

Abstract
Here we present a label-free method for studying the mechanism of surface effects on amyloid aggregation. In this method, spin-coating is used to rapidly dry samples, in a homogeneous manner, after various incubation times. This technique allows the control of important parameters for self-assembly, such as the surface concentration. Atomic force microscopy is then used to obtain high-resolution images of the morphology. While imaging under dry conditions, we show that the morphologies of self-assembled aggregates of a model amyloid-β peptide, Aβ12–28, are strongly influenced by the local surface concentration. On mica surfaces, where the peptides can freely diffuse, homogeneous, self-assembled protofibrils formed spontaneously and grew longer with longer subsequent incubation. The surface fibrillization rate was much faster than the rates of fibril formation observed in solution, with initiation occurring at much lower concentrations. These data suggest an alternative pathway for amyloid formation on surfaces where the nucleation stage is either bypassed entirely or too fast to measure. This simple preparation procedure for high-resolution atomic force microscopy imaging of amyloid oligomers and protofibrils should be applicable to any amyloidogenic protein species.
Keywords: amyloids, protofibrils, fibrils, surface-mediated self-assembly, Aβ
Amyloid fibrils have been considered as causative agents in many neurodegenerative diseases, including Alzheimer’s disease and Parkinson’s disease,1−5 as well as other serious conditions such as diabetes6 and amyloidosis.7 Alzheimer’s disease and Parkinson’s disease are among the 10 leading causes of death in the United States, with healthcare costs totaling more than $200 billion annually.8−11 Understanding the molecular mechanisms responsible for the aggregation of metastable soluble proteins into highly ordered cross-β fibrils is central to the prediction and treatment of these diseases.4,6,12 Indeed, stabilizing nontoxic, soluble forms of these proteins has emerged as a viable and potentially general therapeutic strategy.13,14 Amyloid-β (Aβ) peptide, the main component of insoluble plaques in the brains of Alzheimer’s disease patients,1 is one of the most studied amyloid peptides.15−18 Despite years of study, many questions about the in vivo mechanism of Aβ fibrillization remain unanswered.
In vitro, Aβ peptide undergoes a nucleation-limited polymerization process to form fibrils.19,20 Depending on the solution conditions and the peptide sequence, the nucleation step requires a critical concentration for fibrillization.12,19,21,22 However, the critical concentration is usually much higher than the concentration of Aβ peptide under physiological conditions (nanomolar range).23In vivo, membrane-mediated fibril formation can provide an alternative pathway, with potentially lower barriers for fibrillization. Previous studies indicate that membrane–amyloid interactions can modulate fibrillization by increasing the local concentration and inducing a preferred orientation in the bound proteins.12,24−26 Pronchik et al.(27) demonstrated that the presence of hydrophobic interfaces can accelerate fibrillization. Shen et al.(28) showed that the existence of a mobile fibril precursor near a surface determines fibril formation ability, particularly when the solution concentration is below the critical concentration. These results reveal the importance of interactions of amyloid peptides with surfaces and interfaces in mediating fibril growth at physiologically relevant concentrations. However, the detailed mechanism of surface-mediated fibril growth remains unexplored and merits further investigation.
It has been suggested that Aβ oligomers and protofibrils formed during the nucleation stage are neurotoxic and may be the main cause of Alzheimer’s disease.15,16,29 Due to limitations in existing experimental techniques, it is extremely difficult to determine prefibrillar structures and understand the mechanisms by which surfaces affect the early stages of fibril growth at the nanoscale. Fluorescence-based techniques,20,30−33 solid-state NMR spectroscopy,17 and 2D infrared spectroscopy34,35 are common methods used to study fibril growth, but labeling and staining can potentially alter the interactions at surfaces. Other label-free methods, such as quartz crystal microbalance measurements36 and electron microscopy,37 are only feasible on specific substrates. Atomic force microscopy (AFM) is a powerful imaging technique for studying prefibrillar and fibrillar nanostructures on various surfaces, either in air or in an aqueous environment.28,38−52 For example, Mastrangelo et al.41 measured soluble Aβ42 oligomers adsorbed on mica using high-resolution AFM and Adamcik et al.42 studied fibril structures with statistical analysis. This technique can also be used to study the growth rate of fibrillar structures if they are adsorbed on a surface. For example, Kowalewski et al.(39) and Blackley et al.(40) imaged the time evolution of fibril formation on mica, and Jeong et al.(48) monitored secondary nucleation in an in situ measurement. However, imaging these nanostructures under liquid is not easy due to the instability of the microscope as well as the motion of the fibrils during imaging. Therefore, only relatively large structures can be imaged. Furthermore, if the AFM scanning rate is comparable to or slower than the fibril growth rate, the results may be hard to interpret. Additionally, the peptides may adhere to the AFM tip and reduce the resolution or produce artifacts. On the other hand, drop-casting solutions for imaging under dry conditions can present other types of artifacts, such as rapid deposition of peptides in some areas of the surface and inhomogeneous drying. Therefore, it is imperative to develop sample preparation methods that allow one to control important variables such as surface concentration, deposition rate, and substrate interactions. Here, we present a simple method that allows one to control these variables independently, which allows us to reliably study surface-mediated fibril formation using AFM. We show that on surfaces where the peptide can freely diffuse, amyloid protofibrils rapidly self-assemble, even when the solution concentration is well below the critical concentration for nucleation. Under these conditions, the nucleation stage is bypassed and amyloid protofibrils form through a process that resembles diffusion-limited aggregation.53 We construct the phase diagram of the final fibril length and show that the morphology of the fibrils on the surface is strongly correlated with the local concentration of peptides adsorbed on the surface.
Results and Discussion
Fibrillar Structures Do Not Exist in Solution
In order to demonstrate surface-mediated self-assembly, we chose a simple peptide system, Aβ12–28, a fragment of the wild-type Aβ peptide that contains the main sequence for cross-β aggregation.37,54 Solutions of this Aβ fragment with concentrations between 0.33 and 10 μM were prepared in Milli-Q water (see Materials and Methods section for details). Cryogenic transmission electron microscopy (cryo-TEM) images showed no evidence of fibrillar structure in the 10 μM sample solution (Supporting Information Figure S1). The solutions with varying peptide concentrations were then used to prepare self-assembled fibrils on mica substrates by slowly drying the samples, as shown schematically in the top row of Figure 1. Only the 10 μM sample solution was used in our spin-coating experiments, depicted at the bottom of Figure 1.
Figure 1.

Schematic diagram of different sample preparation methods. Top: Slowly dried sample under ambient conditions. Middle: Samples prepared by spin-coating with a speed of 1000 rpm after 30 or 60 min of incubation. Bottom: Samples prepared by spin-coating with a speed of 3000 rpm after 30 or 60 min of incubation.
Inhomogeneous Morphologies Form on the Surface after Slow Drying
Three 50 μL droplets of the sample solution were deposited on freshly cleaved mica and allowed to slowly dry over about ∼2.5 h. Figure 2A–F shows the various forms of fibrils, monolayers, and multilayers that were observed on the surface of these films (more images are shown in Supporting Information Figure S2). This ensemble of morphologies can be classified based on the fibril length, l, and thickness, h. The structures can then be categorized as short fibrils with l < 1 μm and h < 0.5 nm (Figure 2A), long fibrils with l > 1 μm and h < 0.5 nm (Figure 2B–D), monolayers with random patterns and h < 1 nm (Figure 2E), and multilayers with random patterns and h > 1 nm (Figure 2F). Fibrillar structures were observed on the surface at all initial solution concentrations except for the highest concentrations of 6.67 and 10 μM, where multilayers predominated (see Supporting Information Figure S2).
Figure 2.

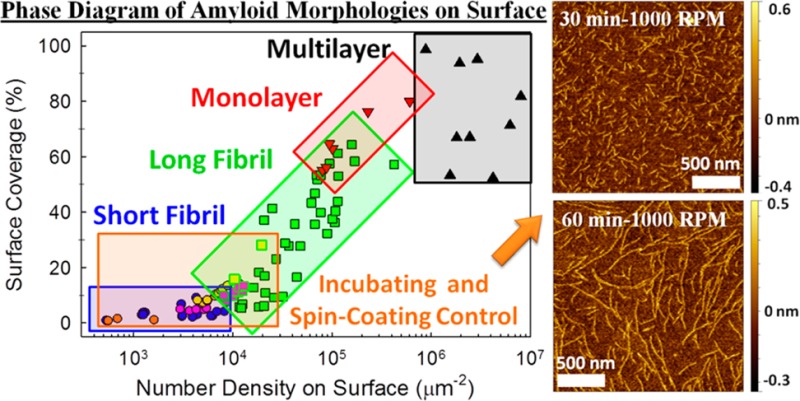
AFM images of Aβ12–28 peptide samples prepared using different drying methods. (A–F) Representative images of slowly dried samples. The solution concentrations are (A) 0.33 μM, (B) 0.67 μM, (C) 0.33 μM, (D) 3.3 μM in, (E) 3.3 μM, and (F) 6.6 μM. (G–L) Representative images of spun-cast samples on mica with different incubation times and spin speeds: (G) 0 min, 1000 rpm; (H) 30 min, 1000 rpm; (I) 60 min, 1000 rpm; (J) 0 min, 3000 rpm; (K) 30 min, 3000 rpm; and (L) 60 min, 3000 rpm. The drying times for spun-cast samples at 1000 and 3000 rpm of 3 min and 30 s, respectively, need to be added to the incubation time. The scale bar is 500 nm for all images. The color frame around each image highlights the observed morphology and corresponds to the color of phases in the phase diagram (Figure 3).
While fibrils were observed on the mica surface using the slow-drying method, we were unable to further study the specific conditions that lead to the rapid fibrillization, due to lack of control over the process. For example, the shrinking of the bulk droplet on the surface during the slow-drying process may gradually increase the peptide’s concentration at the edge of the droplet, resulting in a high local concentration and inhomogeneous distribution of the peptides on the surface. As a result, it was impossible to distinguish structures that formed before drying, when the surface coverage was homogeneous, and structures that formed during drying as a result of the increased local concentration. Other techniques usually used to quickly remove excess water suffer from similar problems. Brief descriptions of these techniques and the resulting morphologies are given in the Supporting Information. Similar to slow drying, the resulting morphologies are not homogeneous and depend strongly on the method used to dry the sample.
Drying Process Can Be Controlled via Spin-Coating
In order to probe the mechanisms leading to self-assembly and fibril formation, important variables such as the surface and bulk concentrations, the deposition rate, and the duration of the deposition need to be controlled independently. We used spin-coating to control the drying process and maintain uniform surface coverage during self-assembly. A combination of certain prespin incubation times and spin rates allowed for precise control of the peptide concentration on the surface and, therefore, the aggregate morphology, as schematically shown in Figure 1. Figure 2G–L shows some of the structures obtained after spin-coating. When the spin rate was slow (1000 rpm), homogeneously distributed fibril-like structures were observed on the surface. The structures grew in length with longer incubation times (Figure 2G–I). In contrast, at the fast spin rate (3000 rpm), globular structures predominated on the surface of all samples. At zero incubation time (Figure 2J), the samples did not contain the other types of nanostructures observed under slow-drying and slow-spinning conditions, while some fibrillar structures were observed underneath the globular structures at longer incubation times (Figure 2K,L). We note that under the two spin rates used in this study, 1000 and 3000 rpm, the drying time was 3 min and 30 s, respectively. This defined the shortest incubation times available in these experiments and effectively increased the incubation times reported in Figure 2.
Rapid Spinning Can Elucidate Bulk Structure
Spin-coating involves dispensing a solution onto a substrate, then spinning to achieve a uniform film or uniform surface coverage. During fast spinning, the centrifugal force is strong enough to break the solution film into many small droplets on the surface, as shown schematically in the bottom of Figure 1. Because of the low bulk concentration, each droplet may contain a dilute mixture of monomers, oligomers, and micelles. Rapid drying under fast-spinning conditions prevents the peptides from further rearrangement and self-assembly and allows us to study the structure of the peptides as they existed in solution (Figure 2J). The fact that fibrils are not observed in these samples further confirms that the original solution is fibril-free. When the samples are incubated for 30–60 min (Figure 2K,L), some peptides are deposited on the surface and have a chance to self-assemble before the droplet is broken and rapidly dried. In contrast to the nonincubated case, the fact that here we observe fibrils underneath globular structures provides strong evidence that the fibrillar structures formed during incubation. This suggests that during the incubation period some peptides were adsorbed on the surface and had a chance to diffuse and self-assemble on the surface before rapid drying by fast spinning.
Local Concentration Determines the Morphology on the Surface
Below, we discuss the details of the assembly process using the data obtained by the slow-drying and the slow-spinning processes. The observation of various morphologies for a single bulk concentration during slow drying indicates that the bulk concentration does not play a major role in the surface-mediated self-assembly of these peptides. The variations in the morphology are instead probably correlated with the local concentration of peptides, which can strongly vary on the surface due to inhomogeneous drying and coffee-stain effects during slow drying,55 as shown schematically in the top of Figure 1. The morphological heterogeneity observed within a sample, such as the 3.33 μM sample with long fibrils, monolayers, and multilayers (Supporting Information Figure S2), implies that the local concentration may determine the observed morphologies. When the local concentration was too high, monolayers or multilayers formed, while at lower local concentrations, elongated fibrillar structures were observed. We note that all of these fibrils rapidly formed at a 0.33 μM concentration, which is much lower than the critical micelle concentration of Aβ42 in buffer solution (17.5 μM).21
Nucleation-Free Fibrillization on the Surface
As shown in Figure 2G, we observed fibrillar structures on the mica surface even for the shortest incubation time possible in our experiment, the ∼3 min drying time for the slow-spinning process. Therefore, the lag time usually observed for the nucleation process in solution is either too short to be observed or entirely absent. Furthermore, fibrils of all lengths and sizes were formed by slow drying at concentrations that are too low for nucleation-limited fibril formation in solution. Therefore, our data suggest that the nucleation step is bypassed during surface-mediated self-assembly and another process is responsible for such unprecedented rapid fibril formation rates. The self-assembly on the two-dimensional surface is governed by the translational motion and conformational rearrangement of peptides during the incubation and drying. Two factors that can affect such self-assembly are the deposition and the surface diffusion rates.53 The local number density depends on the flux of incoming molecules during the incubation period and the adsorption–desorption rate. The strength of the interaction with the surface is important in the adsorption rate of the peptides, which can enhance fibril formation rate. However, the rapid growth of the fibrils, as shown in Figure 2, indicates that the peptides can diffuse on the substrate and form hydrogen bonds after self-assembly, which indicates low barriers for surface diffusion and rearrangement, despite high adsorption rates.
Phase Diagram for the Surface-Mediated Self-Assembly
In order to quantify the relationship between surface coverage and self-assembled morphology, each AFM image taken after slow drying (included these shown in Figure 2A–F and Supporting Information Figure S2) was analyzed as follows. Two important parameters, the surface coverage and the two-dimensional number density, were calculated. The former is defined as the percentage of the image area occupied with nanostructures, and the latter is the average number of molecules per unit area of the same image, that is, the two-dimensional concentration. Comparing these two parameters can distinguish between monolayers, where the two parameters are fully correlated, and multilayered structures, where the surface coverage is at almost 100%. Figure 3 illustrates the correlation between the surface coverage, the number density, and the observed morphologies. As the number density or the surface coverage increases, the morphology changes from short fibrils to long fibrils, to monolayers, and eventually multilayers. In this phase diagram, several boundaries between different types of morphologies can be observed. Two interesting features can be highlighted. First, two distinct types of fibrils are present in this diagram. At low number densities (<104 μm–2), the fibrils are short and scattered, while at intermediate number densities (104–105 μm–2), the fibrils are many microns long, well-packed, and cover a large surface area. Second, the regions of well-packed long fibrils and monolayer overlap around a number density of 105 μm–2. In this region, the deposition rate is high and the initial alignment of the fibrils on the surface probably determines the final morphology.
Figure 3.
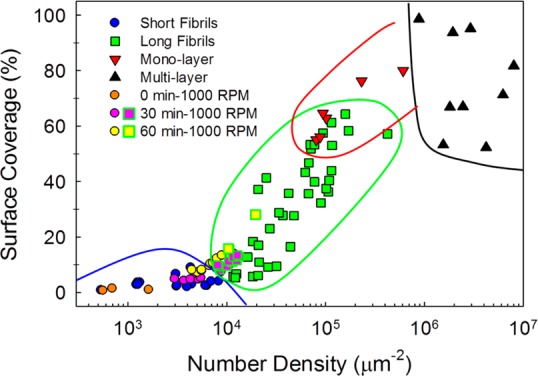
Phase diagram of Aβ12–28 samples, prepared in slow-dried and slow-spinning conditions. Each data point is obtained from a single AFM image based on analysis described in the Supporting Information. The shape of each symbol represents the observed morphology, with circles, squares, downward triangles, and upward triangles representing short fibrils (l < 1 μm, h < 0.5 nm), long fibrils (l > 1 μm, h < 0.5 nm), monolayers (h < 1 nm), and multilayers (h > 1 nm), respectively. The data obtained by spin-coating at 1000 rpm with various incubation times are categorized according to the observed morphology. The solid lines highlight the approximate phase boundaries.
Spin-Coating Can Be Used To Control Fibril Morphology and Phase
Further characterization of the observed morphologies required controlled deposition afforded by spin-coating. In order to compare the results of experiments under slow-drying and slow-spinning conditions, the slow-spinning data were analyzed in the fashion described above and added to the data presented in the phase diagram of Figure 3. These data points strongly follow the same trend and provide additional confirmation that the surface coverage and number density are good indicators of the observed morphologies. In contrast to the slow-drying conditions, the morphology of the slow-spinning aggregates can be precisely controlled using variations of the solution concentration and the incubation time, thereby controlling the deposition rate and local concentration. This provides a unique form of control over important variables for the surface-mediated self-assembly of these peptides. The slow-spinning samples (fibrillar structure in Figure 2H,I and Supporting Information Figure S4 and Figure S5A) can also be compared with samples prepared using conventional methods of drying (Figure S5), as well as the slow-drying samples (multilayers in Figure S2A–C) with the same initial bulk concentrations. These comparisons show the clear advantage of this method over other sample preparation methods in keeping the surface morphology uniform and intact. Under the slow-spinning conditions, the surface is uniformly covered with only one structure and inhomogeneous coverage and coffee-stain effects can be completely prevented.
All Fibrils Formed Are One Monomer Thick
To further understand the surface-mediated fibrillization process, we analyzed the statistics of the height and the length of the fibrils as a function of time. The height of the fibrils, determined from line profiles of the AFM images, is 3.0 ± 1.0 Å (Figure 4B). The mean height of the fibrils can also be derived from fitting two Gaussian functions to the height histogram, as shown in Figure 4C. These measurements are consistent with the image processing and lead to a thickness of 2.2 ± 1.0 Å. Thus, these fibrils can be classified as protofibrils, with a thickness consistent with that of a single peptide. The fibrils never cross each other, which is a strong indication that the rearrangements and fibrillization occurred on the two-dimensional surface and that bulk peptides did not participate in the elongation process. We also analyzed the distribution of fibril lengths using five AFM images on different areas of the substrate. Figure 4D shows the normalized length distributions of the slow-spinning samples after 30 (blue) and 60 min (orange) of incubation, respectively. The length distribution right shifts and broadens as the incubation time is increased. This is consistent with the assumption that the self-assembly mechanism is through a diffusion-limited aggregation (DLA) process on the surface,53 where the adsorbed peptides diffuse on the surface until they adhere to a cluster or to each other. In contrast to a standard DLA process, the anisotropic hydrogen bonding interactions of the peptide backbones lead to the formation of long chains instead of fractal clusters. This will be further explored in our future work.
Figure 4.

(A) Representative AFM image of 60 min/1000 rpm Aβ12–28 sample. (B) Line profile of the three white lines indicated in (A). (C) Normalized height distribution of the image shown in (A). Two Gaussian functions are fit to the data. The Gaussian peak on the left (gray) shows the position of the mica substrate and highlights the roughness of the mica substrate and the AFM noise. The Gaussian peak on the right (green) shows the height distribution of the protofibrils, which is wider than the background noise. (D) Normalized distribution of fibril lengths 30 min/1000 rpm and 60 min/1000 rpm Aβ12–28 samples. The right shift reflects the growth of protofibrils on the surface.
Fibril Diffusion on the Surface Continues Even after Deposition Has Stopped
On the surface of the 30 min/1000 rpm sample, we observed interesting droplet-like patterns on the edge of the mica, as shown in Figure 5A. At the edge of the sample, the uniform film of water broke into small droplets, roughly 10–15 μm in diameter, which dried much later than the rest of the sample. The inner and outer morphologies of the droplet-like patterns were quite different, as shown in Figure 5B. Long, randomly distributed protofibrils can be found everywhere outside the droplets, but parallel-packed bundles of protofibrils can only be seen within the droplets. The difference between the morphologies indicates that, similar to the individual peptides, the protofibrils also diffuse on the surface, even after peptide deposition has stopped. The protofibrils bundle into longer and more packed protofibrils, potentially due to hydrophobic interactions. However, the detailed mechanism of this process merits further studies and will be addressed in the future.
Figure 5.

AFM images of the 30 min/1000 rpm Aβ12–28 sample near the edge of the mica substrate. (A) Large area image. (B) Zoomed-in image near the boundary of the droplet-like structure, highlighted by red square frame in (A). (C) Zoomed-in image inside the droplet-like structure, green square frame in (B).
Conclusions
In summary, we have observed that surface-mediated self-assembly of peptides, due to adsorption and diffusion of peptides on the surface, can lead to the rapid formation of protofibrils even with low bulk concentrations. The morphologies of the fibrils strongly depend on the number density of the peptides on the surface, which in turn depends on the deposition and diffusion rates of the peptides. Using a spin-coating process, we were able to control the fibril formation rates and final morphologies of the fibrils on the surface. We have shown that protofibrils can be made on the surface homogeneously, and the slow-spinning data are consistent with the trend of morphology transitions in the phase diagram. This simple, but reliable technique will allow us to systematically study specific interactions that lead to rapid fibril formation on the surface by using various peptides, buffer conditions, and substrate systems, including larger amyloidogenic proteins such as α-synuclein and more physiologically relevant surfaces such as lipid monolayers. It will also allow us to explore the effect of common protein labels such as fluorophores on aggregation rates and morphologies. Most importantly, our work highlights the need for concern that aggregate morphologies observed using surface imaging techniques may not accurately reflect the populations and structures in solution. Our continued work in this area will further delineate the role of surface effects so that the biochemistry community may better understand how to interpret AFM data in protein misfolding studies.
Materials and Methods
Sample Preparation
The synthetic Aβ12–28 peptide was purchased from rPeptide with high purity, >95%, and directly used without further purification. We dissolved Aβ12–28 in 1% NH4OH(aq) at a concentration of 1 mg/mL. Once dissolved, the solution was sonicated for 2 min and then diluted with Milli-Q water to a concentration of either 1 or 10 μM. The samples were filtered with a 0.22 μm PTFE membrane syringe filter to eliminate large aggregates and then incubated at room temperature without agitation for 2 days. As such, the real concentration was expected to be lower than the reported values. Before preparing dried samples, the 10 μM sample solution was diluted to 3.33 or 6.67 μM by placing 1 or 2 drops of water on the substrate. A 1 μM sample solution was prepared following the same procedure and diluted to 0.33 and 0.67 μM.
Spin-Coating Process
The 10 μM sample solution was used to prepare all spun-cast samples. Three droplets of sample solution were deposited on the mica substrate and incubated for 0, 30, or 60 min and then dried using a spin-coater (WS-650 MZ-23, Laurell Technologies Corp.) with different spinning rates. The drying times for spun-cast samples at 1000 and 3000 rpm were 3 min and 30 s, respectively. However, the fast fibrillar formation reported here can be reproduced using fresh solutions with the same concentration.
Cryogenic Transmission Electron Microscopy (Cryo-TEM)
Vitreous cryo-TEM samples of Aβ12–28 were prepared for imaging as follows. A 5 μL droplet of the amyloid peptide solution (10.0 μM, incubated for 3 days) was placed on a lacy carbon-coated copper grid (Ted Pella, Inc.) in a humidity- and temperature-controlled environment vitrification system (Vitrobot, FEI, Inc.). The humidity was controlled at 100%, and the temperature of the chamber was 22.0 °C. Then, the sample was blotted to remove excess solvent and create a thin film of the peptide on the grid surface using a −2 mm blotting force, a 1.0 s blot time, and a 10 s drain time to equilibrate any stresses in the sample caused by blotting. The resulting sample grid was vitrified by plunge freezing into liquid ethane (−183 °C). A Tecnai T-12 TEM operating at 120 kV was used to obtain the cryo-TEM images in Supporting Information Figure S1.
Atomic Force Microscopy
The dried samples were prepared by depositing three droplets of sample solution on the freshly cleaved mica surface and dried via different methods, as depicted in Figure 1. The morphologies of nanostructures on mica were imaged using an Agilent 5500 AFM (Agilent Technologies) with a multipurpose XYZ closed-loop scanner. Rotated monolithic silicon probes with aluminum reflex coating (BudgetSensors, Tap-300G, resonance frequency ∼300 kHz, tip radius of curvature <10 nm, force constant 40 N/m) were used to record topographic, amplitude, and phase images with 512 × 512 pixel resolution. Imaging under dry conditions allowed us to achieve high resolution in the Z dimension and observe one peptide thick protofibrils. Gwyddion and ImageJ packages were used to analyze the AFM results. Quantitative analysis methods are provided in the Supporting Information.
Acknowledgments
Research reported in this publication was partially supported by the National Institute of Aging of the National Institutes of Health (NIH) under Award Number P30AG010124 (PI: John Trojanowski), the National Institute of Neurological Disorders and Stroke of the NIH under Award Number NS081033 (PI: E.J.P.), and the University of Pennsylvania. We also thank Milton H. Repollet-Pedrosa and Mahesh Mahanthappa from University of Wisconsin—Madison for cryo-TEM experiments, Yanxin Wang, Tomohiro Tanaka, and Yuetong Kang (an exchange student from Tsinghua University, China) for help in initializing this project, Matthew Tucker for ATR-FTIR, Yu-Hsiu Wang for dynamic light scattering, and Amish Patel for valuable discussions.
Supporting Information Available
Quantitative analysis methods, a comparison of the other sample preparation methods, and supporting figures, including supplementary cryo-TEM images and AFM images. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Masters C. L.; Simms G.; Weinman N. A.; Multhaup G.; McDonald B. L.; Beyreuther K. Amyloid Plaque Core Protein in Alzheimer Disease and Down Syndrome. Proc. Natl. Acad. Sci. U.S.A. 1985, 82, 4245–4249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobson C. M. Protein Folding and Misfolding. Nature 2003, 426, 884–890. [DOI] [PubMed] [Google Scholar]
- Soto C. Unfolding the Role of Protein Misfolding in Neurodegenerative Diseases. Nat. Rev. Neurosci. 2003, 4, 49–60. [DOI] [PubMed] [Google Scholar]
- Lashuel H. A.; Overk C. R.; Oueslati A.; Masliah E. The Many Faces of α-Synuclein: From Structure and Toxicity to Therapeutic Target. Nat. Rev. Neurosci. 2013, 14, 38–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert B. J. The Role of Amyloid Beta in the Pathogenesis of Alzheimer’s Disease. J. Clin. Pathol. 2013, 66, 362–366. [DOI] [PubMed] [Google Scholar]
- Jiménez J. L.; Nettleton E. J.; Bouchard M.; Robinson C. V.; Dobson C. M.; Saibil H. R. The Protofilament Structure of Insulin Amyloid Fibrils. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 9196–9201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulite M.; Fakhraai Z.; Li I. T.; Gunari N.; Tanur A. E.; Walker G. C. Imaging Secondary Structure of Individual Amyloid Fibrils of a β2-Microglobulin Fragment Using Near-Field Infrared Spectroscopy. J. Am. Chem. Soc. 2011, 133, 7376–7383. [DOI] [PubMed] [Google Scholar]
- Findley L. J. The Economic Impact of Parkinson’s Disease. Parkinsonism Relat. Disord. 2007, 13 Suppl, S8–S12. [DOI] [PubMed] [Google Scholar]
- Heron M.; Hoyert D. L.; Murphy S. L.; Xu J.; Kochanek K. D.; Tejada-Vera B.. Deaths: Final Data for 2006. National Vital Statistics Reports; CDC: Atlanta, GA, 2009; Vol. 57, pp 1–134. [PubMed]
- Bynum J.Characteristics, Costs & Health Service Use for Medicare Beneficiaries with a Dementia Diagnosis; Dartmouth Institute for Health Policy and Clinical Care, January 2009.
- Bach J.-P.; Riedel O.; Klotsche J.; Spottke A.; Dodel R.; Wittchen H.-U. Impact of Complications and Comorbidities on Treatment Costs and Health-Related Quality of Life of Patients with Parkinson’s Disease. J. Neurol. Sci. 2012, 314, 41–47. [DOI] [PubMed] [Google Scholar]
- Hamley I. W. The Amyloid Beta Peptide: A Chemist’s Perspective. Role in Alzheimer’s and Fibrillization. Chem. Rev. 2012, 112, 5147–5192. [DOI] [PubMed] [Google Scholar]
- Bulawa C. E.; Connelly S.; Devit M.; Wang L.; Weigel C.; Fleming J. A.; Packman J.; Powers E. T.; Wiseman R. L.; Foss T. R.; et al. Tafamidis, a Potent and Selective Transthyretin Kinetic Stabilizer That Inhibits the Amyloid Cascade. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 9629–9634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coelho T.; Maia L. F.; da Silva A. M.; Cruz M. W.; Plante-Bordeneuve V.; Lozeron P.; Suhr O. B.; Campistol J. M.; Conceicao I. M.; Schmidt H. H. J.; et al. Tafamidis for Transthyretin Familial Amyloid Polyneuropathy a Randomized, Controlled Trial. Neurology 2012, 79, 785–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayed R.; Head E.; Thompson J. L.; McIntire T. M.; Milton S. C.; Cotman C. W.; Glabe C. G. Common Structure of Soluble Amyloid Oligomers Implies Common Mechanism of Pathogenesis. Science 2003, 300, 486–489. [DOI] [PubMed] [Google Scholar]
- Cleary J. P.; Walsh D. M.; Hofmeister J. J.; Shankar G. M.; Kuskowski M. A.; Selkoe D. J.; Ashe K. H. Natural Oligomers of the Amyloid-Beta Protein Specifically Disrupt Cognitive Function. Nat. Neurosci. 2005, 8, 79–84. [DOI] [PubMed] [Google Scholar]
- Petkova A. T.; Leapman R. D.; Guo Z.; Yau W. M.; Mattson M. P.; Tycko R. Self-Propagating, Molecular-Level Polymorphism in Alzheimer’s Beta-Amyloid Fibrils. Science 2005, 307, 262–265. [DOI] [PubMed] [Google Scholar]
- Haass C.; Selkoe D. J. Soluble Protein Oligomers in Neurodegeneration: Lessons from the Alzheimer’s Amyloid β-Peptide. Nat. Rev. Mol. Cell Biol. 2007, 8, 101–112. [DOI] [PubMed] [Google Scholar]
- Harper J. D.; Lansbury P. T. Jr. Models of Amyloid Seeding in Alzheimer’s Disease and Scrapie: Mechanistic Truths and Physiological Consequences of the Time-Dependent Solubility of Amyloid Proteins. Annu. Rev. Biochem. 1997, 66, 385–407. [DOI] [PubMed] [Google Scholar]
- Lee J.; Culyba E. K.; Powers E. T.; Kelly J. W. Amyloid-Beta Forms Fibrils by Nucleated Conformational Conversion of Oligomers. Nat. Chem. Biol. 2011, 7, 602–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabaté R.; Estelrich J. Evidence of the Existence of Micelles in the Fibrillogenesis of β-Amyloid Peptide. J. Phys. Chem. B 2005, 109, 11027–11032. [DOI] [PubMed] [Google Scholar]
- Knowles T. P.; Waudby C. A.; Devlin G. L.; Cohen S. I.; Aguzzi A.; Vendruscolo M.; Terentjev E. M.; Welland M. E.; Dobson C. M. An Analytical Solution to the Kinetics of Breakable Filament Assembly. Science 2009, 326, 1533–1537. [DOI] [PubMed] [Google Scholar]
- Seubert P.; Vigo-Pelfrey C.; Esch F.; Lee M.; Dovey H.; Davis D.; Sinha S.; Schlossmacher M.; Whaley J.; Swindlehurst C.; et al. Isolation and Quantification of Soluble Alzheimer’s Beta-Peptide from Biological Fluids. Nature 1992, 359, 325–327. [DOI] [PubMed] [Google Scholar]
- Gorbenko G. P.; Kinnunen P. K. The Role of Lipid–Protein Interactions in Amyloid-Type Protein Fibril Formation. Chem. Phys. Lipids 2006, 141, 72–82. [DOI] [PubMed] [Google Scholar]
- Byström R.; Aisenbrey C.; Borowik T.; Bokvist M.; Lindström F.; Sani M.-A.; Olofsson A.; Gröbner G. Disordered Proteins: Biological Membranes as Two-Dimensional Aggregation Matrices. Cell Biochem. Biophys. 2008, 52, 175–189. [DOI] [PubMed] [Google Scholar]
- Butterfield S. M.; Lashuel H. A. Amyloidogenic Protein–Membrane Interactions: Mechanistic Insight from Model Systems. Angew. Chem. 2010, 49, 5628–5654. [DOI] [PubMed] [Google Scholar]
- Pronchik J.; He X.; Giurleo J. T.; Talaga D. S. In Vitro Formation of Amyloid from α-Synuclein Is Dominated by Reactions at Hydrophobic Interfaces. J. Am. Chem. Soc. 2010, 132, 9797–9803. [DOI] [PubMed] [Google Scholar]
- Shen L.; Adachi T.; Vanden Bout D.; Zhu X. Y. A Mobile Precursor Determines Amyloid-Beta Peptide Fibril Formation at Interfaces. J. Am. Chem. Soc. 2012, 134, 14172–14178. [DOI] [PubMed] [Google Scholar]
- Benilova I.; Karran E.; De Strooper B. The Toxic Aβ Oligomer and Alzheimer’s Disease: An Emperor in Need of Clothes. Nat. Neurosci. 2012, 15, 349–357. [DOI] [PubMed] [Google Scholar]
- Naiki H.; Higuchi K.; Hosokawa M.; Takeda T. Fluorometric Determination of Amyloid Fibrils in Vitro Using the Fluorescent Dye, Thioflavine T. Anal. Biochem. 1989, 177, 244–249. [DOI] [PubMed] [Google Scholar]
- Batjargal S.; Wang Y. J.; Goldberg J. M.; Wissner R. F.; Petersson E. J. Native Chemical Ligation of Thioamide-Containing Peptides: Development and Application to the Synthesis of Labeled α-Synuclein for Misfolding Studies. J. Am. Chem. Soc. 2012, 134, 9172–9182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y.; Lynn D. G.; Berland K. M. Direct Observation of Nucleation and Growth in Amyloid Self-Assembly. J. Am. Chem. Soc. 2010, 132, 6306–6308. [DOI] [PubMed] [Google Scholar]
- Kaminski Schierle G. S.; van de Linde S.; Erdelyi M.; Esbjorner E. K.; Klein T.; Rees E.; Bertoncini C. W.; Dobson C. M.; Sauer M.; Kaminski C. F. In Situ Measurements of the Formation and Morphology of Intracellular Beta-Amyloid Fibrils by Super-resolution Fluorescence Imaging. J. Am. Chem. Soc. 2011, 133, 12902–12905. [DOI] [PubMed] [Google Scholar]
- Kim Y. S.; Liu L.; Axelsen P. H.; Hochstrasser R. M. Two-Dimensional Infrared Spectra of Isotopically Diluted Amyloid Fibrils from Aβ40. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 7720–7725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strasfeld D. B.; Ling Y. L.; Shim S.-H.; Zanni M. T. Tracking Fiber Formation in Human Islet Amyloid Polypeptide with Automated 2D-IR Spectroscopy. J. Am. Chem. Soc. 2008, 130, 6698–6699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buell A. K.; Dhulesia A.; White D. A.; Knowles T. P.; Dobson C. M.; Welland M. E. Detailed Analysis of the Energy Barriers for Amyloid Fibril Growth. Angew. Chem. 2012, 51, 5247–5251. [DOI] [PubMed] [Google Scholar]
- Tjernberg L. O.; Callaway D. J. E.; Tjernberg A.; Hahne S.; Lilliehöök C.; Terenius L.; Thyberg J.; Nordstedt C. A Molecular Model of Alzheimer Amyloid β-Peptide Fibril Formation. J. Biol. Chem. 1999, 274, 12619–12625. [DOI] [PubMed] [Google Scholar]
- Gosal W. S.; Myers S. L.; Radford S. E.; Thomson N. H. Amyloid under the Atomic Force Microscope. Protein Pept. Lett. 2006, 13, 261–270. [DOI] [PubMed] [Google Scholar]
- Kowalewski T.; Holtzman D. M. In Situ Atomic Force Microscopy Study of Alzheimer’s β-Amyloid Peptide on Different Substrates: New Insights into Mechanism of B-Sheet Formation. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 3688–3693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackley H. K.; Sanders G. H.; Davies M. C.; Roberts C. J.; Tendler S. J.; Wilkinson M. J. In-Situ Atomic Force Microscopy Study of Beta-Amyloid Fibrillization. J. Mol. Biol. 2000, 298, 833–840. [DOI] [PubMed] [Google Scholar]
- Mastrangelo I. A.; Ahmed M.; Sato T.; Liu W.; Wang C.; Hough P.; Smith S. O. High-Resolution Atomic Force Microscopy of Soluble Aβ42 Oligomers. J. Mol. Biol. 2006, 358, 106–119. [DOI] [PubMed] [Google Scholar]
- Adamcik J.; Jung J.-M.; Flakowski J.; De Los Rios P.; Dietler G.; Mezzenga R. Understanding Amyloid Aggregation by Statistical Analysis of Atomic Force Microscopy Images. Nat. Nanotechnol. 2010, 5, 423–428. [DOI] [PubMed] [Google Scholar]
- Burke K. A.; Godbey J.; Legleiter J. Assessing Mutant Huntingtin Fragment and Polyglutamine Aggregation by Atomic Force Microscopy. Methods (San Diego, CA) 2011, 53, 275–284. [DOI] [PubMed] [Google Scholar]
- Keller A.; Fritzsche M.; Yu Y. P.; Liu Q.; Li Y. M.; Dong M.; Besenbacher F. Influence of Hydrophobicity on the Surface-Catalyzed Assembly of the Islet Amyloid Polypeptide. ACS Nano 2011, 5, 2770–2778. [DOI] [PubMed] [Google Scholar]
- Wang Q.; Shah N.; Zhao J.; Wang C.; Zhao C.; Liu L.; Li L.; Zhou F.; Zheng J. Structural, Morphological, and Kinetic Studies of β-Amyloid Peptide Aggregation on Self-Assembled Monolayers. Phys. Chem. Chem. Phys. 2011, 13, 15200–15210. [DOI] [PubMed] [Google Scholar]
- Kang S. G.; Li H.; Huynh T.; Zhang F.; Xia Z.; Zhang Y.; Zhou R. Molecular Mechanism of Surface-Assisted Epitaxial Self-Assembly of Amyloid-like Peptides. ACS Nano 2012, 6, 9276–9282. [DOI] [PubMed] [Google Scholar]
- Norlin N.; Hellberg M.; Filippov A.; Sousa A. A.; Gröbner G.; Leapman R. D.; Almqvist N.; Antzutkin O. N. Aggregation and Fibril Morphology of the Arctic Mutation of Alzheimer’s Aβ Peptide by CD, TEM, STEM and In Situ AFM. J. Struct. Biol. 2012, 180, 174–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong J. S.; Ansaloni A.; Mezzenga R.; Lashuel H. A.; Dietler G. Novel Mechanistic Insight into the Molecular Basis of Amyloid Polymorphism and Secondary Nucleation during Amyloid Formation. J. Mol. Biol. 2013, 425, 1765–1781. [DOI] [PubMed] [Google Scholar]
- Kellermayer M. S. Z.; Murvai Ü.; Horváth A.; Lászlóffi E.; Soós K.; Penke B. Epitaxial Assembly Dynamics of Mutant Amyloid B25–35_N27c Fibrils Explored with Time-Resolved Scanning Force Microscopy. Biophys. Chem. 2013, 184, 54–61. [DOI] [PubMed] [Google Scholar]
- Qiang W.; Kelley K.; Tycko R. Polymorph-Specific Kinetics and Thermodynamics of β-Amyloid Fibril Growth. J. Am. Chem. Soc. 2013, 135, 6860–6871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates E. A.; Owens S. L.; Lynch M. F.; Cucco E. M.; Umbaugh C. S.; Legleiter J. Specific Domains of Abeta Facilitate Aggregation on and Association with Lipid Bilayers. J. Mol. Biol. 2013, 425, 1915–1933. [DOI] [PubMed] [Google Scholar]
- Charbonneau C.; Kleijn J. M.; Cohen Stuart M. A. Subtle Charge Balance Controls Surface-Nucleated Self-Assembly of Designed Biopolymers. ACS Nano 2014, 8, 2328–2335. [DOI] [PubMed] [Google Scholar]
- Jensen P.; Barabasi A. L.; Larralde H.; Havlin S.; Stanley H. E. Deposition, Diffusion, and Aggregation of Atoms on Surfaces: A Model for Nanostructure Growth. Phys. Rev. B 1994, 50, 15316–15329. [DOI] [PubMed] [Google Scholar]
- Fraser P. E.; Nguyen J. T.; Surewicz W. K.; Kirschner D. A. pH-Dependent Structural Transitions of Alzheimer Amyloid Peptides. Biophys. J. 1991, 60, 1190–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yunker P. J.; Lohr M. A.; Still T.; Borodin A.; Durian D. J.; Yodh A. G. Effects of Particle Shape on Growth Dynamics at Edges of Evaporating Drops of Colloidal Suspensions. Phys. Rev. Lett. 2013, 110, 035501. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.