

ABSTRACT
Epsilon toxin (ETX), a pore-forming toxin produced by type B and D strains of Clostridium perfringens, mediates severe enterotoxemia in livestock and possibly plays a role in human disease. During enterotoxemia, the nearly inactive ETX prototoxin is produced in the intestines but then must be activated by proteolytic processing. The current study sought to examine ETX prototoxin processing and activation ex vivo using the intestinal contents of a goat, a natural host species for ETX-mediated disease. First, this study showed that the prototoxin has a KEIS N-terminal sequence with a molecular mass of 33,054 Da. When the activation of ETX prototoxin ex vivo by goat small intestinal contents was assessed by SDS-PAGE, the prototoxin was processed in a stepwise fashion into an ~27-kDa band or higher-molecular-mass material that could be toxin oligomers. Purified ETX corresponding to the ~27-kDa band was cytotoxic. When it was biochemically characterized by mass spectrometry, the copresence of three ETX species, each with different C-terminal residues, was identified in the purified ~27-kDa ETX preparation. Cytotoxicity of each of the three ETX species was then demonstrated using recombinant DNA approaches. Serine protease inhibitors blocked the initial proteotoxin processing, while carboxypeptidase inhibitors blocked further processing events. Taken together, this study provides important new insights indicating that, in the intestinal lumen, serine protease (including trypsin and possibly chymotrypsin) initiates the processing of the prototoxin but other proteases, including carboxypeptidases, then process the prototoxin into multiple active and stable species.
IMPORTANCE
Processing and activation by intestinal proteases is a prerequisite for ETX-induced toxicity. Previous studies had characterized the activation of ETX using only arbitrarily chosen amounts of purified trypsin and/or chymotrypsin. Therefore, the current study examined ETX activation ex vivo by natural host intestinal contents. These analyses demonstrated that (i) ETX processing in host intestinal contents occurs in an ordered, stepwise fashion, (ii) processing of prototoxin by host intestinal contents results in higher-molecular-mass material and 3 distinct ~27-kDa ETX species, and (iii) serine proteases, such as trypsin, chymotrypsin, and other proteases, including carboxypeptidases, play a role in the activation of ETX by intestinal contents. These studies provide new insights into the activation and processing of ETX and demonstrate that this process is more complicated than previously appreciated.
INTRODUCTION
The Gram-positive, sporulating, anaerobic bacterium Clostridium perfringens causes many important and diverse diseases in humans and livestock (1). Epsilon toxin (ETX), a pore-forming, single polypeptide, is only produced by toxinotypes B and D of C. perfringens (2–4). Molecular Koch’s postulate analyses showed that ETX production is essential when type D strains cause fatal enterotoxemias in livestock (5). ETX is also a National Institute of Allergy and Infectious Diseases category B priority toxin and a former CDC select toxin because of its extreme potency (50% lethal dose [LD50] of 70 ng/kg of body weight in mice) (4, 6), which ranks ETX as the third most lethal clostridial toxin, behind botulinum and tetanus neurotoxins (7). There were limited reports of human disease involving ETX until a recent study suggested that ETX may trigger multiple sclerosis (8–10).
Enterotoxemia begins when C. perfringens type B or D strains secrete the ~33-kDa ETX prototoxin into the intestinal lumen (4, 11). To exert significant in vivo pathology or in vitro cytotoxic activity, the secreted prototoxin must be proteolytically processed, which increases its activity nearly 1,000-fold (12). Once activated, ETX increases the intestinal mucosal permeability (13), which allows the entry of ETX into the bloodstream, where it can then travel to organs such as the brain and kidney to cause enterotoxemia (14–16).
Purified trypsin or α-chymotrypsin can activate ETX prototoxin in vitro (4, 12, 17). Edman degradation analyses by Minami et al. and others demonstrated that treatment with an arbitrarily chosen amount of purified trypsin removes the 13 N-terminal amino acids from the prototoxin (4, 11). Matrix-assisted laser desorption ionization–time of flight mass spectrometry (MALDI-TOF MS) analyses showed that this trypsin treatment of prototoxin removes the 23 C-terminal amino acids of ETX, while treatment of prototoxin with α-chymotrypsin in the presence of trypsin cleaves away the 29 C-terminal ETX amino acids; this C terminus removal is required for ETX activation (4, 18).
The effects of natural host small intestinal contents on the proteolytic processing/activation of ETX prototoxin have not been evaluated. This issue is important since (i) ETX is secreted by C. perfringens types B and D into the jejunal and ileal lumen but rarely into the colon of naturally infected hosts (mainly goats and sheep) (15, 16, 19), (ii) ETX increases small intestinal permeability in rodent models (13), and (iii) ETX causes intestinal damage in naturally infected goats (15, 19). In addition to trypsin and chymotrypsin, intestinal fluid contains other proteases, including elastase, enteropeptidase, and carboxypeptidases (20), so it is possible those proteases also play a role in ETX activation/proteolytic processing in the intestine.
To address and characterize the proteolytic processing and activation of ETX prototoxin by intestinal proteases at native concentrations, the current study examined the ex vivo effects of goat small intestinal contents on native ETX prototoxin. By amino acid sequencing and mass spectrometry, the processing of prototoxin by goat intestinal contents was examined. In addition, inhibitor studies examined steps in this ex vivo prototoxin processing. These studies provide new insights into the activation of this powerful toxin.
RESULTS
Prototoxin purification and analysis.
ETX prototoxin was purified as previously described (21–23); the purity and identity of this preparation were assessed by SDS-PAGE with Coomassie staining and Western blotting (Fig. 1A and B). Since the precise identity of prototoxin has been unclear, the purified prototoxin was subjected to both Edman degradation amino acid sequencing and liquid chromatography (LC)–electrospray ionization (ESI)-TOF MS for mass determination. Edman sequencing showed that the N terminus of the prototoxin begins with the sequence KEIS (Fig. 1C). LC–ESI-TOF MS of the purified prototoxin detected a protein of 33,054.0 Da, which is within 2 Da of the predicted molecular mass for the ETX prototoxin from C. perfringens type D strain NCTC8346 (GenBank accession number AAA23236.1) (Fig. 1D and E).
FIG 1 .

Purification and characterization of ETX prototoxin. (A and B) The purity and identity of ETX prototoxin were assessed by SDS-PAGE, followed by Coomassie staining (A) or Western blotting (B). (C) Schematics of the N terminus of full-length toxin encoded by the etx open reading frame (ORF), the predicted N-terminal start site of the ETX prototoxin, and the N-terminal start site of the ETX prototoxin as determined by Edman degradation. The N-terminal sequence of the purified prototoxin is shown by boldface and underlining. (D and E) The ETX prototoxin was subjected to LC–ESI-TOF MS to determine its intact molecular weight (M.W.) (D), and the molecular weight from this result is compared against the predicted prototoxin molecular weight to deduce the C terminus (E).
Activation of the ETX prototoxin by goat intestinal contents.
The total trypsinlike protease activity in the intestinal contents was calculated using the Pierce colorimetric protease assay kit, which identified ~1 µg/µl of this activity in intestinal contents. Using this value as a crude benchmark of proteolytic activity, 5 µg of ETX prototoxin was treated for 60 min at 37°C with 10 µg of purified trypsin, 10 µg of purified chymotrypsin, a mixture of 5 µg each of purified trypsin and chymotrypsin, or 10 µl of goat intestinal contents. Following the addition of a broad-range protease inhibitor, each mixture was diluted in Dulbecco’s phosphate-buffered saline (dPBS) and added to a confluent monolayer of MDCK-II cells (60 min at 37°C). Cytotoxicity was measured by lactose dehydrogenase (LDH) release and found to be equivalent among the 4 proteolysis conditions tested, with a value of ~60% cell death (Fig. 2). ETX prototoxin was inactive without protease treatment.
FIG 2 .
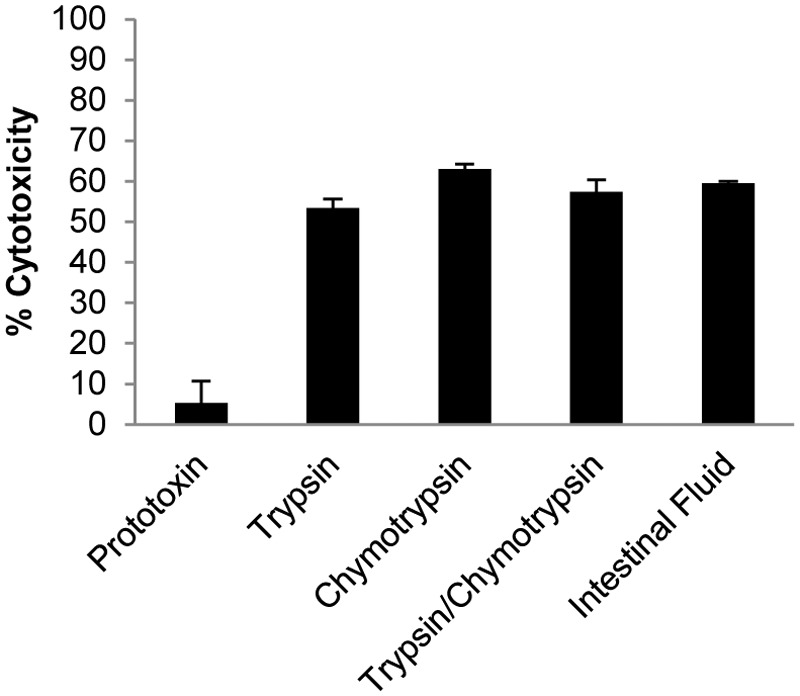
Goat intestinal contents activate ETX. Purified ETX prototoxin was incubated for 60 min with 10 µg of the indicated proteases (total or combined) or with goat intestinal contents as indicated on the x axis. Following incubation, protease inhibitor was added and each mixture was then applied to MDCK-II cells for 60 min. The percent cytotoxicity (y axis) was measured by LDH release. As was expected, cytotoxicity was not observed when MDCK cells were treated with prototoxin alone. Goat intestinal contents or purified proteases alone did not cause MDCK cytotoxicity (not shown). The data represent three independent experiments, and error bars represent the standard errors.
Time course of ex vivo proteolytic processing and activation of ETX prototoxin by goat intestinal contents.
Earlier studies of ETX prototoxin processing/activation used arbitrarily chosen amounts of purified trypsin, with or without chymotrypsin, and a single digestion time that ranged from 30 to 120 min (4). To characterize the ex vivo proteolytic processing/activation of ETX prototoxin in physiologically relevant goat intestinal contents, those processes were observed over 90 min using SDS-PAGE with Coomassie staining, Western blotting, and an MDCK cell cytotoxicity assay (Fig. 3A to C). During the treatment with intestinal contents, proteolysis of prototoxin began within 1 min, while the activation of ETX-induced MDCK cell cytotoxicity began within 5 min. With longer treatment, a ladder-like pattern of ETX cleavage was observed by SDS-PAGE and Western blotting, with approximately 3 intermediate ETX bands eventually observed. The cytotoxic properties of ETX in goat intestinal contents then remained relatively constant until ~30 min of treatment, when there was an ~25% increase in cytotoxic activity. This increase in cytotoxicity correlated with the appearance of a major ETX band of ~27 kDa (Fig. 3A); that band remained present in the goat intestinal contents with incubation up to 120 min, as visualized by SDS-PAGE (data not shown).
FIG 3 .

Goat intestinal contents activate ETX prototoxin in a precise, stepwise pattern. (A and B) ETX prototoxin was incubated with goat intestinal contents (i.c.) for the indicated times, and the resulting processed forms of ETX were visualized using SDS-PAGE and Coomassie blue staining (A) or Western blotting (B), demonstrating a stepwise pattern of processing and activation. Aliquots of the same digestion products (containing 5 µg of toxin) were used to treat MDCK cells, and cytotoxicity was measured using an LDH release assay. This analysis showed that the appearance of an ETX species at 30 min, which then remains stable for a significant period of time, correlates with an ~30% increase in ETX-induced cytotoxicity. (C) Goat intestinal contents or purified proteases alone did not cause MDCK cytotoxicity (not shown). Three experiments were performed independently, and error bars represent the standard errors. (D) Using goat intestinal contents (90 min), physiologic levels of trypsin (90 min), or the amount of trypsin used by Minami et al. (30 min), the stability of ETX was assessed by SDS-PAGE and Coomassie blue staining (left) or Western blotting (right). (E) Alexa Fluor 488-labeled prototoxin was treated with intestinal contents for 90 min at 37°C and subjected to SDS-PAGE and fluorescent imaging.
For comparison, the prototoxin was digested with the same nonphysiological, large amount of trypsin (1:25, µg ETX/µg trypsin) used by Minami et al. (4). Within 30 min of digestion, ETX was undetectable by Coomassie staining and only weakly detectable by Western blotting. However, ETX remained strongly detectable by Coomassie staining or Western blotting after a 90-min treatment with purified trypsin used at an activity equivalent to that measured in goat intestinal content (Fig. 3D).
During this kinetics experiment, we noted that the 90-min sample retained the same cytotoxicity as prototoxin samples digested for 30 min, even though there was apparently much more ETX-related protein in the 30-min sample. However, more high-molecular-weight proteinaceous material was noted in the 90-min versus the 30-min treatment sample (Fig. 3A). To evaluate whether this material might include an ETX species that was not reactive with the ETX polyclonal antibody in Western blot assays, Alexa Fluor 488-labeled ETX was incubated for 90 min with goat intestinal contents. The results of this study showed small amounts of a fluorescent 27-kDa protein, along with fluorescent high-molecular-mass material (Fig. 3E).
The effects of class-specific protease inhibitors on ETX processing and activation.
To better understand the results shown in Fig. 3, we examined which protease classes were important for the activation of the ETX prototoxin by caprine intestinal contents. For this purpose, caprine intestinal contents were incubated with class-specific protease inhibitors for 30 min at room temperature before being mixed with ETX prototoxin for 90 min at 37°C. Only the Bowman-Birk inhibitor, which targets serine proteases (e.g., trypsin and chymotrypsin), blocked MDCK cytotoxicity (Fig. 4A) and prevented most but not all of the initial proteolytic processing of prototoxin by the intestinal contents (Fig. 4B). Pretreatment of the intestinal contents with carboxypeptidase inhibitor prevented the extensive processing of ETX, with no ~27-kDa species detectable (Fig. 4B). Similarly, treatment of the prototoxin with purified trypsin and chymotrypsin resulted in an ETX band of the same size as observed in the presence of intestinal contents and carboxypeptidase inhibitors (Fig. 4C).
FIG 4 .

Characterization of the effects of protease inhibitors on ETX processing and activation in goat intestinal contents. Goat intestinal contents (i.c.) were pretreated with the indicated protease inhibitors for 30 min at room temperature, and ETX was then added for 90 min at 37°C. (A and B) MDCK cell cytotoxicity was measured (n = 3) (A), and the processing was analyzed by SDS-PAGE and Coomassie staining (left) and Western blotting (right) (B). (C) ETX activated in the presence of carboxypeptidase inhibitors was compared to ETX activated by trypsin and chymotrypsin using SDS-PAGE and Coomassie staining (left) and Western blotting (right). SPI, serine protease inhibitor; CPI, carboxypeptidase inhibitor; E64, E-64 cysteine protease inhibitor.
Purification and characterization of the ~27-kDa ETX species produced during the activation of prototoxin by intestinal contents.
Given the presence of the ~27-kDa ETX band after prolonged treatment with goat intestinal contents, this ETX species was analyzed further. The ~27-kDa ETX species was purified, the purity was then demonstrated by SDS-PAGE with Coomassie staining, and the identity of ETX in the preparation was confirmed by Western blotting (Fig. 5A and B). This purified ~27-kDa ETX preparation (5 µg/ml) was biologically active, as tested by cytotoxicity on MDCK-II cells (Fig. 5C). Edman degradation analysis revealed the first 6 amino acids of the ~27-kDa ETX species to be KASYDN, indicating trypsin cleavage of the first 13 N-terminal amino acids from the prototoxin (Fig. 6A).
FIG 5 .
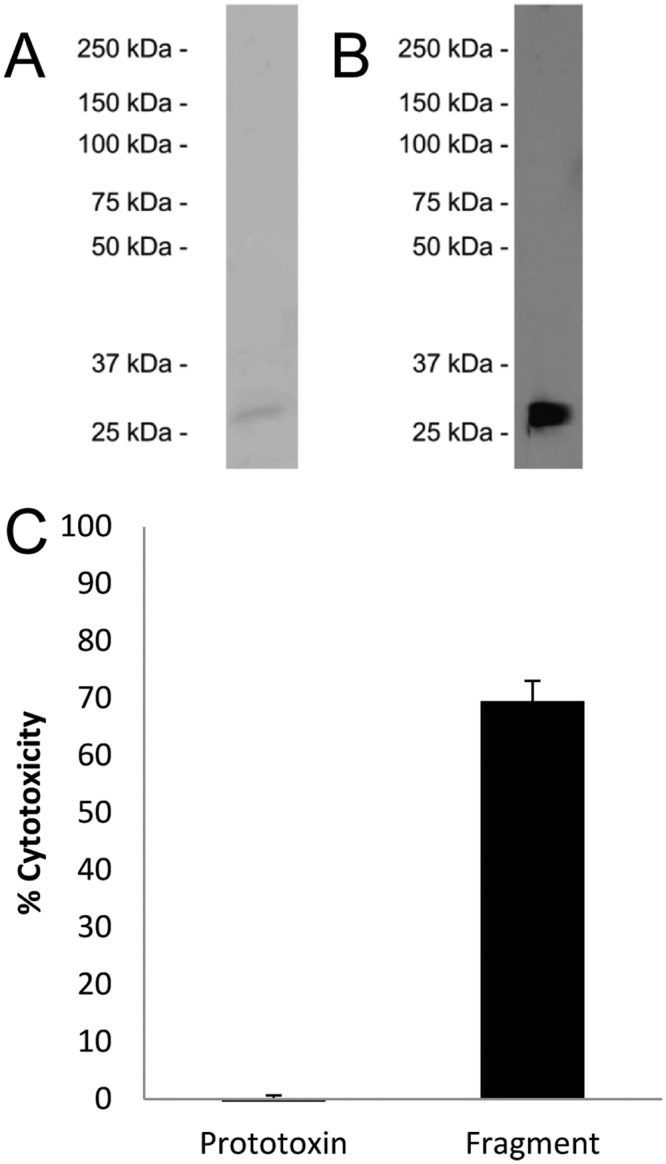
Purification and cytotoxicity of the stable ~27-kDa ETX produced by treatment with goat intestinal contents. (A and B) The stable ~27-kDa ETX identified in the experiments whose results are shown in Fig. 3 was purified by ion-exchange chromatography, and its purity was assessed by SDS-PAGE followed by Coomassie blue staining (A) and its identity confirmed by Western blotting (B). (C) The resulting ETX preparation was used to treat MDCK cells for 1 h, and cytotoxicity was again measured by LDH release, demonstrating that the purified ~27-kDa ETX material is biologically active. Goat intestinal contents or purified proteases alone did not cause MDCK cytotoxicity (not shown). Error bars represent standard errors (n = 3).
FIG 6 .

Characterization of purified ~27-kDa ETX activated by goat intestinal contents (A). The purified ~27-kDa ETX preparation was subjected to Edman sequencing, and the resulting N-terminal sequence was compared to the sequence of the prototoxin N terminus. The N-terminal sequences identified are indicated by boldface and underlining. (B) The purified and processed ~27-kDa ETX preparation was subjected to LC–ESI-TOF MS, which demonstrated the presence of three protein species of 27,688.0, 27901.4, and 27,900.0 Da. (C) Based upon the molecular masses, the predicted molecular weights were used to map the C termini of these species. The identified C termini of the ETX species are shown with the C termini of prototoxin and ETX treated with trypsin or with trypsin and chymotrypsin for reference.
Interestingly, LC–ESI-TOF MS for intact-mass analysis of the purified ~27-kDa ETX species produced after a 90-min treatment with intestinal contents revealed the presence of three distinct ETX species with demonstrated molecular masses of 27,688.0, 27,801.4, and 27,900 Da (Fig. 6B). Molecular mass prediction based on the epsilon toxin sequence revealed that proteins spanning from K14 to N262, N263, or V264 would have molecular masses of 27687.84, 27801.94, or 27901.08 Da, respectively (Fig. 6C).
Evaluation of the cytotoxic activity of the ~27-kDa ETX fragments using recombinant proteins.
The cytotoxic activity of each of the three identified ETX species produced after proteolytic processing by goat intestinal contents was evaluated using a recombinant protein approach. For this purpose, each of the three ETX fragments, along with ETX fragments corresponding to those produced by treatment of native prototoxin with purified trypsin or a combination of purified trypsin and α-chymotrypsin (based upon previous results [4]), was expressed in Escherichia coli as maltose-binding protein (MBP) fusion proteins (Fig. 7A). Following expression and purification of the MBP-rETX (recombinant ETX) fragments, the fragments were liberated from the MBP solubility tag and used to test MDCK cell cytotoxicity (Fig. 7B and C).
FIG 7 .
Recombinant ETX species corresponding to those produced by treatment of prototoxin with goat intestinal contents are cytotoxic in vitro. (A) To confirm whether any of the 3 ~27-kDa ETX species identified following treatment with goat intestinal contents are active, the resulting ETX fragments were cloned, expressed as MBP fusion proteins, and purified from E. coli. (B and C) Following liberation of the ETX species from the MBP tag, the presence of appropriately sized bands was visualized by SDS-PAGE followed by Western blotting (B) or Coomassie blue staining (C). (D) The percent cytotoxicity of MDCK cells treated with the rETX fragments was measured using LDH release. rETX14–295 and MBP alone were used as negative controls to ensure that cytotoxicity was from the resultant rETX fragments and not from contaminating protein. Error bars represent standard errors (n = 3).
By measuring MDCK cell cytotoxicity by the LDH release assay, it was determined that the rETX fragments comprising amino acids 14 to 262, 14 to 263, and 14 to 264 (rETX14–262, rETX14–263, and rETX14–264 fragments) each represent cytotoxic forms of ETX. For comparison, the rETX14–267 and rETX14–273 fragments (corresponding to trypsin–α-chymotrypsin- and trypsin-activated forms, respectively) also possessed cytotoxic activity (4). Furthermore, the cytotoxic properties of the rETX14–267 and rETX14–273 fragments were similar to those of the rETX14–262, rETX14–263, and rETX14–264 fragments. The negative controls, i.e., full-length rETX prototoxin or MBP alone, were inactive, confirming that the observed cytotoxicity was specifically due to the activity of those rETX fragments rather than activity from contaminating E. coli proteins (Fig. 7D).
DISCUSSION
This study reports several important results with substantial import for our understanding of ETX processing and activation. First, it resolves the precise biochemical identity of the secreted ETX prototoxin, which had remained unclear in the literature. Hunter et al. determined by SDS-PAGE that the prototoxin has an approximate molecular mass of ~33 kDa, with a predicted N-terminal sequence of KEISNTVSNEM based upon in silico analysis of the deduced etx open reading frame sequence (11). However, the N-terminal sequence and the precise mass of the prototoxin were not experimentally determined in their study. The prediction of Hunter et al. (11) for the prototoxin N-terminal sequence was in general agreement with earlier amino acid-sequencing results reported by Bhown and Habeeb in 1977, who determined the N-terminal sequence of the ETX prototoxin to be KEICB(N or R)PVSYEM (11, 17). More different were the results reported by Minami et al., where the ETX prototoxin was found to have a molecular mass of 32,307 Da and an N-terminal sequence of VSNEM (4). The results from the current study match exactly the bioinformatic predictions of Hunter et al. regarding the identity of ETX prototoxin (11), i.e., Edman degradation determined that the ETX prototoxin has an N-terminal sequence of KEIS, while LC–ESI-TOF MS detected a molecular mass of 33,054 Da (Fig. 1C to E). These results match the predicted molecular mass of 33,055.99 Da for a protein encoded by the etx open reading frame of type D strain NCTC8346 that has a KEIS N-terminal sequence (GenBank accession number AAA23236.1). It is not clear whether the prototoxin characterization differences between Minami et al. and the other studies are attributable to strain-dependent differences in prototoxin processing during secretion or have another explanation. Any observed differences in the prototoxin characterization results are not due to strain-dependent sequence variation, since the N-terminal amino acid sequence of ETX is identical for all strains examined to date (GenBank accession numbers Q02307, AEH25946, ADF42572, AFQ89891, 1UYJ_A, AFP43229, AFN42326, WP_00345888, and ADU04573).
Previous studies of the proteolytic processing and activation of ETX prototoxin provided considerable insights into the biology of this potent pore-forming toxin (3, 4, 12, 18, 24, 25). For example, earlier studies revealed that the ETX prototoxin is ~1,000 times less active than proteolytically activated ETX (12). However, the nature of prototoxin proteolytic processing and activation during disease has not been clear. Purified λ-protease of C. perfringens can process and activate the ETX prototoxin (4), but the physiologic importance of this effect is under question since many ETX-producing type B and D strains, including strain NCTC8346 used for purifying prototoxin in this study, do not carry the gene encoding λ-protease (26). Furthermore, even those type B and D strains encoding λ-protease typically cannot self-process or activate their prototoxin (26). A single λ-protease-negative type D strain was identified that produces a partially processed ETX using an intracellular protease, but this effect is not shared by other type B and D strains surveyed and only produces a weakly activated ETX (26).
Those findings made it apparent that intestinal proteases play a major role in the processing and activation of prototoxin after its secretion by C. perfringens into the intestinal lumen during type B or D disease. Consistent with this view, several previous studies of ETX prototoxin activation used purified or crude trypsin and α-chymotrypsin preparations to study prototoxin processing and activation (4, 11). While informative, that approach has at least two major limitations. First, those previous experiments used trypsin and α-chymotrypsin, alone or in combination, at apparently arbitrarily chosen concentrations (4). Second, a number of proteases other than trypsin and chymotrypsin are present in the mammalian intestinal lumen and those enzymes may also contribute to ETX prototoxin processing and activation (20). Because of those concerns, we hypothesized that prototoxin processing in the intestinal contents of a host animal may differ from the prototoxin processing reported using the purified intestinal proteases trypsin and chymotrypsin, either alone or in combination.
To address this proposition, the small intestinal contents from a healthy adult goat were used to treat purified prototoxin ex vivo. This experiment revealed that prototoxin incubated with natural small intestinal contents from a healthy adult of a natural host species for the disease causes the characteristic LDH release, nuclear shrinkage, and cellular swelling in MDCK cells, as previously described for active ETX (23, 27). This result demonstrated clearly for the first time that natural intestinal contents can activate the ETX prototoxin (Fig. 2).
Previous studies using purified trypsin and α-chymotrypsin only tested ETX activity after 60 min of prototoxin incubation with those proteases (4), i.e., no time course study was performed to examine the kinetics of ETX processing and activation at various protease treatment times. Therefore, another contribution of the current study involves characterizing prototoxin activation over time. The results of this analysis revealed that, after even very brief (<1-min) contact with host intestinal contents, prototoxin cleavage was detectable. However, when the prototoxin was tested for activity using the MDCK cytotoxicity model, measurable cytotoxicity developed after a 5-min incubation with goat intestinal contents (Fig. 3). This result demonstrates for the first time that natural intestinal contents are extremely efficient at activating the prototoxin. Coupling these activation kinetics with the size of the initially observed ETX species produced upon treatment with intestinal fluid suggests a scenario for initial prototoxin processing, i.e., the first step of ETX prototoxin activation likely involves removal of the N terminus by trypsin, since this effect does not directly trigger cytotoxicity (18). If so, this effect is then rapidly followed by the removal of C-terminal residues, as required for the development of cytotoxicity (4, 18).
Another contribution of this time course study is the first evidence that natural ETX processing and activation follow a stepwise pattern. After a 5-min treatment with intestinal contents, the intact prototoxin was no longer detectable by SDS-PAGE and cytotoxicity had developed. This development of cytotoxic activity was accompanied by the appearance of an ~30-kDa processed ETX species that was smaller than the first ETX species processed. After a 20-min treatment with intestinal contents, an even smaller ETX species of ~27 kDa became detectable by SDS-PAGE. By 30 min of treatment, this ETX species had accumulated significantly, along with a further ~25% increase in MDCK cell cytotoxicity (Fig. 3). The ~27-kDa band persisted in the intestinal contents for up to 120 min (data not shown). It is notable that the stability of this ETX species would not have been appreciated by SDS-PAGE with Coomassie staining if the same high concentrations of purified trypsin or trypsin and chymotrypsin employed in previous studies had been used (4, 18); however, a stable ETX band slightly larger than 27 kDa was also observed by using physiologic concentrations of purified trypsin in this study.
Despite its persistence, the ~27-kDa ETX band diminished by 90 to 120 min of treatment with goat intestinal contents compared to its levels at earlier treatment time points, even though cytotoxicity remained high. This inconsistency was addressed using Alexa Fluor 488-labeled ETX prototoxin, which showed the presence of multiple fluorescently labeled ETX species at molecular masses of between ~150 and >250 kDa (Fig. 3E) after 90 min of incubation with goat intestinal fluid. This shift of some ETX to higher-molecular-mass forms explains the high MDCK cell cytotoxicity at 90 min despite the presence of small amounts of ETX detectable by Western blotting using an anti-ETX polyclonal antibody. The failure of Western blot assays to detect the high-Mr ETX forms after extended treatment with intestinal contents could be due to the epitopes recognized by this antibody being inaccessible in the high-Mr ETX forms. These high-Mr ETX species could represent either spontaneous oligomer formation involving processed ETX or interactions between processed ETX and proteins in the intestinal contents.
Because of its stability in the intestinal contents, the ~27-kDa ETX species was purified to >95% homogeneity as detected by SDS-PAGE, followed by Coomassie staining or Western blotting (Fig. 5A and B). The purified ~27-kDa ETX preparation was active on MDCK cells (Fig. 4C). When Edman sequencing was performed on this species, it identified a single N-terminal sequence, KASYDN, (Fig. 6A), which is consistent with trypsin cleavage between residues 13 and 14 of the prototoxin, as noted previously by Minami et al. and Hunter et al. when prototoxin was treated with purified trypsin in combination with purified chymotrypsin (4, 11).
Removing the C-terminal amino acids from the prototoxin is required for the activation of ETX cytotoxic activity (18), so the C terminus of the ~27-kDa ETX species was deduced by LC–ESI-TOF MS analyses. Those analyses did not detect ETX species with molecular masses of 28,973.3 Da or 28,321.5 Da, as would be expected if the current model (4) were correct that activated ETX species are only produced from cleavage by trypsin or trypsin–α-chymotrypsin, respectively. Instead, three ETX species with molecular masses of 27,688.0, 27,801.4, and 27,900 Da were detected. Those masses correspond precisely to ETX molecules cleaved by trypsin at the N terminus and processed at the C terminus to amino acids N262, N263, and V264 (Fig. 6B and C). This result provided the first evidence that proteases other than trypsin, α-chymotrypsin, or perhaps, occasionally, C. perfringens proteases play a role in ETX activation in the host small intestine.
We hypothesized that, following the initial cleavage of the C terminus by the endopeptidases trypsin and/or α-chymotrypsin (or perhaps another serine endoprotease in the intestinal contents), further C-terminal processing is initiated by intestinal carboxypeptidases. In support of this hypothesis, the results of using protease inhibitors indicated that serine proteases (which include trypsin and chymotrypsin) in goat intestinal contents are the major initial processors and activators of the ETX prototoxin (Fig. 4A and B). However, partial inhibition of late-stage ETX processing was also observed by using a carboxypeptidase inhibitor, producing an ETX band of a size similar to that produced by a 90-min treatment with purified trypsin and chymotrypsin at physiological amounts. These results strongly suggest the involvement of carboxypeptidases in late-stage intestinal content processing to create the ~27-kDa ETX species (Fig. 4C).
Because of the presence of three ETX species in the purified ~27-kDa ETX preparation, it was important to assess which individual ETX species possessed cytotoxic activity. For this purpose, each ETX species was cloned, expressed recombinantly in E. coli, purified, and tested on MDCK cells. The results from these assays indicated that all three individual ETX species are cytotoxic (Fig. 7), supporting a potential pathogenesis role for these ETX fragments during C. perfringens type B and D infections.
In summary, the current studies for the first time investigated the activation and processing of epsilon prototoxin using the intestinal contents of a natural host. Under these conditions, the presence of multiple species of the activated toxin was detected. Future studies should examine the ETX species present in the large complexes identified in this study and whether variations in intestinal contents from different species or animals of different ages and physiologies affect the processing of ETX prototoxin and, in turn, the activity of ETX during in vivo disease, potentially explaining why certain animals do not suffer from disease caused by C. perfringens types B and D. However, the current findings significantly affect the understanding of this important toxin, particularly by identifying a role for other proteases besides trypsin and chymotrypsin in the processing and activation of ETX and by demonstrating that the processing is more complex and heterogenous than previously appreciated.
MATERIALS AND METHODS
Prototoxin purification and labeling with Alexa Fluor 488.
Epsilon prototoxin from C. perfringens type D strain NCTC8346 was purified to electrophoretic homogeneity as previously described (21–23). The prototoxin concentration was determined using a bicinchoninic assay (BCA) kit as recommended by the manufacturer (Pierce). The prototoxin was labeled with Alexa Fluor 488 as described previously (23).
ETX Western blotting.
Samples containing ETX were subjected to Western blotting using polyclonal rabbit anti-ETX antibody (BEI Resources) as described previously (21, 26).
Cell culture.
Madin-Darby canine kidney (MDCK-II) cells were cultured and maintained in a 50/50 (vol/vol) mix of Dulbecco’s modified Eagle’s medium (DMEM, Sigma) and Ham’s F12 nutrient mixture (Sigma) containing 8% fetal bovine serum, 1% glutamine, 100 units/ml of penicillin, and 100 mg/ml streptomycin. Cells were cultured in cell culture flasks at 37°C in 5% atmospheric CO2.
Goat small intestinal contents.
A healthy adult male Anglo Nubian goat was euthanized by an overdose of sodium barbiturate (Beuthanasia; Schering-Plough Animal Health, Kenilworth, NJ), and the contents were harvested from the jejunum and stored at −80°C until use. This procedure was approved by the University of California—Davis IACUC (permit number 16383). The intestinal contents were thawed, and solid material was removed by two rounds of centrifugation at 15,000 × g. The resulting intestinal contents were aliquoted and stored at −80°C until needed. Total protease activity was estimated using a colorimetric protease assay kit (Pierce) and was found to be ~1 µg/µl.
Purification of ETX species after treatment with goat intestinal contents.
ETX prototoxin was treated at 37°C with goat intestinal contents for 90 min. Following this incubation, Complete protease inhibitor cocktail (Roche) was added, the contents containing ETX were diluted in phosphate buffer, and the intestinal content-treated ETX species was purified with an AKTAprime liquid chromatography system, using DEAE support resin. Fractions containing the intestinal content-treated ETX product were pooled and concentrated, and concentrations were determined using the BCA method as described above.
Edman degradation amino acid sequencing of ETX prototoxin and the ~27-kDa ETX species after treatment with goat intestinal contents.
The N-terminal sequences of ETX prototoxin and the ~27-kDa ETX species produced by extended treatment with goat intestinal contents were determined using Edman degradation amino acid sequencing, performed at the University of California—Davis Proteomics Core Facility. A minimum of 4 cycles was used to determine the N-terminal sequence, as well as to confirm the identity of the proteins.
Capillary LC–ESI-TOF MS analysis for molecular-weight determination.
Protein samples were analyzed using capillary LC–ESI-TOF MS. Samples were loaded onto a PRLP-S column (5-µm particle size, 1,000-Å pore size, 300-µm inner diameter by 100 mm; Thermo Fisher) on the LC system (Ultimate 3000; Dionex, Sunnyvale, CA). The LC system was directly coupled to an electrospray ionization time-of-flight mass spectrometer (micrOTOF; BrukerDaltonics, Billerica, MA). Chromatographic separation was performed at a constant flow rate of 3.5 µl/min using a binary solvent system (solvent A, 2.5% acetonitrile, 0.1% formic acid; solvent B, 80% acetonitrile, 0.1% formic acid) and a linear gradient program (0 to 5 min, 5% B; 5 to 10 min, 5 to 30% B; 10 to 30 min, 30 to 75% B; 30 to 35 min, 75 to 100% B; 35 to 45 min, 100 to 5% B; and 45 to 60 min, 5% B). Mass spectra were acquired in positive ion mode over the mass range m/z 50 to 3,000. ESI spectra were deconvoluted to obtain molecular ion masses with DataAnalysis 3.3 (Bruker Daltonics, Billerica, MA) with an accuracy of 1 to 2 Da.
Recombinant ETX expression constructs and recombinant ETX purification.
rETX truncation fragments were constructed based upon the mass spectrometry and Edman degradation results of the current study or from previous results (4, 11) describing ETX processing. PCR was performed using Taq polymerase (New England Biolabs) and the following cycle conditions: 94°C for 5 min; 35 cycles of 94°C for 30 s, 50°C for 30 s, and 68°C for 1 min; and 68°C for 5 min. All primers and constructs are listed in Table 1. The forward primers contain a site for enterokinase cleavage that was used following protein purification. The PCR products and vector pMAL-c2x (New England Biolabs) were digested with EcoRI and BamHI (New England Biolabs), the PCR products were ligated into pMAL-c2x using Instant Sticky-end master mix (New England Biolabs), and the plasmids were transformed into Escherichia coli chi1776 (ATCC) in a manner that met E. coli 2 (EK2) plasmid system standards for biological containment. The sequence of each construct was confirmed by DNA sequencing at the University of Pittsburgh Genomics and Proteomics Core Laboratories.
TABLE 1 .
Primers and expression constructs
| Primer or expression construct | Primer sequence or construct description and primers used |
|---|---|
| Primers | |
| ETX-Forward | CATCAGAATTCGATGACGATGACAAGAAAGCTTCTTATGATAATGTAGATAC |
| ETX-262Reverse | CATCATGGATCCTTAATTTGTATTTAAATTTCTAACCTTAACT |
| ETX-263Reverse | CATCATGGATCCTTAATTATTTGTATTTAAATTTCTAACCTTA |
| ETX-264Reverse | CATCATGGATCCTTATACAATAATTGTATTTAAATTTCTAACC |
| ETX-267Reverse | CATCATGGATCCTTAATATTCTTGTACATTATTTGTATTTAAATTTC |
| ETX-273Reverse | GACCATGGATCCTTATTTTTTATCTACAGGTATTACATATTC |
| ETX-296Reverse | TTTGGATCCTTATTTTATTCCTGGTGCCTTAATAG |
| Constructs | |
| ETX-14/262 | N-terminal site of trypsin activation to Asn262, ETX-Forward/ETX-262Reverse |
| ETX-14/263 | N-terminal site of trypsin activation to Asn263, ETX-Forward/ETX-263Reverse |
| ETX-14/264 | N-terminal site of trypsin activation to Val264, ETX-Forward/ETX-264Reverse |
| ETX-14/267 | N-terminal site of trypsin activation to Tyr267, ETX-Forward/ETX-267Reverse |
| ETX-14/273 | N-terminal site of trypsin activation to Lys273, ETX-Forward/ETX-273Reverse |
| ETX-14/296 | N-terminal site of trypsin activation to Lys296, ETX-Forward/ETX-296Reverse |
Transformants were grown in LB broth supplemented with 50 µg/ml of ampicillin, 100 µg/ml of dl-diaminopimelic acid, and 10 µg/ml of thymidine. For induction, E. coli chi1776 (ATCC) was grown to an optical density at 600 nm (OD600) of 0.6 at 37°C before shifting the culture to room temperature and inducing with 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) for 2.5 h. Cells were harvested by centrifugation and lysed by sonication, and MBP-rETX fusion proteins were purified over amylose resin according to the manufacturer’s instructions (New England Biolabs). Purified MBP-rETX fusion proteins were then cleaved with enterokinase before downstream assays.
Assays of ETX-induced MDCK-II cytotoxicity.
To measure cytotoxicity, 10 µg of ETX prototoxin was treated for 1 h at 37°C with 10 µl of goat intestinal contents, 10 µg of pure trypsin (Sigma), 10 µg of pure chymotrypsin (Sigma), or a mixture containing 5 µg each of trypsin and chymotrypsin. Trypsin-chymotrypsin inhibitor from Glycine max (Sigma) was added to the treated samples, and 5 µg of ETX in dPBS with Ca2+ and Mg2+ (cellgro; Corning) was added to washed MDCK cells. The cells were incubated with ETX at 37°C for 1 h. ETX prototoxin alone (no protease) or (in the absence of prototoxin) goat intestinal contents, purified trypsin, and purified chymotrypsin with protease inhibitors served as negative controls for cytotoxicity. rETX fragments were used at a concentration of 5 µg/ml. For these assays, buffer was used as the negative control. A time course cytotoxicity study was performed using the same ratio of intestinal contents to ETX as described above. Aliquots were removed at the time points indicated in Fig. 2, 3, 4, and 5, and Complete protease inhibitor cocktail (Roche) was added. In all iterations, cytotoxicity was measured using a cytotoxicity detection kit (Roche) that measures lactose dehydrogenase (LDH) release, and the results are expressed as the percentage of total nonviable cells. Experiments were performed 3 times in duplicate; mean values are shown and the standard errors were determined. When broad-range protease inhibitors were utilized, each inhibitor was preincubated with intestinal contents for 30 min at room temperature before the addition of ETX and incubation at 37°C for 90 min. Inhibitors were purchased from Sigma-Aldrich and used at final concentrations as follows: 10 µM pepstatin A (aspartyl protease inhibitor), 10 µM E-64 (cysteine protease inhibitor), 2 µg/µl Bowman-Birk inhibitor (serine protease inhibitor), and 0.3 µg/µl carboxypeptidase inhibitor (mammalian carboxypeptidase inhibitor).
ACKNOWLEDGMENTS
These studies were supported by National Institute of Allergy and Infectious Diseases grant RO1 AI056177 (B.A.M.), MARCE grant 2U54AI057168-08 (Myron Levine, Principal Investigator) awarded to B.A.M., and T32 AI060525 (JoAnne Flynn, Principal Investigator) awarded to J.C.F.
We thank the University of California—Davis Proteomics Core for performing Edman sequencing and Guy T. Uechi from the University of Pittsburgh Genetics and Proteomics Core Laboratories (supported by the UPCI Cancer Proteomics Facility that is supported in part by award P30CA047904) for performing the LC–ESI-TOF MS analysis and for his assistance in data analysis. The following reagent was obtained through BEI Resources, NIAID, NIH: polyclonal anti-epsilon toxin from Clostridium perfringens (immunoglobulin G, rabbit), NR-865.
Footnotes
Citation Freedman JC, Li J, Uzal FA, McClane BA. 2014. Proteolytic processing and activation of Clostridium perfringens epsilon toxin by caprine small intestinal contents. mBio 5(5):e01994-14. doi:10.1128/mBio.01994-14.
REFERENCES
- 1. Uzal FA, Freedman JC, Shrestha A, Theoret JR, Garcia J, Awad MM, Adams V, Moore RJ, Rood JI, McClane BA. 2014. Towards an understanding of the role of Clostridium perfringens toxins in human and animal disease. Future Microbiol. 9:361–377. 10.2217/fmb.13.168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Petit L, Gibert M, Popoff MR. 1999. Clostridium perfringens: toxinotype and genotype. Trends Microbiol. 7:104–110. 10.1016/S0966-842X(98)01430-9 [DOI] [PubMed] [Google Scholar]
- 3. Popoff MR. 2011. Epsilon toxin: a fascinating pore-forming toxin. FEBS J. 278:4602–4615. 10.1111/j.1742-4658.2011.08145.x [DOI] [PubMed] [Google Scholar]
- 4. Minami J, Katayama S, Matsushita O, Matsushita C, Okabe A. 1997. Lambda-toxin of Clostridium perfringens activates the precursor of epsilon-toxin by releasing its N- and C-terminal peptides. Microbiol. Immunol. 41:527–535. 10.1111/j.1348-0421.1997.tb01888.x [DOI] [PubMed] [Google Scholar]
- 5. Garcia JP, Adams V, Beingesser J, Hughes ML, Poon R, Lyras D, Hill A, McClane BA, Rood JI, Uzal FA. 2013. Epsilon toxin is essential for the virulence of Clostridium perfringens type D infection in sheep, goats, and mice. Infect. Immun. 81:2405–2414. 10.1128/IAI.00238-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Miyamoto O, Sumitani K, Nakamura T, Yamagami S, Miyata S, Itano T, Negi T, Okabe A. 2000. Clostridium perfringens epsilon toxin causes excessive release of glutamate in the mouse hippocampus. FEMS Microbiol. Lett. 189:109–113. 10.1111/j.1574-6968.2000.tb09215.x [DOI] [PubMed] [Google Scholar]
- 7. Stiles BG, Barth G, Barth H, Popoff MR. 2013. Clostridium perfringens epsilon toxin: a malevolent molecule for animals and man? Toxins (Basel) 5:2138–2160. 10.3390/toxins5112138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gleeson-White MH, Bullen JJ. 1955. Clostridium welchii epsilon toxin in the intestinal contents of man. Lancet 268:384–385 [DOI] [PubMed] [Google Scholar]
- 9. Kohn J, Warrack GH. 1955. Recovery of Clostridium welchii type D from man. Lancet 268(6860):385. 10.1016/S0140-6736(55)91276-9 [DOI] [PubMed] [Google Scholar]
- 10. Rumah KR, Linden J, Fischetti VA, Vartanian T. 2013. Isolation of Clostridium perfringens type B in an individual at first clinical presentation of multiple sclerosis provides clues for environmental triggers of the disease. PLoS One 8:e76359. 10.1371/journal.pone.0076359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hunter SE, Clarke IN, Kelly DC, Titball RW. 1992. Cloning and nucleotide sequencing of the Clostridium perfringens epsilon-toxin gene and its expression in Escherichia coli. Infect. Immun. 60:102–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Worthington RW, Mülders MS. 1977. Physical changes in the epsilon prototoxin molecule of Clostridium perfringens during enzymatic activation. Infect. Immun. 18:549–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Goldstein J, Morris WE, Loidl CF, Tironi-Farinati C, McClane BA, Uzal FA. 2009. Clostridium perfringens epsilon toxin increases the small intestinal permeability in mice and rats. PLoS One 4:e7065. 10.1371/journal.pone.0007065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Finnie JW. 2004. Neurological disorders produced by Clostridium perfringens type D epsilon toxin. Anaerobe 10:145–150. 10.1016/j.anaerobe.2003.08.003 [DOI] [PubMed] [Google Scholar]
- 15. Uzal FA. 2004. Diagnosis of Clostridium perfringens intestinal infections in sheep and goats. Anaerobe 10:135–143. 10.1016/j.anaerobe.2003.08.005 [DOI] [PubMed] [Google Scholar]
- 16. Uzal FA, Kelly WR. 1998. Experimental Clostridium perfringens type D enterotoxemia in goats. Vet. Pathol. 35:132–140. 10.1177/030098589803500207 [DOI] [PubMed] [Google Scholar]
- 17. Bhown AS, Habeerb AF. 1977. Structural studies on epsilon-prototoxin of Clostridium perfringens type D. Localization of the site of tryptic scission necessary for activation to epsilon-toxin. Biochem. Biophys. Res. Commun. 78:889–896. 10.1016/0006-291X(77)90506-X [DOI] [PubMed] [Google Scholar]
- 18. Miyata S, Matsushita O, Minami J, Katayama S, Shimamoto S, Okabe A. 2001. Cleavage of a C-terminal peptide is essential for heptamerization of Clostridium perfringens epsilon-toxin in the synaptosomal membrane. J. Biol. Chem. 276:13778–13783. 10.1074/jbc.M011527200 [DOI] [PubMed] [Google Scholar]
- 19. Uzal FA, Songer JG. 2008. Diagnosis of Clostridium perfringens intestinal infections in sheep and goats. J. Vet. Diagn. Invest. 20:253–265. 10.1177/104063870802000301 [DOI] [PubMed] [Google Scholar]
- 20. Antalis TM, Shea-Donohue T, Vogel SN, Sears C, Fasano A. 2007. Mechanisms of disease: protease functions in intestinal mucosal pathobiology. Nat. Clin. Pract. Gastroenterol. Hepatol. 4:393–402. 10.1038/ncpgasthep0846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sayeed S, Fernandez-Miyakawa ME, Fisher DJ, Adams V, Poon R, Rood JI, Uzal FA, McClane BA. 2005. Epsilon-toxin is required for most Clostridium perfringens type D vegetative culture supernatants to cause lethality in the mouse intravenous injection model. Infect. Immun. 73:7413–7421. 10.1128/IAI.73.11.7413-7421.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Habeeb AF. 1969. Studies on epsilon-prototoxin of Clostridium perfringens type D. I. Purification methods: evidence for multiple forms of epsilon-prototoxin. Arch. Biochem. Biophys. 130:430–440. 10.1016/0003-9861(69)90055-1 [DOI] [PubMed] [Google Scholar]
- 23. Robertson SL, Li J, Uzal FA, McClane BA. 2011. Evidence for a prepore stage in the action of Clostridium perfringens epsilon toxin. PLoS One 6:e22053. 10.1371/journal.pone.0022053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Worthington RW, Mülders MS, Van Rensburg JJ. 1973. Clostridium perfringens type D epsilon prototoxin. Some chemical, immunological and biological properties of a highly purified prototoxin. Onderstepoort J. Vet. Res. 40:143–149 [PubMed] [Google Scholar]
- 25. Worthington RW, Mülders MS, Van Rensburg JJ. 1973. Enzymatic activation of Clostridium perfringens epsilon prototoxin and some biological properties of activated toxin. Onderstepoort J. Vet. Res. 40:151–154 [PubMed] [Google Scholar]
- 26. Harkness JM, Li J, McClane BA. 2012. Identification of a lambda toxin-negative Clostridium perfringens strain that processes and activates epsilon prototoxin intracellularly. Anaerobe 18:546–552. 10.1016/j.anaerobe.2012.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hambrook JL, Lindsay CD, Hughes N. 1995. Morphological alterations in MDCK cells induced by exposure to Clostridium perfringens epsilon-toxin. Biochem. Soc. Trans. 23:44S. [DOI] [PubMed] [Google Scholar]