

ABSTRACT
The cytokine gamma interferon (IFN-γ), with antimicrobial and immunoregulatory functions, can be produced by T cells following stimulation through their T cell receptors (TCRs) for antigen. The innate cytokines type 1 IFNs and interleukin-12 (IL-12) can also stimulate IFN-γ production by natural killer (NK) but not naive T cells. High basal expression of signal transducer and activator of transcription 4 (STAT4), used by type 1 IFN and IL-12 to induce IFN-γ as well as CD25, contributes to the NK cell responses. During acute viral infections, antigen-specific CD8 T cells are stimulated to express elevated STAT4 and respond to the innate factors with IFN-γ production. Little is known about the requirements for cytokine compared to TCR stimulation. Primary infections of mice with lymphocytic choriomeningitis virus (LCMV) demonstrated that although the elicited antigen-specific CD8 T cells acquired STAT4-dependent innate cytokine responsiveness for IFN-γ and CD25 induction ex vivo, TCR stimulation induced these through STAT4-independent pathways. During secondary infections, LCMV-immune CD8 T cells had STAT4-dependent IFN-γ expression at times of innate cytokine induction but subsequently expanded through STAT4-independent pathways. At times of innate cytokine responses during infection with the antigen-distinct murine cytomegalovirus virus (MCMV), NK and LCMV-immune CD8 T cells both had activation of pSTAT4 and IFN-γ. The T cell IFN-γ response was STAT4 and IL-12 dependent, but antigen-dependent expansion was absent. By dissecting requirements for STAT4 and antigen, this work provides novel insights into the endogenous regulation of cytokine and proliferative responses and demonstrates conditioning of innate immunity by experience.
IMPORTANCE
Understanding the regulation and function of adaptive immunity is key to the development of new and improved vaccines. Its CD8 T cells are activated through antigen-specific receptors to contribute to long-lasting immunity after natural infections or purposeful immunization. The antigen-receptor pathway of stimulation can lead to production of gamma interferon (IFN-γ), a cytokine having both direct antimicrobial and immunoregulatory functions. Natural killer cells can also produce IFN-γ in response to the innate cytokines type 1 IFNs and/or interleukin-12. This work demonstrates that CD8 T cells acquire parallel responsiveness to innate cytokine signaling for IFN-γ expression during their selection and development and maintain this capability to participate in innate immune responses as long-lived memory cells. Thus, CD8 T cells are conditioned to play a role in innate immunity, and their presence under immune conditions has the potential to regulate resistance to either secondary challenges or primary infections with unrelated agents.
INTRODUCTION
Because of its many antimicrobial and immunoregulatory effects (1–5), the cytokine gamma interferon (IFN-γ) is a key player in both innate and adaptive immunity. Resting CD8 T cells of the adaptive immune system and natural killer (NK) cells of the innate immune system can be distinguished by their preparedness to respond with IFN-γ expression under naive conditions. NK cells express high basal levels of signal transducer and activator of transcription 4 (STAT4), and the innate interleukin-12 (IL-12) and type 1 IFN cytokines induce IFN-γ expression through pathways depending on this signaling molecule (6–9). The cells can also use STAT4 to respond with expression of the CD25 molecule required to form a high-affinity receptor for the adaptive IL-2 cytokine (10). Thus, NK cells are equipped through STAT4-dependent mechanisms to activate effector functions in response to innate cytokines early during infections and can use these factors to help extend their participation in endogenous immune responses into periods overlapping adaptive immunity.
In contrast, naive CD8 T cells have relatively low-level expression of STAT4 and low basal responsiveness to type 1 IFNs for pSTAT4 activation (8, 11). They can be stimulated through their T cell receptor (TCR) for antigen to express IFN-γ by using STAT4-independent pathways (12). At times of peak expansion during acute lymphocytic choriomeningitis virus (LCMV) infection in vivo, however, the LCMV-specific CD8 T cells have elevated STAT4 (11) with sensitivity to type 1 IFNs and IL-18 (13), as well as IL-12 (14), for IFN-γ induction ex vivo and respond with IFN-γ in vivo following bacterial endotoxin induction of IL-12 (15, 16). Moreover, at times of viral clearance during acute LCMV infection, systemic IFN-γ depending on antigen-specific CD8 T cells (13, 17), type 1 IFNs, and STAT4 (6, 13) is endogenously produced. Hence, in the context of acute viral infections, antigen-specific CD8 T cells change their responsiveness to innate cytokines and the change is instrumental in eliciting their effector functions. The consequences for innate cytokine responsiveness of long-lived CD8 T cells remain to be defined, with current understanding focused on the role of antigen-dependent stimulation during secondary infections (18) and complex cytokine effects for enhancing primary and maintaining secondary adaptive immunity (19–21).
The studies presented here were undertaken to define the relationship between innate cytokine and antigen stimulation in acutely responding and resulting memory CD8 T cells. Characterization on day 8 of primary LCMV infection showed that CD8 T cells acquired a sensitivity to IL-12, the type 1 IFN IFN-α, and IL-18 stimulation for IFN-γ and CD25 induction. The LCMV-specific CD8 T cells were preferentially responding, and the effects were STAT4 dependent, particularly in regard to IFN-γ expression. However, TCR stimulation induced both IFN-γ and CD25 through STAT4-independent pathways. The sustained LCMV-specific CD8 T cells in immune mice maintained elevated STAT4, responded to IFN-α with pSTAT4 induction in vivo, and had STAT4-dependent induction of IFN-γ expression at times of innate cytokine responses during secondary LCMV infection or primary infection with antigen-independent murine cytomegalovirus (MCMV). In contrast, a dramatic expansion of the LCMV-specific CD8 T cells detected at times subsequent to innate cytokine induction was STAT4 independent but antigen dependent. Thus, because of their ability to rapidly respond to innate cytokines, immune CD8 T cell populations of the adaptive immune system have acquired features of innate immunity while maintaining antigen specificity for expansion. These results demonstrate how innate immunity can be conditioned by CD8 T cell experience. They have important implications for vaccination strategies and mechanisms of defense against infections.
RESULTS
Innate cytokines and TCR responsiveness of CD8 T cells elicited during acute LCMV infection.
Acute infections of wild-type (WT) C57BL/6 (B6) mice with LCMV induce an expansion of LCMV-specific CD8 T cells that peaks at days 7 to 8 after a challenge (18, 22). To examine the responsiveness of in vivo activated CD8 T cells to a range of stimuli, splenic populations were prepared from mice infected intraperitoneally (i.p.) with 1 × 105 PFU of LCMV Armstrong clone 350 (clE350). The CD8 T cells were identified by flow cytometric analyses as positive for CD8 and the TCR for antigen β chain (TCRβ). They were then further analyzed as CD8+ TCRβ+ cells that were LCMV specific or nonspecific based on binding of pools of class 1 major histocompatibility complex (MHC) H2Db tetramers presenting three known immunodominant LCMV epitopes in the H-2b background, i.e., NP396-404 (NP396), GP276-286 (GP276), and GP33-41 (GP33) (11). As expected, the conditions of day 8 infection resulted in a 3-fold increase over the uninfected (day 0) CD8 T cell proportions, with LCMV-Tet+ cells representing ~40% of these (Fig. 1A). The cell populations were cultured overnight in medium only as a control or with the type 1 IFN IFN-α, IL-12, IL-18, or IFN-α combined with IL-18. In comparison with control CD8 T cells from day 0 mice, those from day 8 of LCMV infection had elevated sensitivity for innate cytokine induction of either intracellular expression of IFN-γ or cell surface expression of CD25 (Fig. 1B). Any of the cytokines alone induced changes, but more dramatic induction in terms of both percentages and intensity of expression was observed when IFN-α was added with IL-18. Cytokine responsiveness for either IFN-γ or CD25 expression was much greater in LCMV-Tet+ than in LCMV-Tet− CD8 T cells (Fig. 1C).
FIG 1 .

Changing responses to stimulation in CD8 T cells during acute LCMV infection. WT (A to C) or WT and STAT4−/− (STAT4−) (D to F) B6 mice were left uninfected (day 0) or i.p. infected with 1 × 105 PFU LCMVclE350 for 8 days. (A, D) Flow cytometric analyses were used to identify CD8+ TCRβ+ cells within splenic leukocytes and further analyzed as LCMV specific (LCMV-Tet+) or nonspecific (LCMV-Tet−) on the basis of binding of pools of MHC class 1 tetramers presenting the three known immunodominant LCMV epitopes in the H-2b background, i.e., NP396-404 (NP396), GP276-286 (GP276), and GP33-41 (GP33). Responses to cytokine or TCR stimulation ex vivo were examined as indicated after 24 h in culture under control conditions (medium), after treatment with IFN-α at 1,000 U/ml, IL-12 at 10 ng/ml, or IL-18 at 10 ng/ml alone and combinations of IFN-α and IL-18 (B, C, E), with plate-bound antibodies against CD3 (αCD3ε) (E, F), and with pooled immunodominant LCMV peptides, i.e., NP396, GP276, and GP33 (F). (B, E, F) Induction of IFN-γ (green) was evaluated by intracellular staining, and that of CD25 (red) was evaluated by cell surface staining. Gray-shaded histograms are results of staining under control conditions. (C) Responses of day 8 LCMV-Tet+ (color-shaded histograms) compared to LCMV-Tet− (gray-shaded histograms) CD8 T cells are shown. Histograms are results from an individual mouse. Values are mean results ± SEMs obtained with four mice per group in one experiment. The experiments were repeated twice. Significant differences between the responses of WT and STAT4− groups are indicated as follows: ***, P ≤ 0.0001; **, P ≤ 0.005.
To define the requirement for STAT4 in ex vivo stimulation, the sensitivity of CD8 T cells from B6 WT mice was compared to that of B6 mice rendered STAT4 deficient as a result of genetic mutation (9). The STAT4-deficient mice have CD8 T cell expansion during LCMV infection (11), and in the experiments reported here, both STAT4-deficient and WT mice had increases in the proportions of total and LCMV-Tet+ CD8 T cells on day 8 (Fig. 1D). Although the percentages of LCMV-Tet+ CD8 T cells were similar, the STAT4-deficient cells had significantly reduced ex vivo responsiveness to innate cytokines for IFN-γ or CD25 expression (Fig. 1E). The STAT4 requirement was more complete for induction of IFN-γ than CD25. To evaluate the STAT4 role in responses elicited through the TCR, ex vivo stimulation was carried out with plate-bound antibodies to the CD3ε molecule of the TCR or with pools of the immunodominant LCMV peptides NP396, GP276, and GP33. Responses to the LCMV peptides were less intense, but both stimuli induced IFN-γ and CD25 expression in day 8 CD8 T cells and did so when either WT or STAT4-deficient populations were used (Fig. 1E and F). Thus, CD8 T cells induced during acute LCMV infection acquire the STAT4-dependent IFN-γ and CD25 responses to innate cytokines mimicking those of NK cells, but TCR stimulation can induce these through STAT4-independent pathways.
Characterization of STAT4 levels and type 1 IFN responsiveness for pSTAT4 activation in long-lived CD8 T cells.
The LCMV-specific CD8 T cells induced on day 8 of infection have elevated total STAT4 levels (11). To extend the studies to long-lived antigen-specific CD8 T cells, LCMV-infected B6 mice maintained for periods of ≥2 months postinfection (mpi) were used as a source of immune cells (Fig. 2A). The LCMV-specific CD8 T cell subsets identified by LCMV tetramer staining were below the limit of detection in the age-matched naive control but represented ~5% of the CD8 T cells in the immune mice (Fig. 2B). To evaluate STAT4 expression and in vivo type 1 IFN responsiveness for STAT4 activation, populations were prepared from immune control mice and immune mice that had been IFN-α treated in vivo by intravenous (i.v.) administration of the cytokine at 90 min prior to harvest. The LCMV-Tet+ and Tet− CD8 T cells purified by sorting with a fluorescence-activated cell sorter (FACS) were used in Western blot analysis. In comparison to the Tet− subsets, the LCMV-specific cells were high in STAT4 and activated pSTAT4 upon in vivo exposure to IFN-α (Fig. 2B). Conditions were developed for LCMV tetramer staining followed by methanol fixation to allow flow cytometric analysis of cytoplasmic STAT4 and pSTAT4 (see Materials and Methods). This approach gave comparable results with LCMV-Tet+ CD8 T subsets expressing higher levels of STAT4 and IFN-α-induced pSTAT4 (Fig. 2B). Thus, long-lived, antigen-specific CD8 T cells have elevated total STAT4 and increased sensitivity to type 1 IFN for pSTAT4 activation.
FIG 2 .

Characterization of LCMV-induced long-lived antigen-specific CD8 T cells for STAT4 expression and in vivo responsiveness to IFN-α. To examine CD8 T cells in immune mice, LCMV-infected WT mice were maintained for ≥2 months after acute infection. Splenic leukocytes from uninfected mice, control vehicle-treated immune mice, and immune mice treated with 5 × 105 U of IFN-α administered i.v. at 90 min prior to harvest were prepared (A). The total CD8+ T cells and LCMV-Tet+ and LCMV-Tet− CD8 T cell subsets were isolated. (B) Expression of total STAT4 and type 1 IFN induction of pSTAT4 were examined by Western blot and flow cytometric analyses. Total cells from uninfected mice and LCMV-immune mice that had been treated with the vehicle control (PBS) or CD8 and LCMV-Tet+ and LCMV-Tet− CD8 T cell subsets from mice treated with IFN-α were prepared by FACS sorting. Extracted proteins were analyzed for STAT4, pSTAT4, and β-actin by Western blot analysis. Determination of cytoplasmic STAT4 and pSTAT4 within LCMV-specific CD8 T cells (Tet+ compared to Tet−) were evaluated by staining and flow cytometry. Gray areas in histograms represent results obtained with cells from control-treated immune mice, and the thick black lines represent results obtained with cells from IFN-α-treated immune mice. Results are representative of two experiments.
Innate cytokine and CD8 T cell responses following a secondary challenge in LCMV-immune mice.
Acute LCMV infections result in viral replication for several days and high systemic levels of type 1 IFNs detected early and through day 4 postchallenge (7, 13, 23), with appearance of IL-18 at day 4 but low-to-undetectable IL-12 levels (13, 24). There is a T cell IFN-γ response detected at intermediate times of infection depending on antigen and innate cytokines (13). The systemic induction of type 1 IFNs, i.e., IFN-α and IFN-β, biologically active IL-12p70, IL-18, and IFN-γ, was defined following a challenge of immune mice with LCMV (Fig. 3A). Because the mice were resistant to the virus, infection with a larger dose, 4 × 106 PFU, was initiated i.v. with the more aggressive clone 13 LCMV (cl13LCMV) (18). The immune mice had low-to-undetectable viral titers in the spleen. Nevertheless, accelerated innate cytokine responses, including early IFN-γ production, were detected. The serum IFN levels, quantitated in a bioassay (Fig. 3B) and with an enzyme-linked immunosorbent assay (ELISA) specific for IFN-α (Fig. 3C), were detectable as early as 8 h and through day 1.5 but peaked at 16 h through day 1 postchallenge. Only low levels of IFN-β were measured by ELISA (Fig. 3C), and the IL-12p70 level evaluated by cytometric bead array (CBA) was low or below the limit of detection by the assay (Fig. 3D). Detectable IL-18 was measured, and this was induced with a peak on day 1 (Fig. 3D). Induction of systemic IFN-γ was found as early as 8 h, continued to increase at 16 h, and peaked at day 1 of infection (Fig. 3D). The induced cytokines were returning to basal levels by day 2.5.
FIG 3 .
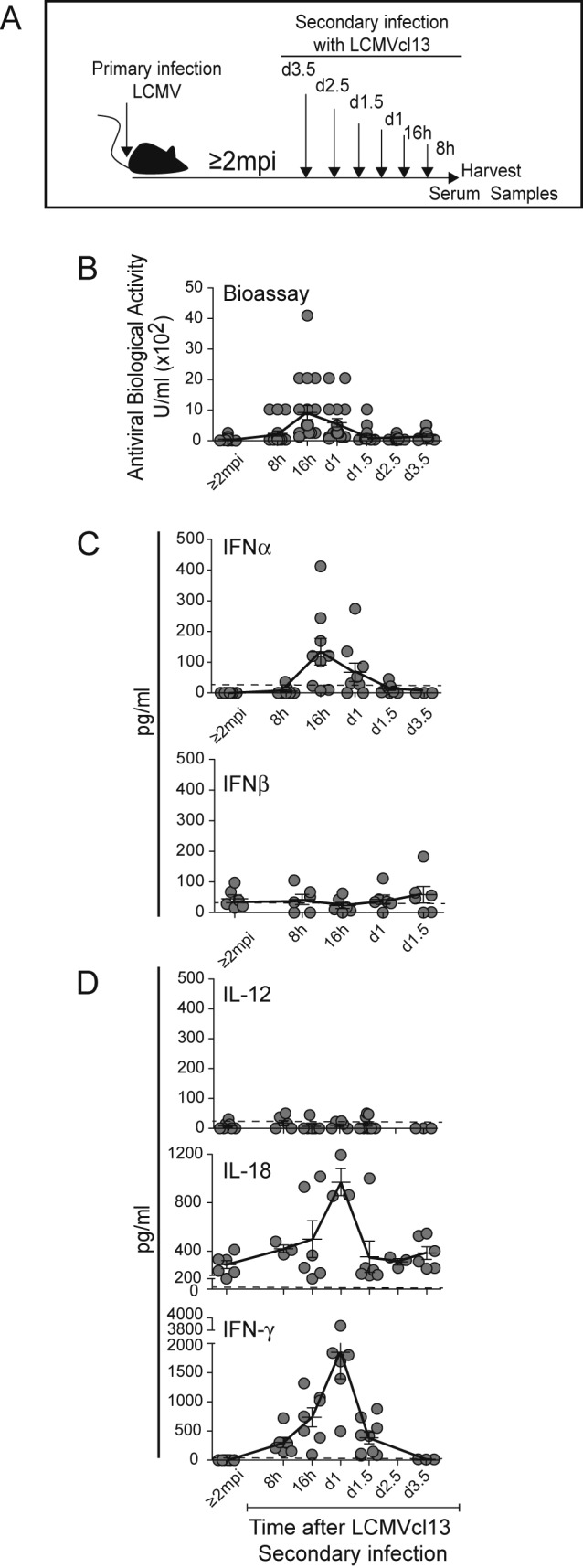
Innate cytokine responses during secondary infections of LCMV-immune mice with LCMVcl13. Serum samples were collected from immune mice and from immune mice after a secondary i.v. infection with 4 × 106 PFU of LCMV clone 13 (LCMVc13) for 8 h, 16 h, 1 day, 1.5 days, 2.5 days, and 3.5 days as indicated (A). Antiviral biological activity was determined in a viral protection assay (B). Circulating levels of IFN-α and IFN-β (C) and IL-18 (D) were measured by ELISA, and those of IL-12 and IFN-γ (D) were measured by CBA as described in Materials and Methods. The samples were collected from eight independent experiments. Symbols represent results from individual mice with three to nine samples from multiple experiments used for each time point. Means ± SEMs (error bars) are shown. Means are connected by solid lines. The limit of detection by the assay is indicated by the dashed line.
The CD8 T cell responses to the secondary challenge were characterized for pSTAT4, IFN-γ, and CD25 expression, as well as percentages and yields of antigen-specific subsets (Fig. 4A). Unchallenged, age-matched, naive (uninfected) and LCMV-immune (≥2 mpi with LCMV) mice were used for comparisons. Consistent with the results shown in Fig. 2, the proportions of LCMV-Tet+ CD8 T cells were detectable in immune but not naive mice (Fig. 4B). After an LCMV challenge, responses were preferentially elicited in LCMV-specific (Tet+) rather than Tet− CD8 T cells. They were all detectable at times overlapping the innate cytokine responses but had different kinetics of peak responses (Fig. 4C). The percentages of LCMV-specific CD8 T cells having pSTAT4 and IFN-γ peaked at 8 h through day 1 of infection. In contrast, CD25 was first detected at 8 h but sustained through day 2.5 (Fig. 4C). There was a delayed expansion of the antigen-specific cells such that >10-fold increases were observed in the LCMV-specific CD8 T cell yields at days 2.5 to 3.5 of infection (Fig. 4D). When the responses were calculated based on cell yields, the numbers of IFN-γ-expressing LCMV-Tet+ CD8 T cells peaked at 8 h to 1 day, but those expressing CD25 were preferentially increased at days 2.5 to 3.5 of infection (Fig. 4D). Thus, under conditions of systemic innate cytokine expression during secondary infection, the antigen-specific CD8 T cells are responding with pSTAT4, IFN-γ, and CD25 expression. Although there is a tight association in the dynamics of the pSTAT4 and IFN-γ responses with circulating cytokines, CD25 expression is sustained, and the antigen-specific CD8 T cell expansion subset is observed at times subsequent to innate cytokine induction.
FIG 4 .

CD8 T cell responses during secondary infections with LCMVcl13. Immune mice at ≥2 mpi with LCMV were i.v. challenged with LCMVcl13 as shown in panel A. Age-matched, uninfected naive and LCMV-immune (≥2 mpi) mice were used as controls. At the infection times indicated, splenic leukocytes were isolated and analyzed for cytoplasmic pSTAT4, cytoplasmic IFN-γ, and cell surface CD25 expression within CD8+ TCRβ+ LCMV-Tet+ and CD8+ TCRβ+ LCMV-Tet− subsets as described in Materials and Methods. Evaluation of pSTAT4 and CD25 was carried out immediately after isolation. Expression of IFN-γ was determined after a 4-h incubation in brefeldin without ex vivo peptide stimulation. (B) Representative flow cytometric analyses of T cell subsets and of pSTAT4, IFN-γ, and CD25 within LCMV-specific T cells (Tet+) compared to nonspecific (Tet−) CD8 T cells at selected time points after a secondary viral infection are shown. Values in plots are percentages of cell subsets in the sample. Histograms are overlaps of results obtained with LCMV-Tet+ (red) and LCMV-Tet− (blue) CD8 T cell subsets. Percentages shown for these are for the LCMV-Tet+ CD8 cells. Percentages (C) and yields (D) of pSTAT4-, IFN-γ-, and CD25-expressing cells within total (white symbols), LCMV-Tet+ (red symbols), and LCMV-Tet− (blue symbols) subpopulations of CD8 T cells after secondary viral infection are shown. Composite data are results obtained with samples from individual mice and are for six mice per group combined from two different experiments. Means ± SEMs (error bars) are shown. Means of LCMV-Tet+ CD8 T cell responses are connected by solid lines.
Requirement for STAT4 in CD8 T cell responses following a secondary challenge.
To evaluate STAT4 function in secondary responses, age-matched naive and LCMV-immune WT and STAT4-deficient mice (≥2 mpi with LCMV) were studied. Controls were left unchallenged and compared to immune mice infected with cl13LCMV at 16 h, 2.5 days, or 3.5 days before harvest (Fig. 5). Consistent with a previous report (25), STAT4 was not required for the development of the long-lived CD8 T cells; the LCMV-Tet+ CD8 T cell basal percentages were similar in the immune STAT4− and WT mice with means ± the standard error of the means (SEMs) of 10 ± 1 for STAT4− and 8 ± 1 for the WT. The proportions and numbers of these cells were also similar in both strains after a cl13LCMV challenge (Fig. 5A), but the peak IFN-γ response was significantly inhibited (P ≤ 0.0001) in the STAT4− populations (Fig. 5B). Dramatic and statistically significant differences in the total yields of IFN-γ-expressing LCMV-Tet+ CD8 T cells were also observed (Fig. 5C). In contrast, the numbers of antigen-specific CD8 T cells remained relatively consistent between the two strains throughout the periods of study and the dramatic expansion of this subset at days 2.5 and 3.5 was elicited in both (Fig. 5C). Except for the absence of IFN-γ in the STAT4-deficient mice, the innate serum cytokine responses were comparable (Fig. 5D). Thus, the STAT4 requirement during secondary infection is for IFN-γ expression but not antigen-specific CD8 T cell expansion.
FIG 5 .

STAT4 dependency of secondary CD8 T cell responses to LCMVcl13. Splenic leukocytes from WT and STAT4− uninfected mice and LCMV-immune (≥2 mpi) mice, either unchallenged or challenged with LCMVcl13, were isolated and examined. (A) Representative flow cytometric analyses of LCMV-Tet+ (red) and LCMV-Tet− (blue) CD8 T cell subsets from one mouse for pSTAT4 and IFN-γ expression at selected time points after a secondary viral infection are shown. Composite data, from the indicated times after secondary infection, of the proportions (B) and yields (C) of IFN-γ-expressing LCMV-Tet+ CD8 T cells, as well as total LCMV-Tet+ CD8 T cell yields (C), are shown, with individual symbols representing results from individual mice. (D) Serum cytokine levels of responses in WT and STAT4− mice measured as described in Materials and Methods. Red symbols represent results from WT mice, and white symbols represent results from STAT4− mice. The results are for three to eight mice per group combined from three different experiments. Means ± SEMs (error bars) are shown. Significant differences between WT and STAT4-deficient groups are identified as follows: ***, P ≤ 0.0001; **, P ≤ 0.005; *, P ≤ 0.05.
Innate cytokine and NK cell responses following a primary MCMV challenge in LCMV-immune mice.
To distinguish between immune responses depending on antigens and cytokines in vivo, innate responses to infection with unrelated MCMV were studied. During infection of naive mice with this virus, innate serum cytokine responses peak at about day 1.5 (36 to 38 h) after infection and include type 1 IFNs, IL-12, and IFN-γ with extended IL-18, and NK cells are stimulated to express IFN-γ (24, 26–30). Following i.p. MCMV infection for 16 h, 1 day, 1.5 days, 2.5 days, or 3.5 days (Fig. 6A), the responses of LCMV-immune mice were similar to those of naive mice, with peak expression of IFN-α, (Fig. 6B), IL-12, and IFN-γ (Fig. 6C) at 1.5 days and a modestly delayed peak in IL-18 (Fig. 6C). The NK cells were identified as NK1.1+ TCRβ− cells (Fig. 6D) and shown to be expressing IFN-γ with a peak induction in proportions (Fig. 6E) and yields (Fig. 6F) of positive cells at 1.5 days of MCMV infection. Thus, LCMV-immune mice have innate cytokine responses to MCMV infection, including NK cell IFN-γ expression, paralleling those of naive mice infected with MCMV.
FIG 6 .

Innate cytokine responses in serum and NK cells during primary MCMV infections of LCMV-immune mice. Serum and splenic leukocyte samples were collected from LCMV-immune (≥2 mpi) mice and from LCMV-immune mice after a primary i.p. infection with 10 to 15,000 PFU MCMV at 8 h, 16 h, 1 day, 1.5 days, and 3.5 days before harvest as indicated (A). Circulating levels of IFN-α and IFN-β (B) and IL-18 (C) were measured by ELISA, and those of IL-12 and IFN-γ (C) were measured by CBA, as described in Materials and Methods. The samples were collected from three independent experiments. The means are connected by solid lines. The limit of detection by the assays is indicated by the dashed line. (D) Splenic leukocytes were analyzed for NK1.1+TCRβ− NK cell populations expressing intracellular IFN-γ. The IFN-γ proportions and yields of NK1.1+ TCRβ− NK cells were determined after 4 h of ex vivo incubation in brefeldin without further stimulation. Representative staining of single samples is shown as plots (D) with histograms of IFN-γ staining within NK cells from uninfected LCMV-immune (≥2 mpi) (blue line) or day 1.5 MCMV-challenged (red line) LCMV-immune mice presented. The percentages (F) and yields (G) of NK cells expressing IFN-γ are shown. Symbols represent results from individual mice with three to nine samples from multiple experiments (B, C) or three mice per group in one experiment (D to G) used for each time point shown. Means ± SEMs (error bars) are shown. Mean NK cell percentages and IFN-γ expression values are connected by solid lines.
Antigen-independent, but cytokine- and STAT4-dependent, IFN-γ expression by LCMV-immune CD8 T cells following primary MCMV infection.
To dissect the T cell IFN-γ response in the absence of antigen, LCMV-immune CD8 T cell responses were evaluated during infection with unrelated MCMV (Fig. 7A). Cells for study were harvested from uninfected mice, LCMV-immune mice without a viral challenge, and LCMV-immune mice infected with MCMV for 16 h, 1.5 days, and 3.5 days. The LCMV-specific (LCMV-Tet+) but not the nonspecific (LCMV-Tet−) CD8 T cells were induced to express both pSTAT4 and IFN-γ during acute primary MCMV infection (Fig. 7B and C). Their proportions (Fig. 7C) and yields (Fig. 7D) peaked at times corresponding to the systemic innate cytokine response, i.e., day 1.5 (see Fig. 6). Despite the increasing number of LCMV-Tet+ CD8 T cells expressing IFN-γ, there was no overall expansion of the LCMV-specific CD8 T cells during MCMV infection (Fig. 7D). This was in contrast to that following an LCMV challenge (kinetics, Fig. 4D) and in a control day 3.5 arm of LCMV infection for these experiments (Fig. 7D). Thus, at times of innate cytokine expression, the LCMV-specific CD8 T cells in immune mice were capable of responding to an unrelated viral infection with pSTAT4 activation and IFN-γ expression but were not induced to expand.
FIG 7 .

Responses of LCMV-specific CD8 T cells of LCMV-immune mice during primary MCMV infection. Uninfected and LCMV-immune (≥2 mpi) mice were used. Immune mice were left untreated or challenged by primary MCMV i.p. infection with 10 to 15,000 PFU at 16 h, 1.5 days, and 3.5 days prior to harvest as shown in panel A. At the postinfection times indicated, splenic leukocytes were isolated and analyzed for total, LCMV-Tet+, and LCMV-Tet− CD8 T cells. Representative results of flow cytometry analyses for pSTAT4 and IFN-γ expression are shown (B). Summary data on the proportions of cells expressing pSTAT4 or IFN-γ (C) and the number of LCMV-Tet+ T cells expressing IFN-γ and total yields (D) are shown. For CD8 T cell yield comparison, LCMV-immune WT mice were challenged with LCMVcl13 at 3.5 days prior to harvest (D). The Tem, Tcm, and effector CD8 T cell subsets were determined by flow cytometric analysis of the relative levels of the CD11a, CD127, CD62L, and KLRG1 markers on cells from immune uninfected mice and mice infected with MCMV for 1.5 days (E, F). The LCMV-Tet− (blue), LCMV-Tet+ (red), and IFN-γ-expressing LCMV-Tet+ (black) cells were evaluated. Representative flow cytometry analyses (E) and summary data (F) are shown. Symbols in the composite data are results obtained with samples from individual mice in one or two experiments with more than three mice per group.
The responding T cells were further characterized as central (Tcm) or effector (Tem) memory cells on the basis of their levels of CD11a, CD127, and CD62L expression (31). The CD8 T cells examined were LCMV-Tet+ compared to LCMV-Tet− cells from LCMV-immune uninfected or day 1.5 MCMV-infected mice and LCMV-Tet+ CD8 T cells expressing IFN-γ from day 1.5 MCMV-infected mice (Fig. 7E and F). The majority, approximately 70%, of the LCMV-immune CD8 T cells were Tem cells, i.e., CD11a high, CD127+, and CD62L low, but approximately 30% were Tcm, i.e., CD11a high, CD127+, and CD62L high, and the LCMV-specific cells expressing IFN-γ were proportionally distributed in these two subsets. KLRG1 expression was used to identify effector and memory cells. Most of the immune T cells, as well as the immune cells expressing IFN-γ during MCMV infection, were negative for KLRG1. Interestingly, the frequencies of KLRG1-positive cells increased during infection such that approximately 20 to 40% of the IFN-γ-expressing cells had the marker under these conditions. Similar results were obtained when pSTAT4 induction during infection was analyzed (not shown). Thus, the LCMV-specific CD8 T cells responding to MCMV infection in the LCMV-immune mice were Tem cells but small proportions were in other cell subsets.
Despite the presence of circulating type 1 IFNs during MCMV infection of naive mice, NK cell IFN-γ expression and serum IFN-γ levels are largely dependent on IL-12 and STAT4 (29). The STAT4 requirement for eliciting the IFN-γ response to MCMV was defined by comparing responses in LCMV-immune STAT4-deficient and WT mice (Fig. 8A and B). The LCMV-Tet+ CD8 T cell IFN-γ response was profoundly inhibited in the absence of the signaling molecule, and the inhibition was significant when the magnitude of the response was calculated as either the proportion or number of IFN-γ-expressing cells (Fig. 8B). The role of IL-12 in the IFN-γ response was demonstrated by using LCMV-immune mice rendered IL-12 unresponsive as a result of mutation in the specific IL-12 receptor chain (IL-12Rβ2−) (Fig. 8C) and WT LCMV-immune mice treated with antibodies neutralizing IL-12 during MCMV infection (Fig. 8D). Both approaches demonstrated that the induction of IFN-γ and pSTAT4 expression by LCMV-specific CD8 T cells was largely dependent on IL-12. Evaluation of the serum cytokine responses demonstrated that the STAT4−, IL-12Rβ2−, and anti-IL-12 antibody-treated mice all had significantly reduced levels of circulating IFN-γ (Fig. 8E). With the exception of the samples taken from anti-IL-12 antibody-treated mice, the IL-12 responses were still detectable and type 1 IFNs were present in all three sets of mice. Taken together, the results show that although specific antigen is required for their expansion, LCMV-immune CD8 T cells are conditioned to respond with IFN-γ expression through antigen-independent but STAT4-dependent pathways during innate cytokine exposure.
FIG 8 .

STAT4 and IL-12 dependency of the immune LCMV-specific CD8 T cell response to primary MCMV infection. Mice were infected as indicated in panel A. Uninfected and LCMV-immune WT (all panels), STAT4− (B, E), and IL-12Rβ2− (C, E) mice were prepared and used as controls or infected with MCMV for 1.5 days. In some experiments, WT immune mice were either treated with an isotype antibody control or given an antibody neutralizing IL-12, anti-IL-12 p40, at 12 h prior to MCMV infection (D, E). Splenic populations were prepared for IFN-γ and pSTAT4 expression analysis within the LCMV-Tet+ CD8 T cells as indicated. Composite data of the proportions (B, C, D) and yields (C) of IFN-γ-expressing LCMV-Tet+ CD8 T cells are shown, with individual symbols representing results from individual mice. Serum cytokine levels were measured in samples from mice infected with MCMV for 1.5 days (E). Red symbols represent results from WT mice, and white symbols represent results from (B) STAT4−, (C) IL-12Rβ2−, or (D) anti-IL-12 antibody-treated mice, and three to seven mice per group in three different experiments were combined. Means ± SEMs (error bars) are shown. Significant differences in reductions between experimental and control groups are identified as follows: ***, P ≤ 0.0001; **, P ≤ 0.005; *, P ≤ 0.05.
DISCUSSION
These studies characterize the acquisition of STAT4-dependent responses to innate cytokines by antigen-specific CD8 T cells. They demonstrate that although IFN-γ and CD25 expression can be induced through STAT4-independent pathways by TCR stimulation, antigen-specific CD8 T cells selected during primary viral infections have elevated STAT4-dependent responsiveness to innate cytokines for IFN-γ and CD25 expression. Moreover, resulting long-lived immune CD8 T cells have increased STAT4 levels, sensitivity to IFN-α for activation of pSTAT4, and preparedness to mediate STAT4-dependent IFN-γ expression at times of innate cytokine responses to secondary infections with the same virus or primary infections with an unrelated virus. Thus, antigen-specific CD8 T cells are equipped during their selection and expansion to participate in innate immune responses when innate cytokines are induced. Because these responses mirror those of NK cells following their first exposure to infection, this study demonstrates that long-lasting CD8 T cells can acquire features of innate immune cells.
The antigen-specific CD8 T cell responses were clearly delineated into two different arms of the immune system; an innate IFN-γ response regulated by STAT4 and innate cytokines and an adaptive arm regulated by antigen exposure and TCR signaling. Although stimulation through either of these two pathways can induce overlapping responses, they had precise dominant functions in vivo. Expansion of the memory LCMV-specific CD8 T cells had the expected antigen dependence (Fig. 4 and 7) (18) but was remarkably STAT4 independent (Fig. 5C). Conversely, although IFN-γ could be elicited through STAT4-independent pathways by stimulation through the TCR ex vivo (Fig. 1) (12), the immune CD8 T cell IFN-γ response during secondary infections with LCMV (Fig. 5) or primary infections with MCMV (Fig. 7) was STAT4 dependent and antigen independent in the context of MCMV. Linking IFN-γ expression to innate cytokine responses and STAT4 suggests that subsets of memory CD8 T cells can mediate all of the functions assigned to innate NK cell IFN-γ. These include the ability to activate antimicrobial defense mechanisms and deliver immunoregulatory effects (1–5). Thus, as their numbers are increased by experience with infections or vaccinations, immune CD8 T cells may replace requirements for NK cell IFN-γ.
The LCMV-immune CD8 T cells responding during MCMV were primarily, but not exclusively, Tem cells (Fig. 7F), and their IFN-γ response was largely dependent on IL-12 and STAT4 (Fig. 8). A question outstanding from this work is whether or not any other CD8 T cell responses, such as expression of tumor necrosis factor, also have innate cytokine pathways for induction. Interestingly, mechanisms might be in place to differentially access particular CD8 T cell cytokines that are concurrently induced by TCR stimulation. Although beyond the scope of this report, the possibilities need to be formally tested.
Because TCR stimulation promotes increased STAT4 levels (11, 32, 33), the CD8 T cells become “NK like” at least in part because of their selection and activation experience during primary responses. This raises questions about the pathways to high basal STAT4 expression in NK cells. Given what has long been described as their “basal” state, this NK cell conditioning may be expected to happen early in life and/or be integrated into cell development. Microbial and host ligands have recently been identified for the semi-invariant TCRs expressed on NKT (iNKT) cells long considered to be part of innate immunity because of their presence prior to a purposeful challenge and stimulation by innate cytokines for responses, including IFN-γ production (34, 35). These iNKT cells also have cytokine- and TCR-dependent responses (36, 37). Thus, iNKT cell conditioning is likely to be induced through their TCRs during early selection and expansion. Once the mechanisms leading to high basal STAT4 expression and innate cytokine responsiveness are defined in NK cells, the parallels of NK, iNKT, and antigen-specific CD8 T cell pathways to “preparedness” for innate responsiveness will be complete. This knowledge will stimulate reexamination of the current definitions of cellular innate and adaptive immunity.
Although NK cell populations respond to innate cytokines, their expansion and maintenance under conditions of sustained MCMV infection are dependent on the expression of an NK-activating receptor used to recognize a viral ligand (38–41). Likewise, iNKT cells arrest and disappear under conditions of IL-12 exposure or MCMV infection (42–44). Thus, activating receptors, be they TCRs or NK-activating receptors, may have important roles in facilitating the homeostatic balance between different lymphocyte populations. In the context of infections encountered throughout the life of a particular individual, a mechanism using all conditioned cells during innate responses but selecting antigen-specific cells for expansion and survival would provide a system for optimizing immediate defense while balancing the overall proportions of different cells to favor subsets most valuable for their potential to deliver additional adaptive immunity. Understanding how to harness the innate and adaptive functions of both NK and T cell populations for the best possible protection against infections in any environment will help in the development of novel vaccines to challenging viruses such as the human immunodeficiency and hepatitis C viruses.
Consistent with another report (25), the comparable proportions and frequencies of antigen-specific CD8 T cells in STAT4− and WT immune mice and during expansion on days 2.5 to 3.5 of secondary infection (Fig. 5C) demonstrated that STAT4 is not critical for T cell expansion and maintenance. During secondary infections with LCMV, the STAT4-dependent stimulation of IFN-γ peaked at 8 h to 1.5 days of infection at times overlapping the innate cytokine responses whereas CD25 expression was sustained such that the peak yields of CD25-expressing LCMV-specific CD8 T cells occurred at the times of cell proliferation, days 2.5 to 3.5 (Fig. 3). Thus, STAT4 in vivo is tightly linked to the IFN-γ response but other pathways depending on TCR selection appear to be the major contributor to CD25 expression, as well as antigen-specific CD8 T cell expansion. This work complements and extends earlier studies examining the role of innate cytokines in promoting naive T cell responses (19, 21, 45, 46) used to suggest a model for activation and expansion which requires three different signals delivered through (i) the TCR, (ii) costimulatory molecules, and (iii) inflammatory cytokines. Innate cytokines have been reported to deliver signal iii to prolong division of activated CD8 T cells by stimulating CD25 expression during immunization under inflammatory conditions (45), and a role for inflammatory monocytes in immunization-induced antigen-independent activation of memory CD8 T cells derived from TCR-transgenic T cells has been reported (47). Both type 1 IFNs and IL-12 activate STAT4, but IL-12 is a more potent inducer and either cytokine can also stimulate the phosphorylation of additional or alternative STAT molecules (48). It is possible, therefore, that a requirement for the TCR in both primary and secondary CD8 T cell IFN-γ responses will be most important under conditions of more modest cytokine stimulation of STAT4 as a consequence of lower levels of available innate cytokines and/or availability of alternative STAT molecules for activation. Additional work is needed to evaluate the role of antigen presentation under the different conditions of stimulation and the importance of other signaling pathways used by the innate cytokines.
New information concerning systemic type 1 IFN, IL-12, IL-18, and IFN-γ responses to infections results from this work. Characteristic type 1 IFN, IL-12, IL-18, and NK cell IFN-γ responses peaking at 36 to 38 h after primary MCMV infection (24, 26–28, 30) were induced in older LCMV-immune mice with similar composition and kinetics of induction, with the possible exception of reduced IL-12 circulating values (Fig. 6). In comparison to the delayed and extended type 1 IFN and IL-18 responses, with antigen-specific CD8 T cell IFN-γ detected on day 4, during LCMV infection in younger naive mice (7, 13, 23), however, responses in LCMV-infected immune mice challenged with LCMVcl13 were diminished in magnitude, had accelerated kinetics, declined more rapidly, and included early IFN-γ (Fig. 3). Given that these differences were associated with enhanced LCMV resistance, they are consistent with the focused type 1 IFN response induced through TLR signaling in plasmacytoid dendritic cells and viral clearance prior to the delayed responses depending on MDA5 sensing and signaling (23). The “immediate early” CD8 T cell IFN-γ response in immune mice does have parallels to one first detected after antigen-specific CD8 T cells are induced on day 4 of primary infections in that it is STAT4 dependent (6, 13). Stimulation through the TCR, however, can act synergistically with innate cytokines for IFN-γ induction, and the endogenous CD8 T cell response to primary infections has a component depending on LCMV antigens (13). Thus, this primary response may occur during a period of transition to the full memory phenotype reported here.
In summary, the work presented here demonstrates an innate IFN-γ response of antigen-specific CD8 T cells in immune mice following viral infection under conditions of secondary exposure to the inducing virus or during infection with an antigen-irrelevant virus. Moreover, it clearly separates the requirements for responses of these cells as innate or adaptive based on early, STAT4-dependent, but antigen-independent induction in the context of other innate cytokines or delayed, antigen-dependent, but STAT4-independent stimulation for expansion. The precise dichotomy of requirements for the in vivo functions suggests that the antigens act to select the cells important to the infection being encountered and then promote their conditioning to utilize innate cytokines. Thus, antigen-specific CD8 T cells acquire the features of innate immunity even when they maintain their adaptive functions. Taken together with other studies of NK and iNKT cells, a picture is emerging of innate and adaptive immunity depending on conditioning for responses to innate cytokines and use of particular activating receptors for antigens or ligands to expand responses into adaptive immunity. Although there are many unanswered questions, these observations challenge the field to consider the definition of cellular innate and adaptive immunity based not simply on the kinetics of responses or the use of activating receptors but on experience.
MATERIALS AND METHODS
Mice.
B6 mice were purchased from Taconic Farms (New York, NY). Breeder pairs of STAT4-deficient mice in the B6 background were obtained from Mark Kaplan (Indiana University School of Medicine), and those of B6 IL-12Rβ2-deficient mice (49) were purchased from The Jackson Laboratory. For examination of acute responses to infection, mice were used at 6 to 12 weeks of age. For long-term studies, 6- to 12-week-old mice were LCMV infected and maintained with age-matched, uninfected controls for at least 2 and up to 8 months. Mouse colonies were housed and maintained at the Brown University Animal Facility. Handling of mice and experimental procedures were done in accordance with institutional guidelines for animal care and use.
Viral infections and antibody treatments.
For acute infections and to establish immunity, mice were infected i.p. with 1 × 105 PFU of Armstrong LCMVclE350 (7, 8). For secondary viral infections at times ≥2 months after primary LCMV infection (≥2 mpi), mice were i.v. challenged with 4 × 106 PFU of LCMVcl13 (18) or infected i.p. with 1 × 104 to 1.5 × 104 PFU of salivary gland-derived MCMV as previously described (34). For neutralization of IL-12, LCMV-immune mice were given 0.75 mg of either anti-IL-12p40 (clone C17.8) or an IgG2a isotype control (clone 2A3), both from BioXCell, at 12 h prior to infection with MCMV.
Sample preparation for flow cytometry.
Spleens were dissociated, and red blood cells were lysed with ACK lysing buffer (Lonza, Walkersville, MD). Lymphocytes were recovered and maintained in 1× phosphate-buffered saline (PBS) containing 2% fetal bovine serum (FBS; Thermo Scientific, South Logan, UT). Pooled biotinylated MHC class 1 monomers (supplied by the NIH Tetramer Core Facility) specific for the LCMV NP396-404/Db, GP276-286/Db, and GP33-41/Db epitopes were formed into tetramers by using streptavidin conjugated to allophycocyanin (APC) or streptavidin-conjugated Alexa 488. Other cell surface staining was performed with the following fluorochrome-conjugated antibodies: phycoerythrin (PE)-CD25, PE-TCRβ, fluorescein isothiocyanate (FITC)-TCRβ, peridinin chlorophyll (PerCP)-TCRβ, PerCP-CD8, FITC-CD8, PerCP-eFluor 710–CD8, APC-NK1.1, FITC-CD11a, FITC-CD127, FITC-CD62L, and FITC-KLRG1 (BD Biosciences or eBioscience, San Diego, CA). For exclusive cell surface marker analysis, cells were fixed with 2% paraformaldehyde. For intracellular cytokine detection, cells were treated with brefeldin A (Sigma, St. Louis, MO) for 4 h, harvested, surface labeled, washed with PBS-FBS, and incubated in Cytofix/CytoPerm buffer (BD Biosciences) for 20 min. Intracellular staining was then performed with FITC–IFN-γ or Alexa 647–IFN-γ (BD Biosciences). For intracellular STAT4 and pSTAT4 detection, the cells were Cytofix/CytoPerm treated, resuspended with 200 µl of prechilled methanol, and incubated on ice for 15 min. Different combinations of MHC class 1 tetramers and antibodies for cell surface determinants were tested for methanol sensitivity. Because the FITC-CD8 antibody was resistant to methanol fixation/permeabilization, it was the fluorochrome of choice and added with APC-LCMV tetramers before fixation. After permeabilization, PE–anti-STAT4 or PE–anti-pSTAT4 (BD Biosciences) was added. Results were acquired with a FACSCalibur (BD Biosciences) and the CellQuest Pro (BD Biosciences) or FlowJo (Tree Star) software. For LCMV tetramer staining and sorting, surface-labeled cells were sorted with a FACSAria Cell Sorting system (Brown University).
Ex vivo stimulation.
Cells were cultured in 96-well plates at 5 × 105/well, incubated under control or stimulation conditions for 24 h, and evaluated for IFN-γ or CD25 expression. The cytokines used were recombinant murine IL-12 (rmIL-12, 10 ng/ml; eBioscience, San Diego, CA), rmIL-18 (10 ng/ml; Medical and Biological Laboratories Co. Ltd., MBL International Corporation, Nagoya, Japan), and rmIFN-α (three) (1,000 U/ml; PBL Interferon Source, Piscataway, NJ). Concentrations were selected on the basis of our previous studies (10, 11, 13). Plate-bound antibody to anti-CD3ε (BD Biosciences) was used at 10 µg/ml in PBS and allowed to adhere to 96-well tissue culture plates overnight, and the supernatant was removed prior to incubation with cells. LCMV peptides NP396-404/Db, GP276-286/Db, and GP33-41/Db were each used at 50 ng/ml.
In vivo treatments.
For in vivo IFN-α treatment, LCMV-immune mice were i.v. injected with 5 × 105 U of IFN-α (PBL Interferon Source) at 90 min before harvest.
Cytokine assays.
For the biological assay, serum samples were serially diluted in a 96-well plate with Vero cells. A day later, when the cells were approximately 90% confluent, they were challenged with vesicular stomatitis virus and incubated for 3 days. Antiviral activity was measured as a ≥50% reduction in cytopathic effects at a defined IFN-α/β dilution of 1 U/ml. Serum samples were also evaluated for IFN-α, IFN-β, and IL-18 by ELISA according to the manufacturer’s instructions (PBL Assay Science, Piscataway, NJ, and MBL International Corporation, Japan). Samples were read with a SpectraMax 250 reader (Molecular Devices, Sunnyvale, CA) at a 405-nm wavelength. The limits of detection by the assays were 25 pg/ml IFN-α, 31 pg/ml IFN-β, and 50 pg/ml IL-18. To measure IL-12p70 and IFN-γ, the BD CBA was used according to the manufacturer’s instructions.
Western blot analysis.
Total splenic leukocytes and FACS-sorted CD8, LCMV-specific (LCMV-Tet+) CD8, and nonspecific (LCMV-Tet−) CD8 T cells were prepared and lysed for Western blot analysis as previous described (11). Mouse monoclonal antibodies against STAT4 (clone 8) and pSTAT4 (clone 38) were purchased from BD Biosciences. A rabbit anti-pSTAT4 polyclonal antibody was also used. A rabbit anti-β-actin polyclonal antibody was obtained from Abcam (Cambridge, United Kingdom). Proteins were resolved by SDS-PAGE, transferred to a polyvinylidene difluoride membrane, and then incubated with antibodies. Reactive bands on Western blots were detected with horseradish peroxidase-coupled secondary antibodies and an enhanced chemiluminescence detection system (GE Healthcare). Anti-STAT4 and -pSTAT4 antibodies were loaded. A rabbit anti-β-actin polyclonal antibody from Abcam was used as a loading control. Control samples were prepared from STAT4-deficient (9) and STAT1-deficient (50) mice.
Statistical analysis.
Unless otherwise stated, the experiments described here were repeated at least twice with three individual animals per group. Significance was determined with an unpaired Student t test (***, P ≤ 0.0001; **, P ≤ 0.005; *, P ≤ 0.05).
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health grant AI05567.
We thank Maria Fragoso, Xin Wang, and Seung-Hwan Lee for help with experiments.
Footnotes
Citation Suarez-Ramirez JE, Tarrio ML, Kim K, Demers DA, Biron CA. 2014. CD8 T cells in innate immune responses: using STAT4-dependent but antigen-independent pathways to gamma interferon during viral infection. mBio 5(5):e01978-14. doi:10.1128/mBio.01978-14.
REFERENCES
- 1. Biron CA, Gazzinelli RT. 1995. Effects of IL-12 on immune responses to microbial infections: a key mediator in regulating disease outcome. Curr. Opin. Immunol. 7:485–496. 10.1016/0952-7915(95)80093-X [DOI] [PubMed] [Google Scholar]
- 2. Boehm U, Klamp T, Groot M, Howard JC. 1997. Cellular responses to interferon-gamma. Annu. Rev. Immunol. 15:749–795. 10.1146/annurev.immunol.15.1.749 [DOI] [PubMed] [Google Scholar]
- 3. Salazar-Mather TP, Hamilton TA, Biron CA. 2000. A chemokine-to-cytokine-to-chemokine cascade critical in antiviral defense. J. Clin. Invest. 105:985–993. 10.1172/JCI9232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Goldszmid RS, Caspar P, Rivollier A, White S, Dzutsev A, Hieny S, Kelsall B, Trinchieri G, Sher A. 2012. NK cell-derived interferon-gamma orchestrates cellular dynamics and the differentiation of monocytes into dendritic cells at the site of infection. Immunity 36:1047–1059. 10.1016/j.immuni.2012.03.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Valentine L, Potts R, Premenko-Lanier M. 2012. CD8+ T cell-derived IFN-gamma prevents infection by a second heterologous virus. J. Immunol. 189:5841–5848. 10.4049/jimmunol.1201679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nguyen KB, Watford WT, Salomon R, Hofmann SR, Pien GC, Morinobu A, Gadina M, O’Shea JJ, Biron CA. 2002. Critical role for STAT4 activation by type 1 interferons in the interferon-gamma response to viral infection. Science 297:2063–2066. 10.1126/science.1074900 [DOI] [PubMed] [Google Scholar]
- 7. Miyagi T, Gil MP, Wang X, Louten J, Chu WM, Biron CA. 2007. High basal STAT4 balanced by STAT1 induction to control type 1 interferon effects in natural killer cells. J. Exp. Med. 204:2383–2396. 10.1084/jem.20070401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mack EA, Kallal LE, Demers DA, Biron CA. 2011. Type 1 interferon induction of natural killer cell gamma interferon production for defense during lymphocytic choriomeningitis virus infection. mBio 2(4):e00169-11. 10.1128/mBio.00169-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kaplan MH, Sun YL, Hoey T, Grusby MJ. 1996. Impaired IL-12 responses and enhanced development of Th2 cells in Stat4-deficient mice. Nature 382:174–177. 10.1038/382174a0 [DOI] [PubMed] [Google Scholar]
- 10. Lee SH, Fragoso MF, Biron CA. 2012. Cutting edge: a novel mechanism bridging innate and adaptive immunity: IL-12 induction of CD25 to form high-affinity IL-2 receptors on NK cells. J. Immunol. 189:2712–2716. 10.4049/jimmunol.1201528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gil MP, Ploquin MJ, Watford WT, Lee SH, Kim K, Wang X, Kanno Y, O’Shea JJ, Biron CA. 2012. Regulating type 1 IFN effects in CD8 T cells during viral infections: changing STAT4 and STAT1 expression for function. Blood 120:3718–3728. 10.1182/blood-2012-05-428672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Carter LL, Murphy KM. 1999. Lineage-specific requirement for signal transducer and activator of transcription (Stat)4 in interferon gamma production from CD4(+) versus CD8(+) T cells. J. Exp. Med. 189:1355–1360. 10.1084/jem.189.8.1355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pien GC, Nguyen KB, Malmgaard L, Satoskar AR, Biron CA. 2002. A unique mechanism for innate cytokine promotion of T cell responses to viral infections. J. Immunol. 169:5827–5837. 10.4049/jimmunol.169.10.5827 [DOI] [PubMed] [Google Scholar]
- 14. Freeman BE, Hammarlund E, Raué HP, Slifka MK. 2012. Regulation of innate CD8+ T-cell activation mediated by cytokines. Proc. Natl. Acad. Sci. U. S. A. 109:9971–9976. 10.1073/pnas.1203543109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nguyen KB, Biron CA. 1999. Synergism for cytokine-mediated disease during concurrent endotoxin and viral challenges: roles for NK and T cell IFN-gamma production. J. Immunol. 162:5238–5246 [PubMed] [Google Scholar]
- 16. Raué HP, Brien JD, Hammarlund E, Slifka MK. 2004. Activation of virus-specific CD8+ T cells by lipopolysaccharide-induced IL-12 and IL-18. J. Immunol. 173:6873–6881. 10.4049/jimmunol.173.11.6873 [DOI] [PubMed] [Google Scholar]
- 17. Liu F, Whitton JL. 2005. Cutting edge: re-evaluating the in vivo cytokine responses of CD8+ T cells during primary and secondary viral infections. J. Immunol. 174:5936–5940. 10.4049/jimmunol.174.10.5936 [DOI] [PubMed] [Google Scholar]
- 18. Murali-Krishna K, Altman JD, Suresh M, Sourdive DJ, Zajac AJ, Miller JD, Slansky J, Ahmed R. 1998. Counting antigen-specific CD8 T cells: a reevaluation of bystander activation during viral infection. Immunity 8:177–187. 10.1016/S1074-7613(00)80470-7 [DOI] [PubMed] [Google Scholar]
- 19. Raué HP, Beadling C, Haun J, Slifka MK. 2013. Cytokine-mediated programmed proliferation of virus-specific CD8(+) memory T cells. Immunity 38:131–139. 10.1016/j.immuni.2012.09.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Thompson LJ, Kolumam GA, Thomas S, Murali-Krishna K. 2006. Innate inflammatory signals induced by various pathogens differentially dictate the IFN-I dependence of CD8 T cells for clonal expansion and memory formation. J. Immunol. 177:1746–1754. 10.4049/jimmunol.177.3.1746 [DOI] [PubMed] [Google Scholar]
- 21. Kolumam GA, Thomas S, Thompson LJ, Sprent J, Murali-Krishna K. 2005. Type I interferons act directly on CD8 T cells to allow clonal expansion and memory formation in response to viral infection. J. Exp. Med. 202:637–650. 10.1084/jem.20050821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cousens LP, Peterson R, Hsu S, Dorner A, Altman JD, Ahmed R, Biron CA. 1999. Two roads diverged: interferon alpha/beta- and interleukin 12-mediated pathways in promoting T cell interferon gamma responses during viral infection. J. Exp. Med. 189:1315–1328. 10.1084/jem.189.8.1315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang Y, Swiecki M, Cella M, Alber G, Schreiber RD, Gilfillan S, Colonna M. 2012. Timing and magnitude of type I interferon responses by distinct sensors impact CD8 T cell exhaustion and chronic viral infection. Cell Host Microbe 11:631–642. 10.1016/j.chom.2012.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Orange JS, Biron CA. 1996. An absolute and restricted requirement for IL-12 in natural killer cell IFN-gamma production and antiviral defense. Studies of natural killer and T cell responses in contrasting viral infections. J. Immunol. 156:1138–1142 [PubMed] [Google Scholar]
- 25. Mollo SB, Ingram JT, Kress RL, Zajac AJ, Harrington LE. 2014. Virus-specific CD4 and CD8 T cell responses in the absence of Th1-associated transcription factors. J. Leukoc. Biol. 95:705–713. 10.1189/jlb.0813429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Orange JS, Biron CA. 1996. Characterization of early IL-12, IFN-alphabeta, and TNF effects on antiviral state and NK cell responses during murine cytomegalovirus infection. J. Immunol. 156:4746–4756 [PubMed] [Google Scholar]
- 27. Ruzek MC, Miller AH, Opal SM, Pearce BD, Biron CA. 1997. Characterization of early cytokine responses and an interleukin (IL)-6-dependent pathway of endogenous glucocorticoid induction during murine cytomegalovirus infection. J. Exp. Med. 185:1185–1192. 10.1084/jem.185.7.1185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pien GC, Satoskar AR, Takeda K, Akira S, Biron CA. 2000. Cutting edge: selective IL-18 requirements for induction of compartmental IFN-gamma responses during viral infection. J. Immunol. 165:4787–4791. 10.4049/jimmunol.165.9.4787 [DOI] [PubMed] [Google Scholar]
- 29. Nguyen KB, Salazar-Mather TP, Dalod MY, Van Deusen JB, Wei XQ, Liew FY, Caligiuri MA, Durbin JE, Biron CA. 2002. Coordinated and distinct roles for IFN-alpha/beta, IL-12, and IL-15 regulation of NK cell responses to viral infection. J. Immunol. 169:4279–4287. 10.4049/jimmunol.169.8.4279 [DOI] [PubMed] [Google Scholar]
- 30. Delale T, Paquin A, Asselin-Paturel C, Dalod M, Brizard G, Bates EE, Kastner P, Chan S, Akira S, Vicari A, Biron CA, Trinchieri G, Brière F. 2005. MyD88-dependent and -independent murine cytomegalovirus sensing for IFN-alpha release and initiation of immune responses in vivo. J. Immunol. 175:6723–6732. 10.4049/jimmunol.175.10.6723 [DOI] [PubMed] [Google Scholar]
- 31. Obar JJ, Lefrançois L. 2010. Early events governing memory CD8+ T-cell differentiation. Int. Immunol. 22:619–625. 10.1093/intimm/dxq053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bacon CM, Petricoin EF, III, Ortaldo JR, Rees RC, Larner AC, Johnston JA, O’Shea JJ. 1995. Interleukin 12 induces tyrosine phosphorylation and activation of STAT4 in human lymphocytes. Proc. Natl. Acad. Sci. U. S. A. 92:7307–7311. 10.1073/pnas.92.16.7307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Watford WT, Hissong BD, Durant LR, Yamane H, Muul LM, Kanno Y, Tato CM, Ramos HL, Berger AE, Mielke L, Pesu M, Solomon B, Frucht DM, Paul WE, Sher A, Jankovic D, Tsichlis PN, O’Shea JJ. 2008. Tpl2 kinase regulates T cell interferon-gamma production and host resistance to Toxoplasma gondii. J. Exp. Med. 205:2803–2812. 10.1084/jem.20081461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rossjohn J, Pellicci DG, Patel O, Gapin L, Godfrey DI. 2012. Recognition of CD1d-restricted antigens by natural killer T cells. Nat. Rev. Immunol. 12:845–857. 10.1038/nri3328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pei B, Vela JL, Zajonc D, Kronenberg M. 2012. Interplay between carbohydrate and lipid in recognition of glycolipid antigens by natural killer T cells. Ann. N. Y. Acad. Sci. 1253:68–79. 10.1111/j.1749-6632.2011.06435.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tyznik AJ, Verma S, Wang Q, Kronenberg M, Benedict CA. 2014. Distinct requirements for activation of NKT and NK cells during viral infection. J. Immunol. 192:3676–3685. 10.4049/jimmunol.1300837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Holzapfel KL, Tyznik AJ, Kronenberg M, Hogquist KA. 2014. Antigen-dependent versus -independent activation of invariant NKT cells during infection. J. Immunol. 192:5490–5498. 10.4049/jimmunol.1400722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Vidal SM, Khakoo SI, Biron CA. 2011. Natural killer cell responses during viral infections: flexibility and conditioning of innate immunity by experience. Curr. Opin. Virol. 1:497–512. 10.1016/j.coviro.2011.10.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dokun AO, Kim S, Smith HR, Kang HS, Chu DT, Yokoyama WM. 2001. Specific and nonspecific NK cell activation during virus infection. Nat. Immunol. 2:951–956. 10.1038/ni714 [DOI] [PubMed] [Google Scholar]
- 40. Lee SH, Kim KS, Fodil-Cornu N, Vidal SM, Biron CA. 2009. Activating receptors promote NK cell expansion for maintenance, IL-10 production, and CD8 T cell regulation during viral infection. J. Exp. Med. 206:2235–2251. 10.1084/jem.20082387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Biron CA. 2010. Expansion, maintenance, and memory in NK and T cells during viral infections: responding to pressures for defense and regulation. PLoS Pathog. 6(3):e1000816. 10.1371/journal.ppat.1000816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Eberl G, MacDonald HR. 1998. Rapid death and regeneration of NKT cells in anti-CD3epsilon- or IL-12-treated mice: a major role for bone marrow in NKT cell homeostasis. Immunity 9:345–353. 10.1016/S1074-7613(00)80617-2 [DOI] [PubMed] [Google Scholar]
- 43. Wesley JD, Tessmer MS, Chaukos D, Brossay L. 2008. NK cell-like behavior of Valpha14i NK T cells during MCMV infection. PLoS Pathog. 4(7):e1000106. 10.1371/journal.ppat.1000106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Velázquez P, Cameron TO, Kinjo Y, Nagarajan N, Kronenberg M, Dustin ML. 2008. Cutting edge: activation by innate cytokines or microbial antigens can cause arrest of natural killer T cell patrolling of liver sinusoids. J. Immunol. 180:2024–2028. 10.4049/jimmunol.180.4.2024 [DOI] [PubMed] [Google Scholar]
- 45. Starbeck-Miller GR, Xue HH, Harty JT. 2014. IL-12 and type I interferon prolong the division of activated CD8 T cells by maintaining high-affinity IL-2 signaling in vivo. J. Exp. Med. 211:105–120. 10.1084/jem.20130901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Curtsinger JM, Mescher MF. 2010. Inflammatory cytokines as a third signal for T cell activation. Curr. Opin. Immunol. 22:333–340. 10.1016/j.coi.2010.02.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Soudja SM, Ruiz AL, Marie JC, Lauvau G. 2012. Inflammatory monocytes activate memory CD8(+) T and innate NK lymphocytes independent of cognate antigen during microbial pathogen invasion. Immunity 37:549–562. 10.1016/j.immuni.2012.05.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kallal LE, Biron CA. 2013. Changing partners at the dance: variations in STAT concentrations for shaping cytokine function and immune responses to viral infections. JAKSTAT 2(1):e23504. 10.4161/jkst.23504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wu C, Wang X, Gadina M, O’Shea JJ, Presky DH, Magram J. 2000. IL-12 receptor beta 2 (IL-12R beta 2)-deficient mice are defective in IL-12-mediated signaling despite the presence of high affinity IL-12 binding sites. J. Immunol. 165:6221–6228. 10.4049/jimmunol.165.11.6221 [DOI] [PubMed] [Google Scholar]
- 50. Durbin JE, Hackenmiller R, Simon MC, Levy DE. 1996. Targeted disruption of the mouse Stat1 gene results in compromised innate immunity to viral disease. Cell 84:443–450 [DOI] [PubMed] [Google Scholar]