to the nucleus, binds to DNA, and activates NF-κB-regulated genes. We recently found that STAT3, constitutively phosphorylated on serine 727 residues in CLL cells, induces the production of STAT3 protein and that CLL cells harbor high levels of U-STAT3 (28). Therefore, we sought to determine whether, as in other cellular systems (29), U-STAT3 activates NF-κB in CLL cells.
Materials and Methods
Cell fractionation
After obtaining Institutional Review Board (IRB)-approved informed consent, we collected peripheral blood (PB) cells from healthy donors and from 22 patients with CLL who were treated at The University of Texas MD Anderson Cancer Center Leukemia Clinic during the years 2006-2010. The clinical characteristics of the patients with CLL whose PB samples were used in the current study are presented in Table 1. To isolate low-density cells, PB cells were fractionated using Ficoll Hypaque 1077 (Sigma-Aldrich, St. Louis, MO). More than 90% of the CLL PB cells were CD19+/CD5+ lymphocytes. To isolate healthy volunteers' CD19+ cells, low-density PB cells were fractionated using micro-immunomagnetic beads (Miltenyi Biotec, Auburn, CA) in accordance with the manufacturer's instructions. Flow cytometry analysis confirmed that ≥ 95% of the fractionated cells were CD19+.
Table 1. Patient characteristics.
| CLL Pt. # |
Sex | Age (Yr) |
WBC (109/l) |
Lymph. % |
Hb. (g/dl) |
Plts. (109/l) |
Rai stage | CD38+/CD19+ % |
β2M (mg/dl) |
VH mutation | ZAP- 70 % |
Status | Cytogenetics | Previous treatment |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | M | 58 | 73.7 | 90 | 13.1 | 128 | 1 | NA | 4.1 | Y | 80.9 | Alive | t 12 | |
| 2 | F | 74 | 102.2 | 88 | 12.3 | 236 | 1 | NA | 1.9 | N | NA | Alive | NA | |
| 3 | F | 88 | 49.1 | 85 | 12.2 | 186 | 0 | NA | 3 | ND | NA | Alive | D13 | |
| 4 | M | 61 | 52.9 | 84 | 14.8 | 129 | 1 | 0.9 | 2.7 | N | Pos. | Alive | del 11q | FCR |
| 5 | M | 63 | 143 | 82 | 13 | 164 | 1 | 97.5 | 3 | Y | 33.3 | Alive | del 11q | FCR |
| 6 | F | 70 | 118 | 94 | 12.5 | 178 | 1 | 62.5 | 3.2 | Y | 0 | Alive | t 12 | Revlimid |
| 7 | M | 60 | 133.5 | 96 | 13.1 | 113 | 0 | NA | 2.8 | N | 0 | Alive | del 13q | |
| 8 | F | 56 | 95.5 | 97 | 13 | 334 | 0 | 2.1 | 2.4 | Y | 0 | Alive | Dip. | |
| 9 | F | 74 | 66.7 | 94 | 12.5 | 231 | 1 | NA | 1.8 | ND | NA | Alive | NA | |
| 10 | F | 75 | 160.7 | 90 | 12.1 | 146 | 1 | 1 | 4 | Y | 44.6 | Alive | del 13q | FCR |
| 11 | F | 72 | 136.6 | 91 | 11.8 | 244 | 2 | 2 | 3.6 | Y | Pos. | Alive | t 12 | Revlimid |
| 12 | M | 63 | 96 | 92 | 14.7 | 155 | 0 | 0.3 | 3.5 | Y | 0 | Alive | del 13q | |
| 13 | M | 60 | 90.6 | 91 | 14.7 | 167 | 2 | NA | 4.8 | Y | 10.8 | Alive | del 13q | FCR |
| 14 | F | 70 | 26.4 | 93 | 10.1 | 101 | 3 | 0.2 | 3.2 | Y | 3.4 | Alive | del 13q | Revlimid |
| 15 | F | 58 | 172.6 | 98 | 13.3 | 212 | 1 | NA | 3.3 | Y | 0 | Alive | NA | |
| 16 | F | 72 | 46.9 | 79 | 9.8 | 272 | 3 | 2 | 4.7 | Y | Pos. | Alive | t 12 | Revlimid |
| 17 | F | 60 | 111.8 | 92 | 12.7 | 219 | 0 | NA | 3.5 | Y | 1.8 | Alive | NA | Ritux. GM |
| 18 | F | 75 | 60.9 | 89 | 12.5 | 179 | 0 | NA | 3.5 | Y | NA | Alive | NA | |
| 19 | M | 47 | 130.9 | 88 | 13 | 258 | 1 | 98.8 | 3.3 | Y | 46.2 | Alive | NA | FCR |
| 20 | M | 48 | 8.7 | 20 | 15.2 | 173 | 0 | NA | NA | N | Pos. | Alive | NA | CFAR |
| 21 | F | 58 | 184.4 | 88 | 13.6 | 216 | 1 | NA | 2.7 | N | Neg. | Alive | NA | |
| 22 | F | 75 | 258.9 | 94 | 11.8 | 140 | 1 | 1.1 | 4.2 | Y | 44.6 | Alive | del 13q |
Abbreviations: Yr., years; WBC, white blood cells; lymph., lymphocytes; Hb., hemoglobin; Plts., platelets; β2M, β2 microglobulin; M, male; F, female; N/A, not available; VH mutation, hypermutation of the immunoglobulin heavy chain gene presented as N (negative; if % derivation from the germline sequence is ≤ 2%) or Y (positive, if % derivation from germline sequence is > 2%); Ritux., Rituxan; GM, granulocyte-macrophage colony-stimulating factor; FCR, cyclophosphamide, fludarabine, and rituxan; CFAR, cyclophosphamide, fludarabine, alemtuzumab, and rituxan; Pos., positive (analyzed by immunohistochemistry only); Neg., negative; t, translocation; del, deletion.
Electrophoretic mobility shift assay (EMSA)
Non-denatured nuclear extracts were prepared using the NE-PER extraction kit (Thermo Scientific, Rockford, IL). Two micrograms of nuclear protein were incubated with a biotin-labeled NF-κB p65 binding-site DNA probe (5′-AGTTGAGGGGACTTTCCCAGGC-3′; synthesized by Sigma Genosys, The Woodlands, TX) in binding buffer for 30 minutes on ice. Following incubation, the samples were separated on a 5% polyacrylamide gel in Tris-borate EDTA, transferred onto a nylon membrane, and fixed on the membrane by ultraviolet cross-linking. The biotin-labeled probe was detected with strepavidin-horseradish peroxidase (Gel–Shift Kit; Panomics, Fremont, CA). As a negative control, we used a probe lacking nuclear extracts. The competition control consisted of up to 7-fold excess unlabeled cold probe combined with the biotin-labeled probes.
Immunoprecipitation
Cell pellets were lysed in ice-cold radioimmunoprecipitation (Ripa) assay buffer containing 1mM sodium orthovanadate, 10 mM sodium fluoride, 100 μM ethylenediaminetetraacetic acid (EDTA), 5 mM β-glycerol phosphate, 1 μg/mL aprotinin, 0.4 mM benzamidine, 1 μg/mL antipain, 1 mM phenylmethylsulphonyl fluoride (PMSF), 1 μg/mL leupeptin, and 1 μg/mL trypsin inhibitor soybean. The lysed cell pellets were incubated on ice for 5 minutes and vortexed for 1 minute. Then, cell debris was eliminated by centrifugation at 14,000 rpm for 15 minutes, supernatants were harvested, and the protein concentration was determined using the Micro BCA protein assay reagent kit (Thermo Scientific, Pierce, Rockford, IL). Whole-cell lysates were incubated with 4 μg of polyclonal rabbit anti–human STAT3 antibodies (Upstate Cell Signaling Solutions/Millipore, Billerica MA) for 16 hours at 4°C. Protein A agarose beads (Upstate Cell Signaling Solutions) were added for 2 hours at 4°C. As a negative control, whole-cell lysates were incubated with either rabbit serum and protein A agarose beads or protein A agarose beads only. After 3 washes with Ripa assay buffer, the beads were resuspended in sodium dodecyl sulfate (SDS) sample buffer, boiled for 5 minutes, and removed by centrifugation. The supernatant proteins were separated by SDS-polyacrylamide gel electrophoresis (PAGE) (as described below in “Western blot analysis”) and subsequently probed with mouse anti-human NF-κB P65 antibodies (Cell Signaling Technology, Beverly, MA).
Western blot analysis
Western immunoblotting was performed as previously described (28). Briefly, cell pellets were lysed in ice-cold Ripa assay buffer, incubated on ice for 5 minutes, and vortexed for 1 minute. Then, cell debris was eliminated by centrifugation at 14,000 rpm for 15 minutes, supernatants were harvested, and the protein concentration was determined using the Micro BCA protein assay reagent kit (Thermo Scientific). Supernatant proteins were denatured by boiling for 5 minutes in SDS, separated by SDS-PAGE using either 5% or 10% density gels, and transferred to a nitrocellulose membrane. After transfer, equal loading was verified by Ponceau staining.
The membranes were blocked with 5% skim milk in Tris-buffered saline and incubated with the following antibodies: monoclonal mouse anti–human STAT3 (BD Biosciences, San Jose, CA); monoclonal mouse anti–human phosphotyrosine STAT3 and polyclonal rabbit anti–human phosphoserine STAT3 (Cell Signaling Technology); and monoclonal mouse anti-human NF-κB p50 (Pierce Biotechnology, Rockford IL) and p65 (Sigma-Aldrich). After binding with horseradish peroxidase-conjugated secondary antibodies, blots were visualized with an enhanced chemiluminescence detection system (GE Healthcare, Piscataway, NJ), and densitometry analysis was performed using an Epson Expression 1680 scanner (Epson America, Long Beach, CA). Densitometry values were normalized by dividing the numerical value of each sample signal by the numerical value of the signal from the corresponding loading control. In some experiments, the membranes were stripped by incubation with stripping buffer (62.5 mM Tris-HCl, pH 6.7, 2% SDS, 100 mM β-mercaptoethanol) for 30 minutes at 50°C, washed, and reprobed with an antibody.
Confocal microscopy
CLL low-density cells were cytospun on poly-L-lysine–coated slides and fixed in 3.7% formaldehyde for 15 minutes at room temperature on a shaker. The slides were then washed 3 times with PBS, incubated with 1% Triton X-100 for 5 minutes at room temperature, and washed 3 more times with PBS before blocking with mouse serum for 1 hour. After blocking, the slides were washed in PBS and incubated overnight with PE-labeled mouse anti–human -STAT3 antibody (BD Biosciences) and AlexaFluor 488-labeled mouse anti-human NF-κB (BioLegend, San Diego, CA). After incubation, the slides were washed three times in PBS and then mounted with Vectashield hard set (Vector Laboratories, Burlingame, CA). The mounted slides were viewed using an Olympus FluoView 500 laser scanning confocal microscope (Olympus America, Houston, TX), and the images were analyzed using FluoView software (Olympus).
Generation of green fluorescence protein (GFP)-lentivirus STAT3 shRNA and infection of cells
293T cells (American type culture collection) were co-transfected with green fluorescence protein (GFP)-lentivirus STAT3 small hairpin RNA (shRNA) or GFP-lentivirus empty vector and the packaging vectors pCMVδR8.2 and pMDG (generously provided by Dr. G. Inghirami, Torino, Italy) using the superfect transfection reagent (Qiagen, Inc., Valencia, CA). 293T cell culture medium was changed after 16 hours and collected after 48 hours. The culture medium was filtered through a 45-μm syringe filter to remove floating cells; the lentivirus was then concentrated by filtration through an Amicon ultra centrifugal filter device (Milipore), and the concentrated supernatant was used to infect CLL cells.
CLL cells (5 × 106/mL) were incubated in 6-well plates (Becton Dickinson, Franklin Lakes, NJ) in 2 mL DMEM supplemented with 10% FCS and transfected with 100 μL of the viral supernatant. Polybrene (10 ng/mL) was added to the viral supernatant at a ratio of 1:1000 (v/v). Transfection efficiency was measured after 48-72 hours and was found to range between 30% and 60% (calculated on the basis of the ratio of propidium iodide (PI)-negative/GFP-positive cells). These experiments were conducted by using the FACSCalibur flow cytometer (Becton Dickinson Immunocytometry Systems, San Jose, CA). Data analysis was performed using CellQuest Pro software (Becton Dickinson). A shift from the control curve was calculated as percent shift beyond control.
Chromatin Immunoprecipitation (ChIP)
The enzymatic ChIP Kit (Cell Signaling Technology) was used. Fractionated CLL cells were cross-linked with 1% formaldehyde for 10 minutes at room temperature, a glycine solution was added to arrest the reaction, and the cells were washed with ice-cold PBS. The cells were pooled, pelleted, and incubated on ice for 10 minutes in cell lysis buffer supplemented with 100 mM phenylmethylsulfonyl fluoride, dithiothreitol (DTT) and a protease cocktail inhibitor mix. Nuclei were pelleted and resuspended in buffer supplemented with DTT, digested by micrococcal nuclease, and homogenized on ice. Following sonication (35 pulses, 20 sec/pulse at 25–30% power) and centrifugation, 12 mg of sheared chromatin was incubated with anti-STAT3 or Rabbit serum (negative control) overnight at 4°C. Then, magnetic-coupled protein G beads were added and the chromatin was incubated for 2 hours in rotation. Antibody-bound protein/DNA complexes were washed, eluted, treated with proteinase digest proteins, and subjected to PCR analyses. An aliquot of chromatin that was not incubated with an antibody was used as the input control sample. The primers to amplify the human STAT3 promoter were F: 3′-CCG AAC GAG CTG GCC TTT CAT-5′ and R: 5′-GGA TTG GCT GAA GGG GCT GTA-3′; to amplify the VEGF C promoter: F: 3′-CCA GAA AGG ATG TGT AGC ATC-5′ and R:5′-TGG TCC TCT GTA ACC TGC TCA-3′; to amplify the CXCR5 promoter: F: 3′-CCT GCC TCA CAA CTC ATC ACT-5′ and R:'-GTT GAG ACA ATT ATT GCC GGG-3′; and to amplify the CCL5 promoter: F: 3′- CTCACACTGTAAATTGAGGCA-5′and R: 5′-AGG TCG CTT AGC AAG TAA ATG-3′. The human RPL30 gene primers were provided by Cell Signaling Technologies. PCR products were resolved on 1.8% agarose gels containing ethidium bromide.
RNA purification and quantitative real-time PCR
RNA was isolated using an RNeasy purification procedure (Qiagen, Inc.). RNA quality and concentration were analyzed with a NanoDrop spectrophotometer (ND-1000; NanoDrop Technologies, Wilmington, Delaware). Ten micrograms of total RNA was used for one-step RT-PCR (Applied Biosystems, Foster City, CA) with the sequence detection system ABI Prism 7700 (Applied Biosystems) using the TaqMan gene expression assay for STAT3, VEGF C, CCL5, and CXCR5 (Hs00374200_m1, Hs01099203_m1, Hs00174575_m1, Hs00173527_m1, respectively), according to the manufacturer's instructions. Samples were run in triplicate, and relative quantification was performed by comparing the values obtained at the fractional cycle number at which the amount of amplified target reaches a fixed (CT) threshold.
Results
NF-κB is constitutively activated in CLL cells
Several investigators have reported that NF-κB is activated, to various degrees, in CLL cells (2-8). To confirm these observations, we obtained PB CLL cells and, by using EMSA, assessed NF-κB-DNA binding. We studied randomly chosen PB samples from 15 patients with CLL who had favorable or unfavorable prognostic factors and different stages of disease (Table 1). As shown in Fig. 1, we found that nuclear extracts from the PB cells of all 15 patients formed complexes with the NF-κB DNA probe, and the addition of excess (5-fold) unlabeled (cold) probe inhibited the binding. These data suggest that NF-κB was constitutively activated in the CLL cells of all studied patients.
Figure 1.
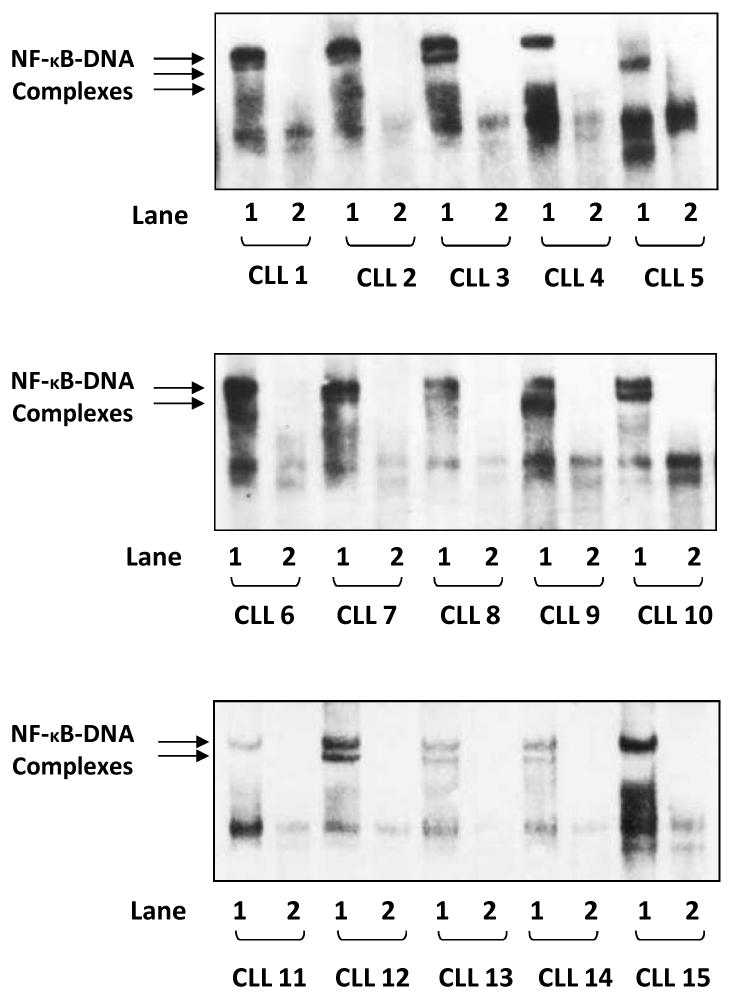
NF-κB is constitutively activated in CLL cells. EMSA studies of PB low-density cell nuclear extracts from 15 different patients with CLL are presented. Lane 1 of each sample depicts binding of CLL cell nuclear extract to a biotin-labeled (hot) NF-κB DNA probe, whereas lane 2 depicts a reduction in binding when an excess of unlabeled probe was added to the biotin-labeled (hot) κB DNA probe (hot + cold). In all 15 samples, NF-κB-DNA (p50/p65) complexes were detected and NF-κB-DNA binding was almost completely abolished by the cold probe.
STAT3 binds NF-κB in CLL cells, and the U-STAT3/NF-κB complex translocates to the nucleus
The p65/p50 heterodimer is the prototype NF-κB transcription factor that is crucial for the expression of genes encoding several cytokines and pro-inflammatory mediators. To determine whether STAT3 binds NF-κB in CLL cells, as previously reported in another cellular system (29), we immunoprecipitated STAT3 protein with rabbit anti–human STAT3 antibodies, and detected NF-κB p65 and p50, and STAT3 proteins in the immunoprecipitate by western blotting using mouse anti-human p65, p50, and STAT3 antibodies, respectively. As shown in Fig. 2A, the NF-κB p65 and p50 proteins were detected in CLL cell STAT3 immunopreciptate obtained from 3 different PB CLL samples. To further delineate this observation, p65 protein was immunoprecipitated with mouse anti–human p65 antibodies, and STAT3, serine pSTAT3, p65, and p50 proteins were detected by western blot analysis. As shown in Fig. 2B, p65, p50, STAT3, but not serine pSTAT3 proteins were detected in CLL cell p50 immunopreciptate obtained from 4 different PB CLL samples, suggesting that U-STAT3 binds NF-κB p50/p65 dimers in CLL cells.
Figure 2.
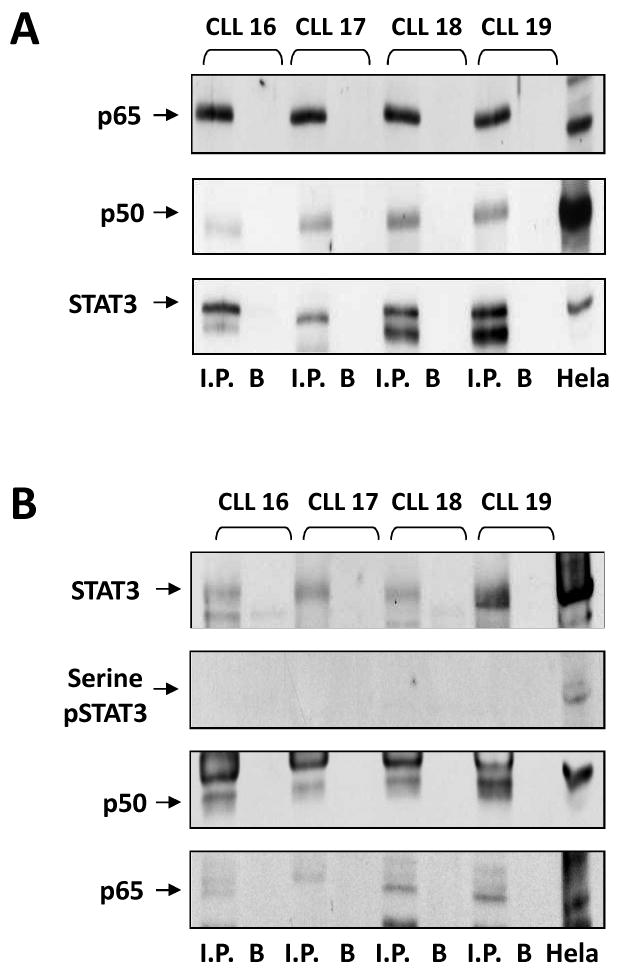
U-STAT3 binds NF-κB p50/p65 dimers. A. PB low-density cell-protein extracts from four different patients with CLL were immunoprecipitated with anti-Stat3 antibodies. As shown, STAT3 and the NF-κB p65 and p50 proteins were detected in the immunoprecipitates of all three PB samples. B. PB low-density cell-protein extracts from the same patients with CLL were immunoprecipitated with anti-p65 antibodies. Hela cell protein extract was used as a positive control. As shown, p65, p50, STAT3, but not serine pSTAT3 proteins were detected in all immunopreciptates. I.P., immunoprecipitate; B, beads (control).
To confirm this observation, we conducted confocal microscopy studies. In normal PB CD19+ cells, faint signals of STAT3 were detected, as we previously reported (28). Scattered signals of the NF-κB p65 protein were detected in the cytoplasm and the nucleus in these control cells (Fig. 3A), likely as a result of the constant shuttling of IκBα/NF-κB complexes between the nucleus and the cytoplasm (30). In contrast, in CLL cells (characterized by an overwhelmingly big nucleus and a thin cytoplasm (28)), dense signals of STAT3 and of the p65 form of NF-κB, as well as STAT3/p65 complexes, were detected mainly in the nucleus and, to a lesser extent, in the cytoplasm (Fig. 3B).
Figure 3.
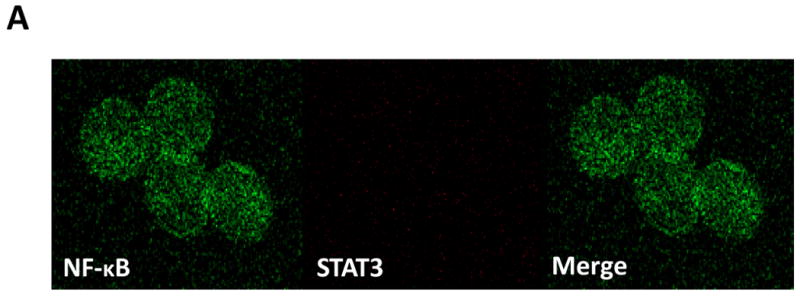
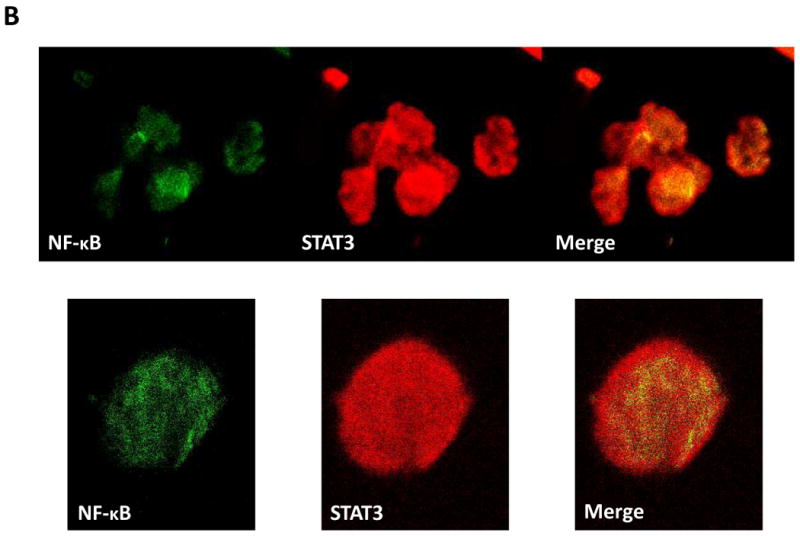
U-STAT3-NF-κB complexes are detected in CLL cells but not in normal CD19+ cells. A. Confocal microscopy studies of normal PB CD19+ cells detected a scattered signal of the NF-κB p65 protein in the cytoplasm and nucleus (left panel; green), faint signals of STAT3 (middle panel; red) but no signal of STAT3-p65 complexes (right panel; yellow). B. Confocal microscopy studies of PB CLL cells detected a dense signal of p65 (green), STAT3 (red), and STAT3-p65 complexes (yellow) in the cytoplasm and mainly in the nucleus. One field of CLL cells (upper panel) and a single CLL cell (lower panel) are shown. Depicted are representative photomicrographs (X 400) obtained from two different samples of normal CD19+ lymphocytes and CLL cells of patients 19 and 20.
U-STAT3/NF-κB complexes bind to DNA and activate NF-κB-regulated genes
To determine whether U-STAT3/NF-κB complexes bind to DNA we isolated nuclear extracts from CLL cells and, using EMSA, assessed binding of CLL cell nuclear proteins to the κB binding site. As shown in Fig. 4A, we found that both anti-p65 and anti-STAT3 antibodies, added to the DNA biotinylated probe, induced a supershift, suggesting that the bands detected by EMSA consisted not only of NF-κB subunits but of STAT3 protein as well. To further delineate this finding, we infected CLL cells with retroviral STAT3-shRNA and, using EMSA examined the binding of CLL cell nuclear extracts to the biotinylated NF-κB p65 binding-site DNA probe. As shown in Fig. 4B, infection of CLL cells with retroviral STAT3-shRNA (previously found to downregulate STAT3 mRNA and protein levels (28)), but not with empty virus, significantly attenuated the binding of NF-κB heterodimers to DNA. To determine whether U-STAT3/NF-κB complexes bind to the promoters of NF-κB genes we used ChIP. As shown in Fig. 5A, anti-STAT3 antibodies coimmunoprecipitated DNA of STAT3, and the NF-κB-regulated genes VEGF C, CCL5 and CXCR5. Then, we asked whether U-STAT3/NF-κB complexes activate NF-κB-regulated genes. As shown in Fig. 5B, STAT3-shRNA downregulated mRNA levels of the NF-κB-regulated genes VEGF C, CCL5, and CXCR5 (which are not regulated by STAT3 (31, 32)) and, as previously reported (28), STAT3-shRNA downregulated STAT3 mRNA. Taken together, these data suggest that U-STAT3/NF-κB complexes bind to DNA and activate NF-κB-target genes.
Figure 4.
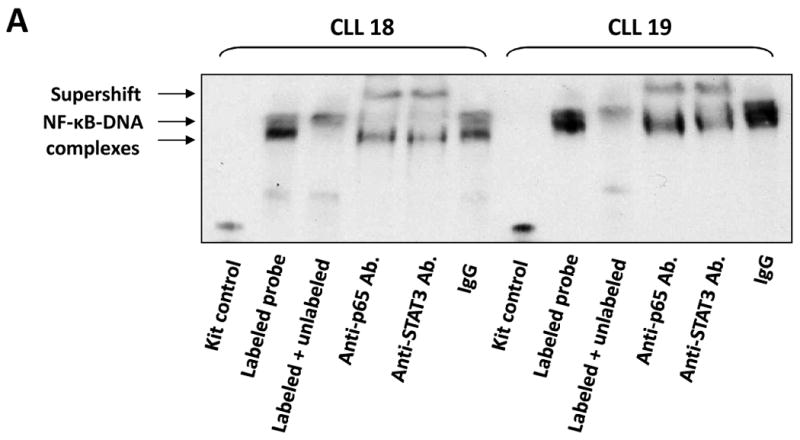
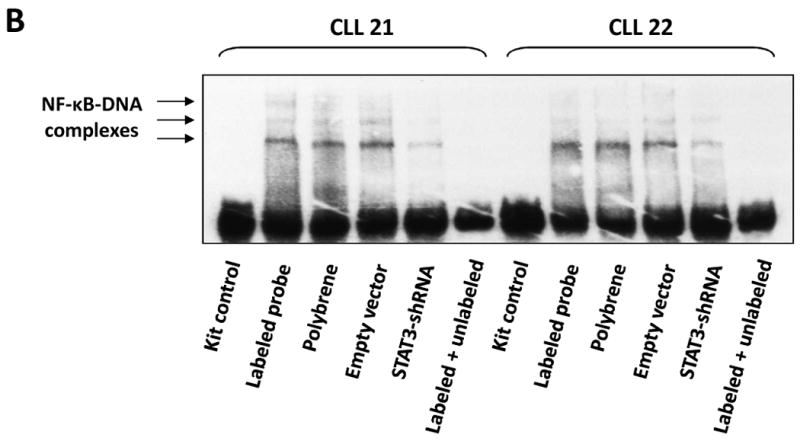
The U-STAT3/NF-κB complex binds to DNA. A. CLL cell nuclear proteins p65 and STAT3 bind to a biotinylated κB-DNA probe. EMSA of CLL cell nuclear extract is depicted. As shown, the addition of anti-p65 or anti-STAT3 antibodies to the κB DNA biotinylated probe induced a supershift, suggesting that p65 and STAT3 bind to DNA. B. STAT3-shRNA attenuates the binding of CLL cell nuclear extract to a biotin-labeled DNA probe. CLL cells were infected either with lentiviral STAT3-shRNA or empty virus. Nuclear protein was extracted and EMSA was performed. As shown, NF-κB-DNA complexes were detected, and excess unlabeled (cold) DNA probe abolished the binding. Furthermore, infections with STAT3-shRNA, but not with empty virus, significantly attenuated NF-κB-DNA binding.
Figure 5.
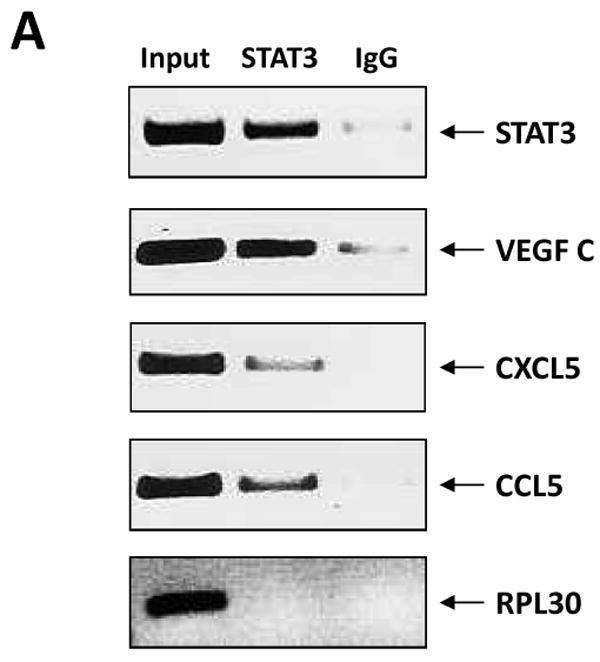

U-STAT3/NF-κB complexes activate NF-κB-target genes. A. STAT3-shRNA downregulated mRNA levels of specific NF-κB-regulated genes. As shown, infection with lentiviral STAT3-shRNA downregulated mRNA levels of VEGF C, CCL5, and CXCR5. As expected, lentiviral STAT3-shRNA downregulated the level of STAT3 mRNA as well. In this experiment we used PB cells from patient 21. B. STAT3 co-immunoprecipitates with DNA of NF-κB-regulated genes. CLL cell-derived chromatin was immunoprecipitated by anti-STAT3 antibodies or rabbit serum (IgG control). The co-immunoprecipitated DNA was amplified by PCR using promoter constructs of STAT3, VEGF C, CCL5, CXCR5, or the control gene RPL30. As shown, DNA of STAT3, VEGF C, CCL5 and CXCR5 but not the control RPL30 gene, co-immunoprecipitated with STAT3 protein. STAT3, VEGF C, CCL5, or RPL30 DNA was detected in whole cell chromatin-extracted DNA (Input).
Discussion
In the current study we show that NF-κB was constitutively activated in CLL cells from 15 patients with CLL, regardless of disease stage or treatment status. Several groups have demonstrated that NF-κB is activated in CLL (2-11) and that its activation is mediated by different, mostly extracellular, stimuli (4, 13-26). Usually, the activation of NF-κB is transient and cyclical in the presence of a continuous stimulus. For example, in mouse fibroblasts maintained by tumor necrosis factor (TNF), NF-κB-DNA binding appears and disappears every 30 to 60 minutes owing to repeated degradation and re-synthesis of IκB and the consequent activation and inactivation of NF-κB, respectively (33). Because NF-κB is constitutively activated in CLL, we assumed that the activation of NF-κB is most likely induced by an uninterrupted intracellular mechanism.
We recently found that CLL cells harbor high levels of U-STAT3, the production of which is induced by phosphoserine STAT3, constitutively upregulated in unstimulated CLL cells (28). STAT3 dynamically shuttles in and out of the nucleus independent of its phosphorylation status (34, 35). The shuttling of STAT3 from the cytoplasm to the nucleus requires binding of the coiled-coil domain of STAT3 to an importin-α-importin-β dimer to mediate passage through the nuclear pore complex. U-STAT3 also is constitutively exported out of the nucleus through a yet unknown mechanism. Similar to its phosphorylated forms, U-STAT3 can also affect NF-κB-regulated gene expression indirectly by binding to NF-κB and mediating its nuclear import (29, 36). Our data are in agreement with those observations. We found that U-STAT3 binds NF-κB and that the U-STAT3/NF-κB complex shuttles to the nucleus, binds to DNA, and activates NF-κB-regulated genes. Our data suggest that STAT3 plays a key role in the constitutive activation of NF-κB in CLL cells.
A broad array of stimuli activates NF-κB, including both endogenous and exogenous ligands and physical and chemical stresses (37). The protein components of pathways that culminate in IKK activation are not yet completely understood. Typically, the binding of cytokines such as TNF-α (38, 39) or interleukin-1 (40) to their corresponding cell surface receptors (e.g., TNF-receptor (TNFR) or a Toll-like receptor) recruits adaptor proteins (e.g., TNFR-associated factors (TRAFs) and receptor-interacting proteins) to the receptor's cytoplasmic domain, and these molecules, clustered at the receptor, activate the IKK complex. IKK then phosphorylates IκB at two serine residues, which leads to its ubiquitination and degradation by the proteasome; NF-κB, free of its inhibitor, then enters the nucleus to turn on target genes (12). This established model appears to be more complex than initially thought. The crystal structure of IκBα bound to the p65/p50 heterodimer reveals that the IκBα protein masks only the nuclear localization sequence (NLS) of p65, whereas the NLS of p50 remains exposed. The exposed NLS of p50 coupled with nuclear export sequences (NES) in IκBα and p65 leads to constant shuttling of IκBα/NF-κB (p50/p65) complexes between the nucleus and the cytoplasm, despite steady state localization that appears almost exclusively cytosolic (30). Our confocal microscopy data showing NF-κB in the nucleus of normal B lymphocytes agree with this observation. Degradation of IκBα drastically alters the dynamic balance between cytosolic and nuclear localization signals to favor nuclear localization of NF-κB. Apparently, NF-κB might be activated without the degradation of IκB (29, 36), as we now show in CLL cells.
Several investigators have suggested that NF-κB should be considered a target for therapy in CLL (9-11). We have recently reported that STAT3-shRNA reduced STAT3 mRNA and protein levels and induced apoptosis in CLL cells (28). In the current study, we demonstrate that STAT3-shRNA inhibits the activity of NF-κB. Whether the STAT3 inhibitors currently being studied in clinical trials in CLL would also inhibit NF-κB activity remains to be determined.
Acknowledgments
We thank Susan Lerner and Susan C. Smith for retrieving the patients' clinical data and Dawn Chalaire for editing the manuscript.
Grant support: This study was supported by a grant from the CLL Global Research Foundation
Footnotes
Author contributions: Performed the experiments, and analyzed the data: Z.L., I. H-H. Performed retroviral infection studies, confocal microscopy studies, and analyzed the data: D.H. Performed quantitative PCR analysis and analyzed the data: P.L. Treated the studied patients, provided clinical samples and analyzed the data: A.F., S.F., M.J.K. Initiated, designed and supervised the research, and wrote the manuscript: Z.E.
Disclosure of potential Conflicts of Interest: The authors declare that no conflict of interest exists.
References
- 1.Yee KW, O'Brien SM. Chronic lymphocytic leukemia: diagnosis and treatment. Mayo Clin Proc. 2006;81:1105–29. doi: 10.4065/81.8.1105. [DOI] [PubMed] [Google Scholar]
- 2.Schuh K, Avots A, Tony HP, Serfling E, Kneitz C. Nuclear NF-ATp is a hallmark of unstimulated B cells from B-CLL patients. Leuk Lymphoma. 1996;23:583–92. doi: 10.3109/10428199609054868. [DOI] [PubMed] [Google Scholar]
- 3.Sembries S, Pahl H, Stilgenbauer S, Dohner H, Schriever F. Reduced expression of adhesion molecules and cell signaling receptors by chronic lymphocytic leukemia cells with 11q deletion. Blood. 1999;93:624–31. [PubMed] [Google Scholar]
- 4.Furman RR, Asgary Z, Mascarenhas JO, Liou HC, Schattner EJ. Modulation of NF-kappa B activity and apoptosis in chronic lymphocytic leukemia B cells. J Immunol. 2000;164:2200–6. doi: 10.4049/jimmunol.164.4.2200. [DOI] [PubMed] [Google Scholar]
- 5.Bernal A, Pastore RD, Asgary Z, et al. Survival of leukemic B cells promoted by engagement of the antigen receptor. Blood. 2001;98:3050–7. doi: 10.1182/blood.v98.10.3050. [DOI] [PubMed] [Google Scholar]
- 6.Cuni S, Perez-Aciego P, Perez-Chacon G, et al. A sustained activation of PI3K/NF-kappaB pathway is critical for the survival of chronic lymphocytic leukemia B cells. Leukemia. 2004;18:1391–400. doi: 10.1038/sj.leu.2403398. [DOI] [PubMed] [Google Scholar]
- 7.Rodriguez A, Martinez N, Camacho FI, et al. Variability in the degree of expression of phosphorylated IkappaBalpha in chronic lymphocytic leukemia cases with nodal involvement. Clin Cancer Res. 2004;10:6796–806. doi: 10.1158/1078-0432.CCR-04-0753. [DOI] [PubMed] [Google Scholar]
- 8.Hewamana S, Alghazal S, Lin TT, et al. The NF-kappaB subunit Rel A is associated with in vitro survival and clinical disease progression in chronic lymphocytic leukemia and represents a promising therapeutic target. Blood. 2008;111:4681–9. doi: 10.1182/blood-2007-11-125278. [DOI] [PubMed] [Google Scholar]
- 9.Pickering BM, de Mel S, Lee M, et al. Pharmacological inhibitors of NF-kappaB accelerate apoptosis in chronic lymphocytic leukaemia cells. Oncogene. 2007;26:1166–77. doi: 10.1038/sj.onc.1209897. [DOI] [PubMed] [Google Scholar]
- 10.Lopez-Guerra M, Colomer D. NF-kappaB as a therapeutic target in chronic lymphocytic leukemia. Expert Opin Ther Targets. 14:275–88. doi: 10.1517/14728221003598930. [DOI] [PubMed] [Google Scholar]
- 11.Hertlein E, Wagner AJ, Jones J, et al. 17-DMAG targets the NF-{kappa}B family of proteins to induce apoptosis in CLL: clinical implications of HSP90 inhibition. Blood. doi: 10.1182/blood-2010-01-263756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008;132:344–62. doi: 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- 13.Edelmann J, Klein-Hitpass L, Carpinteiro A, et al. Bone marrow fibroblasts induce expression of PI3K/NF-kappaB pathway genes and a pro-angiogenic phenotype in CLL cells. Leuk Res. 2008;32:1565–72. doi: 10.1016/j.leukres.2008.03.003. [DOI] [PubMed] [Google Scholar]
- 14.Munzert G, Kirchner D, Stobbe H, et al. Tumor necrosis factor receptor-associated factor 1 gene overexpression in B-cell chronic lymphocytic leukemia: analysis of NF-kappa B/Rel-regulated inhibitors of apoptosis. Blood. 2002;100:3749–56. doi: 10.1182/blood.V100.10.3749. [DOI] [PubMed] [Google Scholar]
- 15.Kern C, Cornuel JF, Billard C, et al. Involvement of BAFF and APRIL in the resistance to apoptosis of B-CLL through an autocrine pathway. Blood. 2004;103:679–88. doi: 10.1182/blood-2003-02-0540. [DOI] [PubMed] [Google Scholar]
- 16.Endo T, Nishio M, Enzler T, et al. BAFF and APRIL support chronic lymphocytic leukemia B-cell survival through activation of the canonical NF-kappaB pathway. Blood. 2007;109:703–10. doi: 10.1182/blood-2007-04-081786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nishio M, Endo T, Tsukada N, et al. Nurselike cells express BAFF and APRIL, which can promote survival of chronic lymphocytic leukemia cells via a paracrine pathway distinct from that of SDF-1alpha. Blood. 2005;106:1012–20. doi: 10.1182/blood-2004-03-0889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Karin M, Lin A. NF-kappaB at the crossroads of life and death. Nat Immunol. 2002;3:221–7. doi: 10.1038/ni0302-221. [DOI] [PubMed] [Google Scholar]
- 19.Romano MF, Lamberti A, Tassone P, et al. Triggering of CD40 antigen inhibits fludarabine-induced apoptosis in B chronic lymphocytic leukemia cells. Blood. 1998;92:990–5. [PubMed] [Google Scholar]
- 20.Muzio M, Scielzo C, Bertilaccio MT, Frenquelli M, Ghia P, Caligaris-Cappio F. Expression and function of toll like receptors in chronic lymphocytic leukaemia cells. Br J Haematol. 2009;144:507–16. doi: 10.1111/j.1365-2141.2008.07475.x. [DOI] [PubMed] [Google Scholar]
- 21.Rosati E, Sabatini R, Rampino G, et al. Constitutively activated Notch signaling is involved in survival and apoptosis resistance of B-CLL cells. Blood. 2009;113:856–65. doi: 10.1182/blood-2008-02-139725. [DOI] [PubMed] [Google Scholar]
- 22.Hammadi A, Billard C, Faussat AM, Kolb JP. Stimulation of iNOS expression and apoptosis resistance in B-cell chronic lymphocytic leukemia (B-CLL) cells through engagement of Toll-like receptor 7 (TLR-7) and NF-kappaB activation. Nitric Oxide. 2008;19:138–45. doi: 10.1016/j.niox.2008.04.017. [DOI] [PubMed] [Google Scholar]
- 23.Blonska M, Lin X. CARMA1-mediated NF-kappaB and JNK activation in lymphocytes. Immunol Rev. 2009;228:199–211. doi: 10.1111/j.1600-065X.2008.00749.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pekarsky Y, Palamarchuk A, Maximov V, et al. Tcl1 functions as a transcriptional regulator and is directly involved in the pathogenesis of CLL. Proc Natl Acad Sci U S A. 2008;105:19643–8. doi: 10.1073/pnas.0810965105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ougolkov AV, Bone ND, Fernandez-Zapico ME, Kay NE, Billadeau DD. Inhibition of glycogen synthase kinase-3 activity leads to epigenetic silencing of nuclear factor kappaB target genes and induction of apoptosis in chronic lymphocytic leukemia B cells. Blood. 2007;110:735–42. doi: 10.1182/blood-2006-12-060947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen SS, Raval A, Johnson AJ, et al. Epigenetic changes during disease progression in a murine model of human chronic lymphocytic leukemia. Proc Natl Acad Sci U S A. 2009;106:13433–8. doi: 10.1073/pnas.0906455106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang J, Liao X, Agarwal MK, Barnes L, Auron PE, Stark GR. Unphosphorylated STAT3 accumulates in response to IL-6 and activates transcription by binding to NFkappaB. Genes & development. 2007;21:1396–408. doi: 10.1101/gad.1553707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hazan-Halevy I, Harris D, Liu Z, et al. STAT3 is constitutively phosphorylated on serine 727 residues, binds DNA, and activates transcription in CLL cells. Blood. 2010;115:2852–63. doi: 10.1182/blood-2009-10-230060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang J, Chatterjee-Kishore M, Staugaitis SM, et al. Novel roles of unphosphorylated STAT3 in oncogenesis and transcriptional regulation. Cancer Res. 2005;65:939–47. [PubMed] [Google Scholar]
- 30.Ghosh S, Karin M. Missing pieces in the NF-kappaB puzzle. Cell. 2002;109 Suppl:S81–96. doi: 10.1016/s0092-8674(02)00703-1. [DOI] [PubMed] [Google Scholar]
- 31.Pahl HL. Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene. 1999;18:6853–66. doi: 10.1038/sj.onc.1203239. [DOI] [PubMed] [Google Scholar]
- 32.Bourillot PY, Aksoy I, Schreiber V, et al. Novel STAT3 target genes exert distinct roles in the inhibition of mesoderm and endoderm differentiation in cooperation with Nanog. Stem cells (Dayton, Ohio) 2009;27:1760–71. doi: 10.1002/stem.110. [DOI] [PubMed] [Google Scholar]
- 33.Hoffmann A, Natoli G, Ghosh G. Transcriptional regulation via the NF-kappaB signaling module. Oncogene. 2006;25:6706–16. doi: 10.1038/sj.onc.1209933. [DOI] [PubMed] [Google Scholar]
- 34.Reich NC, Liu L. Tracking STAT nuclear traffic. Nat Rev Immunol. 2006;6:602–12. doi: 10.1038/nri1885. [DOI] [PubMed] [Google Scholar]
- 35.Liu L, McBride KM, Reich NC. STAT3 nuclear import is independent of tyrosine phosphorylation and mediated by importin-alpha3. Proc Natl Acad Sci U S A. 2005;102:8150–5. doi: 10.1073/pnas.0501643102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang J, Stark GR. Roles of unphosphorylated STATs in signaling. Cell Res. 2008;18:443–51. doi: 10.1038/cr.2008.41. [DOI] [PubMed] [Google Scholar]
- 37.Gilmore TD. Introduction to NF-kappaB: players, pathways, perspectives. Oncogene. 2006;25:6680–4. doi: 10.1038/sj.onc.1209954. [DOI] [PubMed] [Google Scholar]
- 38.Brown K, Gerstberger S, Carlson L, Franzoso G, Siebenlist U. Control of I kappa B-alpha proteolysis by site-specific, signal-induced phosphorylation. Science. 1995;267:1485–8. doi: 10.1126/science.7878466. [DOI] [PubMed] [Google Scholar]
- 39.Chen Z, Hagler J, Palombella VJ, et al. Signal-induced site-specific phosphorylation targets I kappa B alpha to the ubiquitin-proteasome pathway. Genes & development. 1995;9:1586–97. doi: 10.1101/gad.9.13.1586. [DOI] [PubMed] [Google Scholar]
- 40.Estrov Z, Shishodia S, Faderl S, et al. Resveratrol blocks interleukin-1beta-induced activation of the nuclear transcription factor NF-kappaB, inhibits proliferation, causes S-phase arrest, and induces apoptosis of acute myeloid leukemia cells. Blood. 2003;102:987–95. doi: 10.1182/blood-2002-11-3550. [DOI] [PubMed] [Google Scholar]