

Abstract
Proteasomes are ATP-dependent protein degradation machines present in all archaea and eukaryotes, and found in several bacterial species of the order Actinomycetales. Mycobacterium tuberculosis (Mtb), an Actinomycete pathogenic to humans, requires proteasome function to cause disease. In this chapter, we describe what is currently understood about the biochemistry of the Mtb proteasome and its role in virulence. The characterization of the Mtb proteasome has led to the discovery that proteins can be targeted for degradation by a small protein modifier in bacteria as they are in eukaryotes. Furthermore, the understanding of proteasome function in Mtb has helped reveal new insight into how the host battles infections.
Introduction
Mycobacterium tuberculosis (Mtb) is the causative agent of tuberculosis (TB) and kills nearly two million people every year (http://www.who.int/). The infectious process starts with the inhalation of air-borne droplets containing Mtb bacilli. Bacteria replicate in professional phagocytes in the lungs where they must combat numerous anti-microbial molecules. If the host cannot control the infection, Mtb growth will result in the destruction of lung tissues and, ultimately, the death of the host.
Despite the astounding mortality caused by TB, most individuals infected with Mtb can control mycobacterial growth for much of their lives. Among the host’s arsenal of antimicrobial effectors is nitric oxide (NO), which is produced by activated macrophages and is toxic to numerous microbes [1]. Evidence that supports the notion that NO is critical to controlling Mtb has come from mouse studies. Inactivation of the macrophage associated inducible NO synthase, (iNOS) also known as NOS2, dramatically sensitizes mice to Mtb infections [2]. The cytotoxic effects of NO are likely to be dependent on the formation of highly reactive nitrogen intermediates (RNIs). It is thought that in host cells NO is oxidized to nitrite, which can be protonated to nitrous acid in the phagosomes of activated macrophages. Nitrous acid dismutates to reform NO, which can penetrate bacterial membranes and cell walls to combine with reactive oxygen intermediates (ROIs) such as superoxide to generate peroxynitrite. RNIs and ROIs can induce lethal injuries including DNA and protein damage as well as lipid peroxidation [3, 4].
Regardless of the apparent protective effects of host-produced NO during Mtb infections, humans, as well as experimentally infected animals, are rarely sterilized of Mtb [1, 2]. This observation was the basis for the hypothesis that Mtb encodes proteins required for resistance to NO toxicity. Due to the emergence of multi-drug resistant (MDR) and extensively-drug resistant (XDR) Mtb strains, researchers around the world are looking for novel ways to target TB. Drugs that inhibit bacterial defenses against mammalian antimicrobial effectors like NO could help the host win the war against this disease.
It has long been a technical challenge to identify and characterize pathways important for the pathogenesis of Mtb, a highly infectious and slow growing (doubling time ~20 h) Actinobacterium with a high GC-content. Over the last 20 years, improved molecular genetic tools and bio-safety provisions have greatly facilitated studies into understanding the pathogenesis of this challenging organism. With the advent of efficient transposon mutagenesis, it became feasible to perform a screen to identify genes required for NO resistance in vitro. After screening over 10,000 transposon mutants for NO sensitivity, Nathan and colleagues identified five Mtb mutants of the virulent laboratory strain H37Rv with independent insertions in Rv2115c and Rv2097c, two genes that were predicted to be associated with proteasome function [5]. Rv2115c was named mpa (Mycobacterium proteasomal ATPase) due to its high similarity with eukaryotic and archaeal proteasomal ATPases [6]. Mpa is 81 % identical to ARC (AAA ATPase forming ring-shaped complexes) of Rhodococcus erythropolis, the first biochemically characterized bacterial proteasomal ATPase [7]. In contrast to Rv2115c/Mpa, Rv2097c did not exhibit similarity to any known proteins at the time, however, was proposed to participate in proteasomal function, and as such was named paf for proteasome accessory factor (later termed pafA). Importantly, mutations in mpa and pafA severely attenuate Mtb virulence in mice [5].
Proteasomes are multi-subunit barrel-shaped protease complexes that were first discovered in eukaryotes over 20 years ago [8]. In eukaryotes the 26S proteasome is composed of two functionally distinct sub-complexes: the 20S core particle (CP), required for degradation of the substrate, and a 19S regulatory particle (RP) located at either or both ends of the CP, responsible for substrate unfolding and translocation into the CP [9, 10]. The RP is composed of numerous proteins, the composition of which varies depending on its function. The 19S RP contains 19 subunits, including a ring of six distinct AAA (ATPases associated with different cellular activities) proteins that contact the CP, and non-ATPase subunits, which function in various aspects of substrate recognition and processing [11]. The CP is composed of four stacked rings with catalytic activity located within the central rings. The two inner rings are composed of seven distinct catalytic β-subunits sandwiched between two outer rings composed of seven distinct α-subunits [10]. The β-subunits have several proteolytic activities that allow the proteasome to cleave most types of peptide bonds. Protein fragments are estimated to range in size from 8 to 10 residues [12]. The α-subunit rings form a gated channel that controls the passage of substrates and cleaved peptides, and also serves as a docking surface for protein complexes such as the RP [10, 13].
Proteasomes are enzymatically and structurally distinct from the ATP-dependent, chambered bacterial proteases ClpP, Lon, FtsH and HslV [14, 15]. The first clue that bona fide proteasomes were present in prokaryotes came from electron microscopy studies on the thermoacidophilic archaeon, Thermoplasma acidophilum, in which CP-like particles were obtained from T. acidophilum lysates [16]. Ultimately, T. acidophilum CPs were purified and crystallized, and shown to be highly similar in structure to eukaryotic CPs [17]. The first bacterial proteasome to be characterized was the R. erythropolis proteasome [18]. Later, 20S proteasomes were characterized from Mycobacterium smegmatis [19], Streptomyces coelicolor [20] and Frankia [21]. Additionally, genomic sequencing revealed the presence of proteasomal genes in the pathogens Mtb [22] and Mycobacterium leprae [23]. Bacterial proteasomes were thought to be confined to Actinobacteria until studies from the Banfield group (reviewed in [24]), discovered two actinomycete-like proteasome genes clusters in a non-culturable Gram-negative bacterium called Leptospirillum. To date, Actinomycetes and Leptospirillum are the only known bacterial lineages with a proteasome system, and may have acquired this protease complex via lateral gene transfer events [24, 25]. In contrast to the eukaryotic CPs, most prokaryotic CPs are composed of homo-heptameric rings; two β-subunits (PrcB) rings, flanked by two α-subunit (PrcA) rings (reviewed in [26, 27]). For the most part, the presence of only one type of β-subunit limits the proteolytic activity of the prokaryotic 20S CP to chymotryptic activity.
Since the initial identification by Darwin et al. of genes required for NO resistance in Mtb [5], it was later shown (using two separate prcBA mutant Mtb strains) that the CP was also needed for resistance to NO [28, 29]. This provided evidence of a functional link between Mpa, PafA and the CP. However, when compared to the wild type, mpa or pafA Mtb strains, the prcBA mutants grow much more slowly in rich broth (~20–30 % lower optical density at stationary phase) and take longer to form colonies on solid media [28, 29]. This growth defect in these genetically manipulated strains is similar to that observed for wild type Mtb strains treated with a mammalian proteasome inhibitor, N-(4-morpholine)carbonyl-b-(1-naphthyl)-L-alanine-L-leucine boronic acid (MLN-273) or epoxomicin [5]. Collectively, these data suggest that the Mtb CP can also degrade proteins in an Mpa/PafA independent manner; or that the CP has other, possibly protease-independent functions important for growth (to be discussed later). The notion that the Mtb CP is needed for normal growth is supported by a study that attempted to delineate genes essential for Mtb growth in vitro. In this study, Rubin and colleagues found that prcBA, but not mpa or pafA, were required for normal growth in vitro [30], therefore it is, not surprising that prcBA-defective Mtb strains are also highly attenuated in a mouse model of infection [28, 29].
The identification of a bacterial proteasome associated with a virulence phenotype piqued the interest of numerous laboratories to better characterize this protease. Here, we summarize what is currently understood about the structure and function of the Mtb proteasome and discuss its potential roles in pathogenesis.
Mycobacterium tuberculosis Proteasome Structure and Function
Structure of the Mtb 20S Core Particle
The overall architecture of the Mtb 20S CP is similar to the CP of archaeal [31] and eukaryotic [32] proteasomes. All CPs form a barrel-shaped structure consisting of four stacked, seven-subunit rings that are arranged into two central β-rings, flanked by an α-ring at either end. Like other prokaryotic proteasomes, Mtb CPs are arranged into a four-ringed α7β7β7α7 cylinder composed of 14 identical α-subunits and 14 identical β-subunits, ~150 Å in height with a diameter of ~115 Å (Fig. 10.1a, b). The Mtb proteasome shares modest sequence identity with the archaeal CP from Thermoplasma (~32 % identity for both α- and β-subunits) and high identity with the bacterial CP from Rhodococcus (~65 % identity). Despite this, with the exception of helix 2 in both the α- and β-subunits, the three-dimensional structures of all three prokaryotic CPs are virtually super-imposable [33, 34]. Although, only the relative position of helix 2 is altered in the bacterial CPs, this small upward tilt of ~10° creates a wider axial substrate channel in the bacterial CPs when compared to the archaeal CPs [33].
Fig. 10.1. Structure of the Mtb 20S core particle (CP).

(a) Side view of the Mtb 20S CP structure rendered in cartoon view (PDB ID 2FHG). The two α-rings at the top and the bottom are displayed in green, and the two catalytic middle β-rings in blue. (b) The same side view as in (a), but the structure is rendered in surface display. Only a central slab of 30 Å is shown such that the two anterior chambers and one central chamber of the CP are visible. The red circles mark the proteolytic sites
The proteolytic active sites of the CP are housed in the β-subunits (in eukaryotes only three of the seven β-subunits contain functional sites: β-1, -2, and -5 [35]). These active subunits are synthesized with N-terminal pro-peptides, the autocleavage of which exposes the catalytic nucleophile, an N-terminal threonine (Thr-1) [36]. Similarly, in Rhodococcus, there are two β-subunits (β1 and β2) both of which are translated with long propeptides (65 and 59 residues respectively). In this case, recombinant Rhodococcus α- and β-subunits only assemble into an active proteasomes when all subunits are combined, while separately expressed components remain monomeric [34, 37–39]. Collectively, these findings suggest that the β-subunit propeptide not only facilitates the formation of the first assembly intermediate (the α/β heterodimer) but also shields the catalytic Thr residue during proteasomal assembly, preventing undesired protein degradation [37, 40]. In Thermoplasma, the eight amino acid propeptide of the β-subunit seems to be dispensable for assembly of the CP, as the α-subunits can assemble spontaneously into seven subunit rings when produced in Escherichia coli [41]. In contrast the α-ring appears to serve as a template for assembly of the β-ring in the formation of active CP in Rhodococcus. In Mtb the 56-residue propeptide of PrcB appears to inhibit rather than promote CP assembly [42], as a cryo-EM study of the Mtb half proteasome revealed that the propeptide is located outside of the β-ring rather than between the α- and β-rings [43]. Because assembly of the mature CP is a result of the apposition of two half-proteasomes, it seems that the Mtb β-propeptides, which are auto-catalytically removed, could be a barrier to the assembly process. However, it may be that simply more time is required to overcome this barrier.
Chamber of Doom: Core Protease Activity
Although several prokaryotic CPs characterized so far have significant peptidase activity [17, 40, 44, 45], the in vitro peptidase activity of Mtb CP is relatively low [42]. This low activity suggests that the substrate gate is closed in a manner similar to the eukaryotic CP. In the eukaryotic CP, the substrate entrance to the α-ring is closed to prevent uncontrolled proteolytic activity [46]. In the eukaryotic CP, the gate to the catalytic chamber is blocked by the N-terminal sequences of the seven different α-subunits, which adopt different conformations and seal the entry portal [46]. In Mtb, a high-resolution crystal structure of the CP containing a mutant β-subunit (PrcB T1A, which prevents propeptide cleavage) revealed that the seven identical N-terminal peptides that form the gate were ordered, but exhibit three different conformations to tightly seal its substrate entrance at the seven-fold symmetry axis [43] (Fig. 10.2a, b). Deletion of the N-terminal residues 2–9 of PrcA results in an “open gate” conformation that increases peptidolytic activity to small peptides in vitro [42] (Fig. 10.2c).
Fig. 10.2. Top view of the Mtb 20S CP.
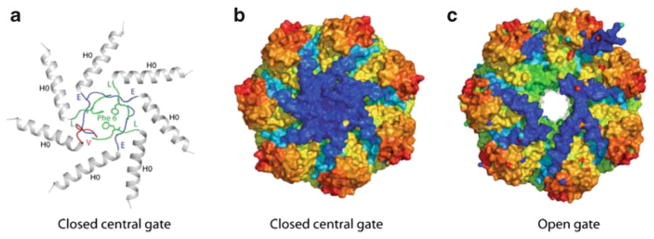
(a) and (b) Closed gate conformation (PDB ID 3MKA). The gate formed by the α-subunits is ordered and closed with a mechanism different to the eukaryotic CP. The seven subunits are chemically identical but adopt a total of three different conformations at their N-termini as indicated by the different colors in (a). A phenylalanine side chain from each of the green octapeptides contributes to the gate closure. (c) Deletion of the N-terminal residues 2–9 results in the open gate conformation (PDB ID 3MFE)
All CPs are N-terminal Thr hydrolases [14, 35, 36]. While most prokaryotic CPs seem to exclusively hydrolyze hydrophobic small synthetic peptide substrates (so-called “chymotryptic” activity), the Mtb CP has broad substrate specificity, targeting not only hydrophobic targets but also basic (“tryptic” activity) and acidic peptides (“peptidyl-glutamyl-peptide-hydrolyzing”, “caspase-like” or “post-acidic” activity) [42]. Unlike eukaryotic CPs in which substrate preference is determined by several different β-subunits [35, 47, 48], the Mtb 20S CP contains a single type of β-subunit, raising the question as to how it displays such broad substrate specificity. Structural analysis revealed that the substrate-binding pocket in the Mtb proteasome combines features found in different eukaryotic β-subunits: a hydrophobic upper surface similar to that of formed by a eukaryotic β5 subunit, and a hydrophilic lower surface similar to eukaryotic β1 and β2 subunits [33]. This composite feature of the substrate-binding pocket in the Mtb CP likely accounts for its broad substrate specificity [42].
Mpa: Gateway to Doom
Like other chambered proteases, proteasomal proteolysis requires an ATP-dependent chaperone to unfold structured proteins for delivery into the proteasome core where they are degraded. In archaea the best-characterized proteasomal ATPase is Methanococcus jannaschii PAN (proteasome activating nucleotidase) [49]. PAN can facilitate the degradation of artificial substrates by CPs [6, 49] but it remains to be determined how PAN recognizes archaeal proteins. Mpa forms homo-hexamers and is homologous to PAN. Like other AAA ATPases, Mpa has characteristic Walker A and B motifs for ATP binding and hydrolysis, respectively [50]. Mpa has relatively low ATPase activity: ATP hydrolysis is about four times slower than that of ARC, the Mpa orthologue in Rhodococcus (Vmax of 62 versus 268 pmol min−1 μg−1) [7, 51], or PAN [49]. As predicted (and will be discussed in detail below), a major function of Mpa is to deliver proteins into the CP for destruction.
The full-length structure of Mpa is currently unknown, however, crystal structure analysis of partial Mpa polypeptides has yielded highly informative insight into its activity. Mpa and ARC each contain two domains of the oligosaccharide/oligonucleotide-binding (OB) fold in tandem, with the second OB fold appearing to play a major role in Mpa oligomerization [52, 53] (Fig. 10.3a). In contrast, the archaeal PAN has only one OB domain [55], and it is not clear why Mpa and ARC have two. Immediately preceding the OB folds in Mpa is a 75 Å long α-helix [54] (Fig. 10.3b). Remarkably, helices from neighbouring subunit pairs form a coiled coil, thereby reducing the six-fold symmetry at the intermediate OB domain region to three-fold. This structural feature may be important for the specific protein recognition and unfolding activity of this class of ATPases.
Fig. 10.3. Comparison of Mpa with PAN.
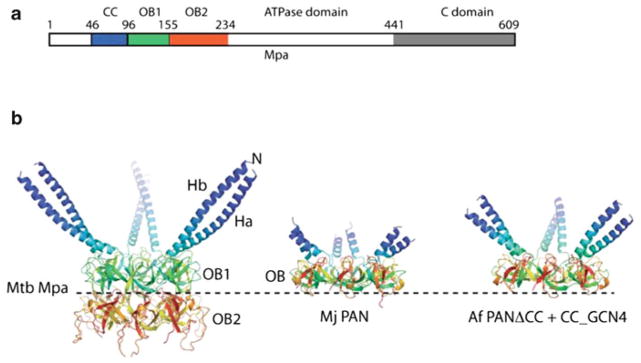
(a) Linear map of the domain structure of Mpa. CC = coiled coil; OB = oligosaccharide/oligonucleotide binding domain. (b) The structures of Mtb Mpa1-234 (PDB ID 3M9B), Archaeoglobus fulgidus PAN-ΔCC_CCGCN4 hybrid (PDB ID 2WG5), and M. jannaschii PAN (PDB ID 3H43) are aligned and displayed individually (Figure adapted from [54])
By analogy to the eukaryotic 26S proteasome, the prokaryotic proteasomal ATPases are also expected to physically interact with the CP in order to couple protein unfolding with delivery into the CP. A major distinction between the prokaryotic ATPases and eukaryotic 19S RP is that the 19S binds the CP with an affinity strong enough for the entire 26S complex (19S RP + 20S CP) to be co-purified. In stark contrast, prokaryotic ATPases, despite strong phenotypic associations with CP activity in vivo, only bind CPs either weakly or transiently in vitro [52, 56]. Methanococcus PAN weakly interacts with Thermoplasma CPs [56], and Mpa can directly associate, although flexibly and weakly, with open-gate mutant Mtb CPs [52] and can degrade proteins [57]. Because Mpa only weakly interacts with an altered proteasome in vitro, the precise nature of how any bacterial proteasome interacts with its cognate ATPase in vivo is a mystery. It is notable that proteasomal ATPases from bacteria to mammals contain a “HbYX (hydrophobic amino acid-tyrosine-X) motif” at their C-termini, and this motif is crucial for degradation but not ATPase activity [51, 56, 58, 59]. This motif is needed for PAN-mediated activation of proteolysis [60] and is implicated in proteasome assembly in yeast [59]. Mpa also has a HbYX-like motif, which is critical for the degradation of proteins in Mtb [51, 58]. Like PAN, Mpa lacking this motif is impaired in its interaction with the Mtb 20S CP in vitro [57].
A Pup-y Tale
In eukaryotes, proteins that are destined for proteasomal degradation are usually post-translationally modified with the small protein ubiquitin (Ub) (reviewed in [61]). Ubiquitin is synthesized as part of a precursor protein, processed to form a 76 amino acid protein containing a C-terminal diglycine motif (Gly-Gly), with a compact β-grasp fold [62–64]. The C-terminal Gly is subject to a series of reactions that result in the conjugation of Ub to a Lys residue on the target protein [65–67]. In the first step, the C-terminal Gly of Ub is adenylated by an Ub activating enzyme (E1) using ATP. Ub is then transferred to the active site Cys residue on the E1 enzyme. Next, Ub is transferred to an Ub conjugating enzyme (E2) and delivered to an Ub ligase (E3), which catalyzes the formation of an isopeptide bond with a Lys residue on the target protein (Fig. 10.4). In eukaryotes there are numerous E2 activating enzymes and E3 ligases (which contain a variety of substrate binding activities) to provide substrate specificity to the Ub proteasome system (UPS) (reviewed in [68, 69]). In general, proteins that are targeted to the proteasome have several Ub molecules added to the substrate, usually resulting in the formation of polyubiquitin (polyUb) chains [66, 70, 71]. The polyUb chains are recognized by the RP of the proteasome, and removed by proteasome-associated deubiquitinases (DUBs) for recycling of Ub. The deubiquitylated substrate is then delivered to the CP for destruction (reviewed in [11]).
Fig. 10.4. Eukaryotic ubiquitin-proteasome system.

Ubiquitin (Ub) is encoded as part of a larger polypeptide. Proteases expose a C-terminal Gly-Gly motif that is activated by adenylation with an E1 enzyme. The E1 enzyme transfers Ub to an E2 enzyme, where a thioester bond is formed. The E2 then transfers Ub to any number of E3 ligases. The E3 ligase family can be sub-divided into HECT and RING domain ligases: RING ligases interact with both the E2 and substrate, and facilitate the direct transfer of Ub from the E2 to the substrate. In contrast, HECT ligases form a thioester bond with Ub prior to transfer to a substrate Lys. E3 ligases dictate the type of Ub linkages that are formed. Ubiquitylated protein with Lys48 (K48) linked Ub chains are targeted for degradation by the 26S proteasome. Other types of Ub linkages (mono- and poly-K63 and others) generally do not result in degradation but serve other functions
Despite the presence of archaeal and bacterial CPs that are almost biochemically indistinguishable from the eukaryotic CP, an Ub-like system for the degradation of protein substrates was not found in prokaryotes for many years. A major hurdle at this time was an inability to reconstitute the proteolytic activity of bacterial proteasomes. This strongly suggested that other co-factors were required for proteolysis or that specific model substrates were required. It was speculated that bacterial proteasomal substrates only required intrinsic signals for degradation, as there was no evidence for the existence of any post-translational small protein modifiers in bacteria. To examine this, Darwin and colleagues set out to identify natural Mtb proteasome substrates by comparing the steady-state proteomes of wild type and mpa Mtb strains using two-dimensional polyacrylamide electrophoresis [58]. Although the proteomic profiles of untreated and NO-treated stationary phase cultures showed limited differences between the two Mtb strains, two proteins, FabD (malonyl CoA-acyl carrier protein transacylase) and PanB (ketopantoate hydroxymethyltransferase), accumulate significantly in the mpa strain. Importantly, the transcript levels fabD and panB are nearly identical in both wild type and mpa Mtb strains, which supported the notion that Mpa is required for FabD and PanB turnover, and not fabD or panB expression.
To further test if FabD and PanB were potential proteasome substrates, Mtb fabD and panB were expressed from a heterologous M. bovis hsp60 promoter in Mtb [72]. In addition to ruling out potential differences in gene expression, this system would also address the possibility that fabD and panB mRNA had differences in translation initiation in the mpa or pafA strains. Each recombinant gene also encoded a FLAG and His6 epitope tag at the N- and C-termini, respectively. Similar to that observed in 2D-PAGE analysis, both FLAG-FabD-His6 and FLAG-PanB-His6 accumulate in the mpa and pafA mutants [58]. Consistently, treatment of wild type Mtb with a eukaryotic proteasome inhibitor stabilized FabD and PanB [58] and deletion of the 20S CP permitted PanB accumulation [29]. Collectively, these data supported the idea that Mpa and PafA were required for the degradation of FabD and PanB by the CP.
In addition to the identification of FabD and PanB as proteasomal substrates, an unexpected observation was made during these studies: Mpa itself was also identified as a putative proteasomal substrate [58]. Chemical inhibition of the CP results in the accumulation of Mpa in Mtb. This finding was consistent with an earlier observation that mutations in mpa, which disrupt the ATPase activity or the HbYX motif, increase the steady-state levels of Mpa [51]. Similarly, Mpa accumulates in a pafA mutant strain, supporting a role for PafA in substrate degradation [58]. Thus it appears that the proteasome may “cannibalize” its ATPase to regulate its levels and hence activity.
Despite the identification of these putative substrates, their degradation, using purified Mpa and CP, could not be reconstituted in vitro. Therefore it was proposed that other factors were needed to facilitate proteasomal degradation. To identify these factors a bacterial two-hybrid screen [73] was used to search for proteins that bind to Mpa, with the reasoning that Mpa interacts not only with substrates but also with other proteins that promote proteolysis. From a library of ~100,000 Mtb genomic DNA fragments, Darwin and colleagues identified a protein encoded upstream of the CP genes prcBA, termed Rv2111c [74]. Importantly, recombinant Mpa and Rv2111c interacted in vitro, however, the addition of purified Rv2111c to CP and Mpa was unable to stimulate degradation of FabD.
The inability to reconstitute proteasomal degradation in vitro suggested that additional co-factors were still needed for proteolysis. Because E. coli does not encode a proteasome system it was reasoned that these co-factors were likely missing and possibly Mycobacterium-specific. A mycobacterial two-hybrid system [75] was thus used to interrogate interactions between proteasome subunits, substrates and the newly identified Rv2111c. In this system, M. smegmatis (Msm), a non-pathogenic relative of Mtb, was used as the host organism to identify protein-protein interactions. An unexpected interaction was detected between the substrate FabD and Rv2111c. To validate the genetic result, recombinant fabD and Rv2111c were co-expressed in Msm and found to co-purify as a heat stable complex. Mass spectrometry revealed that the C-terminal residue on Rv2111c formed an isopeptide bond with the side chain of Lys173 in FabD [74]. However, in contrast to Ub and related modifiers, Rv2111c did not have a C-terminal Gly-Gly motif, but instead has a Gly-Gly-Gln motif. Moreover, the C-terminal Gln was deamidated to Glu, before conjugation to FabD [74]. In a subsequent study, an orthologue of Rv2111c in Msm (MSMEG_3896) was identified as the protein modifier in that species. Thus, these two studies showed that this post-translational protein modification is conserved in both pathogenic and non-pathogenic mycobacteria [74, 76].
To determine if Rv2111c targeted proteins for degradation, Darwin and co-workers mutated the modified lysine residue (Lys173) in FabD [74]. Consistent with the idea that attachment with Rv2111c was the signal for degradation by the Mtb proteasome, mutagenesis of Lys173 to alanine in Mtb FabD stabilized the protein substrate. Based on its functional similarity to Ub, Rv2111c was named “Pup” for prokaryotic ubiquitin-like protein. Polyclonal antibodies to Pup recognize numerous proteins in Mtb H37Rv demonstrating that “pupylation” is widespread. Immunoblot analysis of the pafA mutant shows no anti-Pup reactive proteins, suggesting that PafA was the only Pup ligase in Mtb [74]. This result was somewhat unexpected as there are several hundred different Ub ligases in eukaryotes. In a subsequent study, Weber-Ban and colleagues demonstrated that PafA and ATP were sufficient to conjugate deamidated Pup to proteasomal substrates in vitro [77]. Interestingly, in contrast to Ub and other Ub-related modifiers, which all form a compact β-grasp fold, Pup is an intrinsically disordered protein with a propensity for helicity [78–80].
Lack of a Pupylation Motif
Although sequence recognition motifs for Ub ligases and other accessory factors have been identified, there is currently no known sequence motif surrounding the Lys on which Ub is attached (reviewed in [81]). In contrast, SUMO (small ubiquitin-like modifier) often attaches to a Lys residue that is part of a tetrapeptide motif, ΨKxD/E, where Ψ is a large hydrophobic residue and x is any amino acid (reviewed in [67]). The identification of a sequence that could predict pupylation could be useful for understanding how Pup regulates proteins. Therefore, to identify a possible pupylation motif, the “pupylome” was determined by several independent groups by purifying an epitope tagged Pup from Mtb [82] or Msm [83, 84]. In the Mtb study 604 proteins, representing ~15 % of the total predicted proteome, were identified, but only 55 proteins, including Mpa, were confirmed to harbor a site of Pup attachment [82]. In Msm, two independent studies identified 103 and 243 proteins, with 52 and 41 proteins, respectively, having confirmed Pup attachment sites [83, 84]. In all cases, Pup was attached to Lys, and there was little to no evidence of Pup chains, although Pup contains three Lys residues. In one study, Song and colleagues observed pupylation of Lys31 and Lys61 on Pup [84]; however, the authors of this study speculated this might not be physiologically relevant as Pup was overproduced. The authors also noted that pupylation was dynamic and changed depending on the growth condition examined. In all of the “pupylome” studies most, if not all, of the proteins identified are involved in housekeeping functions or stress responses.
Despite the successful identification of numerous pupylation targets, a motif is yet to be identified. It has however, been speculated that an intrinsic sequence is required to signal pupylation because PafA has a much higher affinity for at least one proteasome substrate, PanB, than for free Lys [85]. Nevertheless, it is hard to imagine how PafA specifically recognizes its targets because so many different proteins can be pupylated. In E. coli, a bacterial species that does not encode a Pup-proteasome system, pupylation can be reconstituted by expressing Pup with a C-terminal Glu (PupGlu) and PafA. Over 50 E. coli proteins can be pupylated using only PafA and a Pup mutant containing a C-terminal Glu (PupGlu), suggesting the notion that an intrinsic Mycobacterium specific sequence is required for pupylation is unlikely [86]. Consistently, PtsI (an E. coli pupylated substrate) was pupylated by a native mycobacterial system when produced in Msm [86]. Thus, signals for PafA target recognition are not expected to be Mycobacterium specific.
Based on these E. coli studies, it appears that pupylation is partly stochastic. However, it seems unlikely that mere over-expression of pup and pafA could determine the fate of so many proteins. Firstly, not all Lys containing proteins are pupylated, e.g. pupylation of the Mtb protein DlaT (dihydrolipoamide acyltransferase), which contains 27 Lys residues, has not been observed in either Mtb [74] or E. coli [86]. Secondly, not all Lys residues within the target protein are modified. Strikingly, Mtb FabD is preferentially modified on a single Lys residue, Lys173 [74, 82], despite the fact that Mtb FabD contains eight surface exposed Lys residues [87]. Although, two additional Lys residues can, to a lesser extent, be pupylated in Mtb FabD when produced in E. coli [86]. A simple explanation may be that over-production of Mtb FabD in the E. coli system merely gives PafA access to other Lys residues. Alternatively, these data may suggest that other factors regulate how and when FabD is pupylated in mycobacteria. Indeed, Darwin and colleagues speculated that the Lys residues in FabD may be involved in interactions with other enzymes in the fatty acid synthesis II (FASII) pathway [86] protecting them from modification. Interestingly, most enzymes in the FASII pathway, several of which are encoded in an operon with fabD, are pupylation targets [82, 83]. However, under normal culture conditions, only some of the proteins in this pathway appear to be proteasome substrates in Mtb. Recombinant FabG (3-ketoacyl-Acp-reductase), KasA and KasB (3-oxoacyl-Acp-synthases 1 and 2, respectively) do not accumulate in mpa or pafA mutants under routine culture conditions [82]. It is possible that FabG, KasA, and KasB are degraded by the proteasome under different conditions or are degraded more slowly than FabD. Taken together, there may still be additional Mycobacterium specificity factors that regulate pupylation or the delivery of certain pupylated proteins to the proteasome.
Pupylation: An Enzymatic Process That Resembles Glutamine Synthesis
At the time of its identification, PafA did not resemble any protein of known function [5]. Shortly after the discovery that PafA was involved in pupylation, Aravind and colleagues performed a detailed bioinformatic analysis that predicted PafA to have structural similarity to glutamine synthetase (GS) and glutamine cysteine synthetase (GCS) [88]. GS catalyzes the formation of Gln from Glu and ammonia, while GCS catalyzes the formation of γ-glutamyl-cysteine from Glu and Cys; both processes occur via a phosphorylated Glu intermediate. It was therefore proposed that the side chain carboxylate group of the C-terminal Glu in Pup would be phosphorylated or “activated” by PafA. This phosphorylated intermediate would then be primed for nucleophilic attack by the ε-amino group of a side chain Lys on a substrate, resulting in an isopeptide bond between C-terminal Glu of Pup and an internal Lys on the substrate. Indeed, as predicted by Iyer et al. [88], Weber-Ban and colleagues could show that Pup ~ substrate conjugates are generated via activation of the carboxylate group on the C-terminal Glu of Pup [77]. Consistently, site directed mutagenesis of residues in PafA predicted to coordinate ATP or Mg2+ disrupted PafA function both in vivo [89] and in vitro [77].
But how is Pup deamidated prior to activation by PafA? The first evidence came from the Weber-Ban group, who identified a homologue of PafA in H37Rv, (Rv2112c) near the proteasome core genes prcBA [77]. They demonstrated, using purified recombinant proteins, that Rv2112c was responsible for Pup deamidation, rendering Pup competent for ligation to FabD or PanB by PafA. Rv2112c was therefore named Dop for deamidase of Pup [77] (Fig. 10.5). In contrast to PafA, which needs ATP to phosphorylate Pup, Dop can deamidate Pup in the presence of ATP, ADP or non-hydrolyzable ATP analogues, suggesting ATP/ADP are allosteric activators of deamidation. Pupylation could also be achieved, in the absence of Dop, when Pup was replaced with a mutant form of Pup (PupGlu, that does not require deamidation for activation), obviating the need for Dop in vitro [77]. This demonstrated that Dop and PafA catalyze independent reactions: deamidation of Pup and conjugation of Pup, respectively. Curiously some Dop-containing bacteria encode PupGlu, presumably eliminating the need for Dop, which suggests that Dop may play a different role in these organisms. Nevertheless, for mycobacteria, deamidation is required for pupylation to take place as disruption of dop impairs pupylation and proteasomal substrate degradation [89, 90].
Fig. 10.5. Overview of the Pup-proteasome system in mycobacteria.
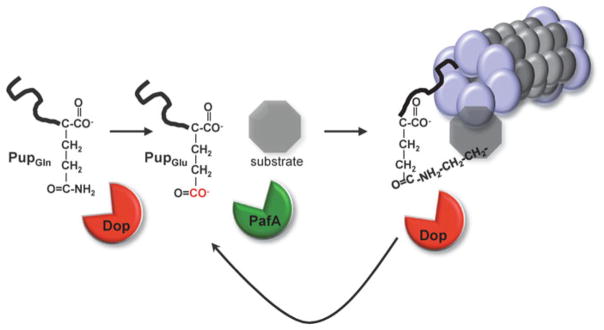
Pup is deamidated at the C-terminal Gln by Dop. PafA phosphorylates the α-carboxylate of the C-terminal Glu of Pup, priming it for attack by the ε-amino group of a substrate Lys. Pup can be removed by Dop prior to degradation to potentially rescue a substrate from destruction or possibly facilitate its degradation
Degradation by the Mtb Proteasome: End of the Road…or Is It?
How are pupylated proteins recognized by Mpa prior to degradation? In eukaryotes, Ub receptors are present in the RP of the 26S proteasome, ready to receive ubiquitylated proteins (reviewed in [11]). To date, an equivalent Pup receptor has not been found, however, Pup has a strong affinity for Mpa both in vitro and in vivo [74] and pull-down experiments using Pup-decorated beads showed that the N-terminal coiled-coil domain of Mpa interacts with Pup [79]. Unlike Ub, Pup is a mostly unstructured, intrinsically disordered protein [78–80], which raised the question: how does Mpa specifically recognize Pup?
A series of in vitro and in vivo experiments determined that Pup is a two-part degron where the N-terminal ~30 residues are required for Mpa to start the unfolding process and the C-terminal ~30 residues, which have the propensity for helicity as determined by NMR, are needed to interact with Mpa [57, 91]. Darwin and colleagues showed that the N-terminal half of Pup is essential for degradation, but dispensable for pupylation in vivo [91], suggesting the C-terminal half of Pup is necessary and sufficient for interaction with PafA and Dop. Weber-Ban and colleagues showed that Mpa could unfold green fluorescent protein if fused to Pup, and this required the N-terminal half of Pup [57]. Importantly, this study also showed that a pupylated substrate can be degraded, albeit somewhat slowly, by Mpa and the proteasome. Interestingly, Pup itself is degraded with the substrate in this system, showing removal of Pup is not essential for degradation.
The molecular details of the interactions required for substrate degradation were ultimately revealed when the three-dimensional structure of the Pup-Mpa complex was solved [54]. Analysis of this complex revealed that the central part of Pup (residues 21–51) becomes ordered upon binding to Mpa [54]. Indeed, the central part of Pup forms an α-helix, using the coiled-coil region of Mpa as a template (Fig. 10.6a). This interaction positions the disordered N-terminus of Pup towards the central channel of the hexameric ATPase, apparently priming the initial threading of Pup into the narrow unfolding pore [54] (Fig. 10.6b). This is consistent with previous studies that suggested the N-terminal half of Pup is needed to facilitate substrate unfolding and degradation [57, 91]. Both hydrophobic and electrostatic interactions are formed between Pup and Mpa, and disruption of either type of interaction abolishes Pup-mediated degradation by the proteasome in Msm [54]. Furthermore, consistent with other AAA proteases, mutagenesis of the conserved hydrophobic “pore loop” (Val342Ala) in Mpa abolishes degradation [52].
Fig. 10.6. Binding of Pup to Mpa induces a helical structure at the C-terminus of Pup.

(a) Top view of the crystal structure of the hexameric Mpa1-234 complex with Pup (PDB ID 3M9D). The binding-induced Pup helix is shown in the cartoon view in red, and the Mpa coiled-coil and OB domains are shown in surface view in green. (b) Model for the targeting of pupylated proteins for degradation by Mpa and CP. The Pup:Mpa1-234 complex structure (red and green) was placed over the homologous PAN AAA+domain structure (PDB ID 3H4M, magenta), which was overlaid on the Mtb CP (PDB ID 2FHH, yellow). A vertical central slice of the complex structure is shown for clarity. Pup is in red, and a model substrate in green
Importantly, although there are three coiled-coils in the Mpa hexamer (each potentially capable of interacting with Pup or a pupylated substrate) it appears that only one Pup associates per Mpa hexamer [54, 78, 79]. This arrangement would prevent multiple substrates from being recruited to the same Mpa hexamer at any one time, and hence would eliminate potential substrate aggregation or jamming at the proteasome.
Depupylation: What Goes On, Must Come Off
In the eukaryotic UPS, DUBs play an important role in protein degradation. Some DUBs, by removing Ub, are responsible for reversing the fate of a protein destined for degradation, while other DUBs located at the 19S RP, remove Ub to facilitate degradation (reviewed in [92]). DUBs are also responsible for the recycling of Ub to permit new ubiquitylation reactions. It was therefore predicted that pupylation would also be reversible. In a series of elegant experiments the Pup deamidase (Dop) was ultimately identified as a “depupylase” (DPUP) in both Mtb [91] and Msm [93]. DPUP activity was first demonstrated by Darwin and colleagues, using purified pupylated substrates (Pup ~ FabD and Pup ~ Ino1) with lysates from wild type Mtb. Although wild type Mtb lysates demonstrated DPUP activity, the dop mutant strain did not [91]. These data suggested that Dop was responsible not only for deamidation of Pup, but also for depupylation (Fig. 10.5). Indeed, Dop-mediated DPUP activity was then confirmed in vitro using a variety of pupylated substrates [91, 93]. It was then shown by Weber-Ban and colleagues that Dop cleaves specifically the isopeptide bond between Pup and the proteasomal substrate Lys and that PupGlu is released from the depupylation reaction, suggesting Pup could be recycled [93].
Dop is a strict isopeptidase because it cannot remove Pup from a longer polypeptide or linear fusion protein [91, 93]. This is in contrast to some Ub processing enzymes (DUBs) that remove Ub from larger polypeptides [64]. In contrast to DUBs, which tend to be either cysteine or zinc metalloproteases (reviewed in [92]), the catalytic motif of Dop is currently unknown; it does not have a nucleophilic Cys in its partially modeled active site [89], and is not known to require zinc for function. Like PafA, Dop is predicted to adopt a GS/GCS fold [88]. Dop binds Pup well in vitro unless Pup’s penultimate Gly-Gly motif is mutated, or additional amino acids are added to the C-terminus. Interestingly, however the Gly-Gly motif is not required for substrate attachment [89]. Taken together, it seems that the Gly-Gly motif is important for access to the active site of Dop but not for conjugation to substrates.
It is challenging to assess the role of Dop’s DPUP activity in vivo because Dop is required for Pup deamidation prior to pupylation in Msm and Mtb [89, 93]. Weber-Ban and colleagues fully restored pupylation in an Msm dop mutant by ectopic expression of pupGlu [93]. In striking contrast, however, expression of pupGlu does not restore the pupylome in Mtb [89]. However, the Mtb pupylome can be restored in the dop strain expressing pupGlu if treated with a proteasome inhibitor. This result suggests that Pup, along with its conjugated proteins, is directly degraded by the proteasome in Mtb lacking Dop. Importantly, this observation strongly suggests depupylation is needed to maintain Pup levels for normal pupylation. Thus, it seems likely that a critical function of Dop in Mtb is to act as a DPUP, an activity that is essential for Pup recycling. Furthermore, DPUP activity could also be used to regulate protein stability by altering the fate of a once doomed pupylated substrate. As with PafA and pupylation, it is not known how Dop selects substrates for depupylation. Finally, in bacteria that encode PupGlu, the primary function of Dop must be as a DPUP. It remains to be determined if certain bacteria have evolved to use Pup deamidation as a regulatory step in pupylation. Additionally, we do not yet understand why dop mutants in Msm and Mtb have such different phenotypes.
A curious observation was made in a study that examined the stability of Ino1 in Msm [94]. Over-expression of pup in wild type Msm results in virtually undetectable levels of endogenous Ino1, presumably due to its accelerated turnover. Interestingly, over-expression of pup in a prcBA mutant results in the accumulation of unpupylated Ino1. This finding suggests that in the prcBA mutant either pupylation of Ino1 is inhibited or depupylation prevents detection of Pup ~ Ino1. In contrast, a follow up study showed that Pup ~ Ino1 accumulates dramatically in an Msm mpa mutant overproducing Pup [91], which led to the hypothesis that Mpa helps to unfold a pupylated substrate in order for it to be depupylated. Consistent with this idea, Weber-Ban and co-workers showed that Mpa increases depupylation of a substrate in vitro [93]. Interestingly, it has been noted that corynebacteria, a distant relative of mycobacteria, encode pup, mpa, pafA, and dop orthologues but do not have proteasomes [90]. It is tempting to speculate that pupylated proteins are degraded by a different protease, or that pupylation-depupylation is involved in regulating protein activity in this bacterial genus.
The notion that protein unfolding by Mpa is coupled to depupylation poses some challenges to the current model of proteasome-mediated degradation in mycobacteria. It is well established that unfolding of proteasome substrates starts with the engagement of Pup with Mpa [54, 57]. Presumably, Pup is threaded through the channel in Mpa and, as previously shown, can itself be destroyed by the proteasome along with the substrate in vitro [57, 89]. However it also appears that substrates can be depupylated prior to degradation, presumably as they exit from the proximal end of the Mpa hexamer. This scenario implies that Dop interacts with Mpa or substrates at the interface between Mpa and the CP where the unfolded protein is being funnelled into the degradation chamber. One wonders if the conserved, but poorly understood, symmetry mismatch between the six-fold ATPase and the seven-fold CP evolved to prevent tight binding, and allow a gap for the removal of Pup by Dop. Clearly, much needs to be done in order to understand how, when and where Dop coordinates depupylation with degradation.
In eukaryotes, DUBs, ATPases, ligases and other proteins are associated with the eukaryotic 19S RP to remodel or recycle Ub chains on substrates. The Mycobacterium proteasome system appears to have been streamlined in such a way that Mpa plays multiple roles in the Pup-proteasome pathway by acting as a substrate receptor, unfoldase and a facilitator of depupylation. It remains to be determined if the Mtb proteasome requires additional co-factors to catalyze proteolysis. Because the in vitro degradation rate of a pupylated protein seems unusually slow [57, 91], it seems likely that other undetermined factors may be needed to facilitate degradation in Mtb.
Proteasomes and Pathogenesis
As discussed earlier in this chapter, mpa and pafA were identified as genes required for NO resistance and virulence in an animal model of infection [5]. Later studies (to be discussed below) identified additional components of the Pup-proteasome system that are also needed for Mtb pathogenesis. How does the Mtb proteasome protect against NO toxicity and promote TB pathogenesis? Perhaps the proteasome provides bacteria with a critical pool of amino acids through protein degradation during the chronic phase of infection. Alternatively, the bacterial proteasome may modify the host machinery to its advantage by degrading proteins that alter the recognition of the pathogen by the host’s immune system. In the next section we will attempt to address these complex questions by discussing the in vitro and in vivo phenotypes of proteasome associated mutants in more detail.
Characterization of Proteasome Pathway Mutants
The mouse model of TB is characterized by two phases, an acute phase, during which Mtb replicates exponentially within the lungs for about 3 weeks and a chronic or latent phase that is brought about by the emergence of acquired cell-mediated immunity. During the chronic phase bacterial numbers are stabilized in the lungs. Eventually, all mice that are experimentally inoculated with wild type Mtb die of TB, in contrast to humans that are naturally infected with Mtb. In a low dose aerosol model of Mtb infection, mice can survive for more than a year before succumbing to TB; in contrast, mice infected with either an mpa or pafA mutant show no symptoms of TB [51, 58]. Similar to the mpa and pafA mutants, an Mtb dop mutant is sensitive to RNI in vitro and severely attenuated in mice [89]. The degree of attenuation in mice (bacterial load and histopathology) is similar among the dop, mpa and pafA mutants, consistent with the notion that pupylation and Mpa-dependent proteolysis are functionally linked [5, 89]. The attenuation of symptoms is likely due to the presence of 100–1,000 times fewer recoverable mutant bacilli in the lungs, spleens and livers during the persistent phase of infection [5]. However, it is also possible that the Mtb proteasome regulates one or more factors that affect the host’s response to infection.
Targeted gene disruptions in the Mtb CP genes dramatically slow Mtb growth on solid media [28, 29]. In C57BL/6 mice infected with either ΔprcBA::hyg or PtetO-prcBA Mtb strains, the number of bacilli recovered from the lungs is approximately 100-fold lower (compared to wild type or non-silenced Mtb) after 3 weeks of infection and continues to decline after this time. This is not completely surprising based on the severe in vitro growth defects associated with Mtb CP mutations. In contrast to the prcBA strains, mpa, pafA and dop Mtb mutants grow more similarly to wild type Mtb in rich broth [5, 89]. These observations suggest that the CP may have critical functions independent of pupylation-dependent proteolysis in Mtb. Interestingly, the mpa, pafA and prcBA-defective Mtb strains are more resistant to hydrogen peroxide than wild type bacteria, suggesting there is an increase in activity or expression of one or more anti-oxidant pathways in the absence of proteasome function [5, 28]. However, it is currently not understood how loss of proteasome activity could lead to increased resistance to ROIs.
Because proteasome pathway mutants are sensitive to NO in vitro, Nathan and colleagues questioned if mice defective in NO production would be more susceptible to infection with mpa or pafA mutants. In mice and humans NO is produced by three different isoforms of nitric oxide synthase (NOS): endothelial NOS (eNOS), neuronal NOS (nNOS) and inducible or immune NOS (iNOS). iNOS is expressed in activated macrophages and is critical for the control of numerous microbial infections (reviewed in [1]). In comparison to wild type mice, mice genetically inactivated for iNOS (iNOS−/−) or treated with chemical inhibitors of NO production are extremely susceptible to Mtb [2]. Low dose aerosol infection of iNOS−/− mice with wild type Mtb (~200 bacteria/mouse) results in death within 3 months [5, 51, 58]. In contrast, iNOS−/− mice live significantly longer when infected with an mpa or pafA Mtb strain (~200–500 days post-infection) compared to infection with wild type Mtb (~60–80 days post-infection) [51, 58]. Because disruption of iNOS does not fully restore the virulence of the mpa and pafA mutants, it appears that the role of the Mtb proteasome extends beyond protection against RNIs in vivo.
In another study Bishai and colleagues identified three clones from a collection of random transposon insertion mutants in CDC1551 (a clinical isolate of Mtb) that were consistently smaller than the wild type strain [95]. All three independent mutants contained insertions in MT2175, which is identical to mpa in Mtb H37Rv. The CDC1551 mutants grow slower (doubling time of ~22 h) than the wild type strain (doubling time of 18 h) in standard 7H9 medium and fail to reach the same final culture density as wild type Mtb. Complementation of this CDC1551 mutant strain with mpa restores wild type colony morphology. Infection of BALB/c mice with a CDC1551 mpa mutant results in similar infection profiles as previously observed with the H37Rv mpa mutant. During the chronic phase of infection, bacterial numbers gradually decrease. Mice infected with CDC1551 mpa mutants survive without any signs of disease until 180 days (the latest time point assessed), while in contrast, mice infected with wild type Mtb succumb within 70 days. Lungs of mice infected with CDC1551 mpa mutants have attenuated pathological symptoms, such as less inflammation and fewer granuloma-like foci, and no weight loss compared to mice infected with wild type Mtb. Interferon gamma (IFN-γ) production fails to rise after 3 weeks of infection with the mutant compared to the wild type Mtb strain, hence mpa mutants seem to elicit a milder Th1 immune response in mice [95].
Is Mtb Proteasome Protease Activity Necessary for All Phenotypes?
Ehrt and colleagues made the puzzling observation that proteasomes containing a mutation in the active site can complement a prcBA null mutation in Mtb for RNI sensitivity and slow growth, but were unable to rescue impaired bacterial persistence in mice [29]. It is unclear how CPs that are proteolytically inactive could restore certain defects but not others. However, it may be possible that the CP has activities that have not yet been identified by routine biochemical assays. For example, the CP may act as a dock or scaffold for other proteins in order to function in specific stress responses. Taken together, these results suggest that the CP, proteolytically active or not, has a broader role for normal cell growth in virulent mycobacteria compared to its non-pathogenic relative Msm in which the CP appears to be dispensable under all conditions tested so far [19] (K.H.D., unpublished observations). The genome of Msm (7 Mb for Msm mc2155) is considerably larger than that of Mtb (4.4 Mb for Mtb H37Rv) and, unlike Mtb, Msm encodes another ATP-dependent compartmentalized protease (Lon protease) that may be able to compensate for deletion of prcBA in Msm (reviewed in [14]).
Proteasome Function and NO Resistance: An Unsolved Mystery
Although it is clear that the lack of proteasome function is a disadvantage for Mtb fitness during an infection, it remains to be established how proteasomal proteolysis is linked to pathogenesis. It seems likely that the inability to regulate proteins through degradation compromises bacterial survival when adapting to a new environment, i.e. within activated macrophages. There are several hypotheses that could explain why proteasome function is protective against RNI stress and important for survival in an animal host. Perhaps the simplest explanation is that the proteasome degrades damaged proteins. Oxidative and nitrosative damage of proteins can result in misfolding and aggregation, which is potentially lethal to cells. This damage could possibly be a signal for pupylation. It is also possible that specific accumulated proteasome substrates are particularly dangerous in the presence of NO. For example, iron-sulfur (Fe-S) clusters or copper (Cu) in metal binding proteins can be displaced by NO [96]. The liberation of Fe2+ or Cu+ is highly toxic to the cell as it can catalyze Fenton chemistry, resulting in the production of ROI. The observation that mpa, pafA and prcBA mutants are hyper-resistant to hydrogen peroxide, suggests that other anti-oxidant pathways may already be induced in an attempt to compensate for loss of the Pup-proteasome system. Currently, however, there is no evidence for the presence of increased amounts of metal-binding or damaged proteins in proteasomal degradation-defective mutants treated with NO.
Regulation of Transcription: Meddling with Metals
Another potential function of the proteasome is in transcriptional regulation. Almost all (if not all) compartmentalized proteases have been shown to regulate gene expression (reviewed in [97]). A microarray study comparing wild type Mtb with mpa and pafA mutants grown under standard culture conditions revealed that a common set of genes was differentially regulated (Table 10.1) [98]. Notably, none of the identified genes appears to be associated with the NO sensitive phenotype of the mpa and pafA mutants. Among the up-regulated genes in the mpa and pafA mutants were members of the zinc uptake regulator (Zur) regulon. In the presence of Zn, Zur is released from operators in at least three promoters in Mtb, and gene expression is induced [99]. One of the Zur-regulated promoters identified in the microarray drives the expression of the esx-3 (ESAT-6, region 3) operon. The esx-3 locus is, for the most part, essential for the growth of Mtb under normal culture conditions and is proposed to encode a type VII secretion system that is involved in zinc and iron acquisition [100, 101]. In addition, Zur regulates the expression of genes that encode homologues of Zn-binding ribosomal proteins. Ribosomes are comprised of numerous small proteins, several of which bind Zn. Under Zn-limiting conditions, these Zn-binding proteins are thought to be replaced with non-metal binding components [102, 103], allowing the bacteria to gain access to a large pool of zinc. If mutations in mpa or pafA result in deregulation of the Zur regulon in vivo as they do in vitro, these data would suggest that metal homeostasis is critical during infection and an inappropriate increase in expression of the Zur regulon during infection may also have deleterious affects on bacterial survival for other reasons.
Table 10.1.
Zur and RicR regulons are differentially regulated in mpa and pafA mutants when compared to wild type Mtb (Adapted from [98])
| CDC1551a | H37Rva | Gene | Name/functionb | mpac | pafAc | |
|---|---|---|---|---|---|---|
| Zur regulond | MT0115 | Rv0106 | Conserved hypothetical protein. Unknown function | 3.25 | 4.21 | |
| MT0292 | Rv0280 | ppe3 | PPE family protein. Unknown function. Unknown function | 3.85 | 4.1 | |
| MT0293 | Rv0281 | Conserved hypothetical protein. Possible methlytransferase | 1.94 | 2.46 | ||
| MT0295 | Rv0282 | Conserved hypothetical protein. Unknown function | 2.15 | 2.49 | ||
| MT0296 | Rv0283 | Possible conserved membrane protein. Unknown function | 2.06 | 2.34 | ||
| MT0297 | Rv0284 | Possible conserved membrane protein. Unknown function | 2.33 | 2.51 | ||
| MT0298 | Rv0285 | pe5 | PE family protein. Unknown function | 1.91 | 2.19 | |
| MT0299 | Rv0286 | ppe4 | PPE family protein. Unknown function | 1.85 | 2.16 | |
| MT0300 | Rv0287 | esxG | ESAT-6 like protein. Unknown function | 1.61 | 2.1 | |
| MT0302 | Rv0289 | Conserved hypothetical protein. Unknown function | 2 | 2.2 | ||
| MT0303 | Rv0290 | Probable conserved transmembrane protein. Unknown function | 1.94 | 1.96 | ||
| MT035 | Rv0292 | Probable conserved transmembrane protein. Unknown function | 1.89 | 2.13 | ||
| MT2115 | Rv2055c | rpsR2 | Probable ribosomal protein S18. involved in translation, amino-acyl tRNA binding | 2.65 | 3.81 | |
| MT2117 | Rv2056c | rpsN2 | Probable ribosomal protein S14. Involved in translation | 2.43 | 4.21 | |
| MT2117.1 | Rv2057c | rpmG1 | Probable ribosomal L33. Involved in translation | 2.91 | 4.2 | |
| MT2118 | Rv2058c | rpmB2 | Probable 50S ribosomal protein L28. Involved I ribosome activity | 3.47 | 6.12 | |
| MT2119 | Rv2059 | Conserved hypothetical protein. Unknown function | n.i | 2.17 | ||
| MT2428 | Rv2359 | zur | Zinc uptake regulator (formally furB) | 0.54 | 0.94 | |
| RicR regulone | MT0196 | Rv0186a | mymT | Metallothionein | 0.71 | 0.47 |
| MT0200 | Rv0190 | ricR | Regulated in copper repressor | n.i. | n.i. | |
| MT0870 | Rv0847 | lpqS | Probable lipoprotein. Unknown function | 0.52 | 0.33 | |
| socA | small ORF. Unknown function | n.i. | n.i. | |||
| MT1746.1 | socB | small ORF. Unknown function | n.i. | n.i. | ||
| MT3039 | Rv2963 | Probable integral membrane protein. Unknown function | 0.6 | 0.38 |
Locus number in Mtb strains CDC1551 and H37Rv
Functional annotations are taken from http://genolist.pasteur.fr/TubercuList/
Numbers represent expression in pafA and mpa strains relative to wild type Mtb H37Rv grown to early stationary phase in 7H9 media, as determined by microarray analysis. n.i: not-identified in the microarray [98]
Zur regulon is described in detail elsewhere [99]
RicR regulon is described in detail elsewhere [98]
Transcriptome analysis also identified a set of genes repressed in the mpa and pafA mutants that are regulated by copper [98]. Several of these genes form a copper-inducible regulon, which is under the control of RicR (regulated in copper repressor). During copper depleted conditions, RicR represses five promoters that drive the expression of ricR itself, mymT (a copper methallothionein) and several genes of unknown function (Table 10.1). Disruption of ricR results in hyper-resistance of Mtb to normally toxic levels of copper, presumably due to the constitutive expression of one or more copper resistance genes like mymT [96]. It is worth noting that several of the RicR-regulated genes (mymT, lpqS, socAB) are unique to pathogenic mycobacteria, suggesting that copper regulation is important for virulence. Thus, the attenuated phenotype of Pup-proteasome pathway mutants may in part be explained by the incomplete derepression of the RicR regulon during infection. These data support an emerging notion that copper has an important antimicrobial role during TB infection and possibly other infections [96, 98, 104–107].
It is interesting that two metal-dependent regulons are deregulated in mpa and pafA mutants. In both cases, the mutant bacteria appear to be responding to low metal concentrations. These data also suggest Mtb (and possibly other bacteria) need to adapt to changes in metal homeostasis in the host. As with Zur, it is currently not understood how the proteasome affects the expression of the RicR regulon.
Remaining Questions
As with other organisms, regulated proteolysis is critical for numerous aspects of TB biology. Mtb possesses a proteasome highly similar to those found in other domains of life, and uses it to resist host derived stresses like NO and regulate pathways that may be needed for pathogenesis. Proteasomal proteolysis is controlled, in part, by pupylation, which is functionally, if not biochemically, similar to ubiquitylation. The characterization of proteasome biochemistry and biology will undoubtedly allow researchers to gain valuable insight into the lifestyle of one of the most successful human pathogens. Among the numerous questions that remain to be asked of the young field of bacterial proteasome biology include:
How are proteins selected for pupylation and depupylation?
How does Mpa interact with the 20S CP? Why is Mpa itself a proteasome substrate?
Does pupylation have functions independent of targeting proteins to the proteasome?
How is proteasome function linked to NO resistance?
Are misfolded or damaged proteins degraded in a proteasome dependent manner?
Why and how are the Zur and RicR regulons affected by proteasome activity?
Acknowledgments
We are grateful to Nadine Bode and Andrew Darwin for critical review of this manuscript. K.H.D is supported by NIH grants AI065437 and HL92774, the Irma T. Hirschl Trust, and holds an Investigators in the Pathogenesis of Infectious Disease Award from the Burroughs Wellcome Fund. H.L. is supported by NIH grant AI070285 and Brookhaven National Laboratory LDRD grant 10-016.
Contributor Information
Marie I. Samanovic, Department of Microbiology, New York University School of Medicine, 550 First Avenue, MSB 236, New York, NY 10016, USA
Huilin Li, Department of Biochemistry and Cell Biology, Stony Brook University, Stony Brook, NY 11794, USA. Brookhaven National Laboratory Biology Department, Brookhaven National Laboratory, 50 Bell Ave, Upton, Brookhaven, NY 11973-5000, USA.
K. Heran Darwin, Email: heran.darwin@med.nyu.edu, Department of Microbiology, New York University School of Medicine, 550 First Avenue, MSB 236, New York, NY 10016, USA.
References
- 1.Nathan C, Shiloh MU. Reactive oxygen and nitrogen intermediates in the relationship between mammalian hosts and microbial pathogens. Proc Natl Acad Sci U S A. 2000;97(16):8841–8848. doi: 10.1073/pnas.97.16.8841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.MacMicking J, Xie QW, Nathan C. Nitric oxide and macrophage function. Annu Rev Immunol. 1997;15:323–350. doi: 10.1146/annurev.immunol.15.1.323. [DOI] [PubMed] [Google Scholar]
- 3.Alvarez B, Radi R. Peroxynitrite reactivity with amino acids and proteins. Amino Acids. 2003;25(3–4):295–311. doi: 10.1007/s00726-003-0018-8. [DOI] [PubMed] [Google Scholar]
- 4.Szabo C. Multiple pathways of peroxynitrite cytotoxicity. Toxicol Lett. 2003;140–141:105–112. doi: 10.1016/s0378-4274(02)00507-6. [DOI] [PubMed] [Google Scholar]
- 5.Darwin KH, Ehrt S, Gutierrez-Ramos JC, Weich N, et al. The proteasome of Mycobacterium tuberculosis is required for resistance to nitric oxide. Science. 2003;302(5652):1963–1966. doi: 10.1126/science.1091176. [DOI] [PubMed] [Google Scholar]
- 6.Zwickl P, Ng D, Woo KM, Klenk HP, et al. An archaebacterial ATPase, homologous to ATPases in the eukaryotic 26S proteasome, activates protein breakdown by 20S proteasomes. J Biol Chem. 1999;274(37):26008–26014. doi: 10.1074/jbc.274.37.26008. [DOI] [PubMed] [Google Scholar]
- 7.Wolf S, Nagy I, Lupas A, Pfeifer G, et al. Characterization of ARC, a divergent member of the AAA ATPase family from Rhodococcus erythropolis. J Mol Biol. 1998;277(1):13–25. doi: 10.1006/jmbi.1997.1589. [DOI] [PubMed] [Google Scholar]
- 8.Arrigo AP, Tanaka K, Goldberg AL, Welch WJ. Identity of the 19S ‘prosome’ particle with the large multifunctional protease complex of mammalian cells (the proteasome) Nature. 1988;331(6152):192–194. doi: 10.1038/331192a0. [DOI] [PubMed] [Google Scholar]
- 9.Pickart CM, Cohen RE. Proteasomes and their kin: proteases in the machine age. Nat Rev Mol Cell Biol. 2004;5(3):177–187. doi: 10.1038/nrm1336. [DOI] [PubMed] [Google Scholar]
- 10.Coux O, Tanaka K, Goldberg AL. Structure and functions of the 20S and 26S proteasomes. Annu Rev Biochem. 1996;65:801–847. doi: 10.1146/annurev.bi.65.070196.004101. [DOI] [PubMed] [Google Scholar]
- 11.Finley D. Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu Rev Biochem. 2009;78:477–513. doi: 10.1146/annurev.biochem.78.081507.101607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rock KL, Goldberg AL. Degradation of cell proteins and the generation of MHC class I-presented peptides. Annu Rev Immunol. 1999;17:739–779. doi: 10.1146/annurev.immunol.17.1.739. [DOI] [PubMed] [Google Scholar]
- 13.Schmidt M, Hanna J, Elsasser S, Finley D. Proteasome-associated proteins: regulation of a proteolytic machine. Biol Chem. 2005;386(8):725–737. doi: 10.1515/BC.2005.085. [DOI] [PubMed] [Google Scholar]
- 14.Butler SM, Festa RA, Pearce MJ, Darwin KH. Self-compartmentalized bacterial proteases and pathogenesis. Mol Microbiol. 2006;60(3):553–562. doi: 10.1111/j.1365-2958.2006.05128.x. [DOI] [PubMed] [Google Scholar]
- 15.Gur E, Ottofuelling R, Dougan DA. Machines of destruction – AAA + proteases and the adaptors that control them. In: Dougan DA, editor. Regulated proteolysis in microorganisms. Vol. 66. Springer, Subcell Biochem; 2013. pp. 3–33. [DOI] [PubMed] [Google Scholar]
- 16.Dahlmann B, Kopp F, Kuehn L, Niedel B, et al. The multicatalytic proteinase (prosome) is ubiquitous from eukaryotes to archaebacteria. FEBS Lett. 1989;251(1–2):125–131. doi: 10.1016/0014-5793(89)81441-3. [DOI] [PubMed] [Google Scholar]
- 17.Lowe J, Stock D, Jap B, Zwickl P, et al. Crystal structure of the 20S proteasome from the archaeon T. acidophilum at 3.4 A resolution. Science. 1995;268(5210):533–539. doi: 10.1126/science.7725097. [DOI] [PubMed] [Google Scholar]
- 18.Tamura T, Nagy I, Lupas A, Lottspeich F, et al. The first characterization of a eubacterial proteasome: the 20S complex of Rhodococcus. Curr Biol. 1995;5(7):766–774. doi: 10.1016/s0960-9822(95)00153-9. [DOI] [PubMed] [Google Scholar]
- 19.Knipfer N, Shrader TE. Inactivation of the 20S proteasome in Mycobacterium smegmatis. Mol Microbiol. 1997;25(2):375–383. doi: 10.1046/j.1365-2958.1997.4721837.x. [DOI] [PubMed] [Google Scholar]
- 20.Nagy I, Tamura T, Vanderleyden J, Baumeister W, et al. The 20S proteasome of Streptomyces coelicolor. J Bacteriol. 1998;180(20):5448–5453. doi: 10.1128/jb.180.20.5448-5453.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pouch MN, Cournoyer B, Baumeister W. Characterization of the 20S proteasome from the actinomycete Frankia. Mol Microbiol. 2000;35(2):368–377. doi: 10.1046/j.1365-2958.2000.01703.x. [DOI] [PubMed] [Google Scholar]
- 22.Lupas A, Zuhl F, Tamura T, Wolf S, et al. Eubacterial proteasomes. Mol Biol Rep. 1997;24(1–2):125–131. doi: 10.1023/a:1006803512761. [DOI] [PubMed] [Google Scholar]
- 23.Cole ST, Brosch R, Parkhill J, Garnier T, et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393(6685):537–544. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- 24.De Mot R. Actinomycete-like proteasomes in a Gram-negative bacterium. Trends Microbiol. 2007;15(8):335–338. doi: 10.1016/j.tim.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 25.Gille C, Goede A, Schloetelburg C, Preissner R, et al. A comprehensive view on proteasomal sequences: implications for the evolution of the proteasome. J Mol Biol. 2003;326(5):1437–1448. doi: 10.1016/s0022-2836(02)01470-5. [DOI] [PubMed] [Google Scholar]
- 26.Groll M, Bochtler M, Brandstetter H, Clausen T, et al. Molecular machines for protein degradation. Chembiochem. 2005;6(2):222–256. doi: 10.1002/cbic.200400313. [DOI] [PubMed] [Google Scholar]
- 27.Cerda-Maira F, Darwin KH. The Mycobacterium tuberculosis proteasome: more than just a barrel-shaped protease. Microbes Infect. 2009;11(14–15):1150–1155. doi: 10.1016/j.micinf.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gandotra S, Schnappinger D, Monteleone M, Hillen W, et al. In vivo gene silencing identifies the Mycobacterium tuberculosis proteasome as essential for the bacteria to persist in mice. Nat Med. 2007;13(12):1515–1520. doi: 10.1038/nm1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gandotra S, Lebron MB, Ehrt S. The Mycobacterium tuberculosis proteasome active site threonine is essential for persistence yet dispensable for replication and resistance to nitric oxide. PLoS Pathog. 2010;6(8):e1001040. doi: 10.1371/journal.ppat.1001040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sassetti CM, Boyd DH, Rubin EJ. Genes required for mycobacterial growth defined by high density mutagenesis. Mol Microbiol. 2003;48(1):77–84. doi: 10.1046/j.1365-2958.2003.03425.x. [DOI] [PubMed] [Google Scholar]
- 31.Maupin-Furlow JA. Archaeal proteasomes and sampylation. In: Dougan DA, editor. Regulated proteolysis in microorganisms. Vol. 66. Springer, Subcell Biochem; 2013. pp. 297–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Buchberger A. Roles of Cdc48 in regulated protein degradation in yeast. In: Dougan DA, editor. Regulated proteolysis in microorganisms. Vol. 66. Springer, Subcell Biochem; 2013. pp. 195–222. [DOI] [PubMed] [Google Scholar]
- 33.Hu G, Lin G, Wang M, Dick L, et al. Structure of the Mycobacterium tuberculosis proteasome and mechanism of inhibition by a peptidyl boronate. Mol Microbiol. 2006;59(5):1417–1428. doi: 10.1111/j.1365-2958.2005.05036.x. [DOI] [PubMed] [Google Scholar]
- 34.Witt S, Kwon YD, Sharon M, Felderer K, et al. Proteasome assembly triggers a switch required for active-site maturation. Structure. 2006;14(7):1179–1188. doi: 10.1016/j.str.2006.05.019. [DOI] [PubMed] [Google Scholar]
- 35.Groll M, Ditzel L, Lowe J, Stock D, et al. Structure of 20S proteasome from yeast at 2.4 A resolution. Nature. 1997;386(6624):463–471. doi: 10.1038/386463a0. [DOI] [PubMed] [Google Scholar]
- 36.Seemuller E, Lupas A, Baumeister W. Autocatalytic processing of the 20S proteasome. Nature. 1996;382(6590):468–471. doi: 10.1038/382468a0. [DOI] [PubMed] [Google Scholar]
- 37.Zuhl F, Seemuller E, Golbik R, Baumeister W. Dissecting the assembly pathway of the 20S proteasome. FEBS Lett. 1997;418(1–2):189–194. doi: 10.1016/s0014-5793(97)01370-7. [DOI] [PubMed] [Google Scholar]
- 38.Zuhl F, Tamura T, Dolenc I, Cejka Z, et al. Subunit topology of the Rhodococcus proteasome. FEBS Lett. 1997;400(1):83–90. doi: 10.1016/s0014-5793(96)01403-2. [DOI] [PubMed] [Google Scholar]
- 39.Mayr J, Seemuller E, Muller SA, Engel A, et al. Late events in the assembly of 20S proteasomes. J Struct Biol. 1998;124(2–3):179–188. doi: 10.1006/jsbi.1998.4068. [DOI] [PubMed] [Google Scholar]
- 40.Kwon YD, Nagy I, Adams PD, Baumeister W, et al. Crystal structures of the Rhodococcus proteasome with and without its pro-peptides: implications for the role of the pro-peptide in proteasome assembly. J Mol Biol. 2004;335(1):233–245. doi: 10.1016/j.jmb.2003.08.029. [DOI] [PubMed] [Google Scholar]
- 41.Zwickl P, Kleinz J, Baumeister W. Critical elements in proteasome assembly. Nat Struct Biol. 1994;1(11):765–770. doi: 10.1038/nsb1194-765. [DOI] [PubMed] [Google Scholar]
- 42.Lin G, Hu G, Tsu C, Kunes YZ, et al. Mycobacterium tuberculosis prcBA genes encode a gated proteasome with broad oligopeptide specificity. Mol Microbiol. 2006;59(5):1405–1416. doi: 10.1111/j.1365-2958.2005.05035.x. [DOI] [PubMed] [Google Scholar]
- 43.Li D, Li H, Wang T, Pan H, et al. Structural basis for the assembly and gate closure mechanisms of the Mycobacterium tuberculosis 20S proteasome. EMBO J. 2010;29(12):2037–2047. doi: 10.1038/emboj.2010.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Groll M, Huber R. Substrate access and processing by the 20S proteasome core particle. Int J Biochem Cell Biol. 2003;35(5):606–616. doi: 10.1016/s1357-2725(02)00390-4. [DOI] [PubMed] [Google Scholar]
- 45.Zhang F, Wu Z, Zhang P, Tian G, et al. Mechanism of substrate unfolding and translocation by the regulatory particle of the proteasome from Methanocaldococcus jannaschii. Mol Cell. 2009;34(4):485–496. doi: 10.1016/j.molcel.2009.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Groll M, Bajorek M, Kohler A, Moroder L, et al. A gated channel into the proteasome core particle. Nat Struct Biol. 2000;7(11):1062–1067. doi: 10.1038/80992. [DOI] [PubMed] [Google Scholar]
- 47.Groll M, Heinemeyer W, Jager S, Ullrich T, et al. The catalytic sites of 20S proteasomes and their role in subunit maturation: a mutational and crystallographic study. Proc Natl Acad Sci U S A. 1999;96(20):10976–10983. doi: 10.1073/pnas.96.20.10976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Heinemeyer W, Fischer M, Krimmer T, Stachon U, et al. The active sites of the eukaryotic 20 S proteasome and their involvement in subunit precursor processing. J Biol Chem. 1997;272(40):25200–25209. doi: 10.1074/jbc.272.40.25200. [DOI] [PubMed] [Google Scholar]
- 49.Benaroudj N, Goldberg AL. PAN, the proteasome-activating nucleotidase from archaebacteria, is a protein-unfolding molecular chaperone. Nat Cell Biol. 2000;2(11):833–839. doi: 10.1038/35041081. [DOI] [PubMed] [Google Scholar]
- 50.Walker JE, Saraste M, Runswick MJ, Gay NJ. Distantly related sequences in the alpha- and beta-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO J. 1982;1(8):945–951. doi: 10.1002/j.1460-2075.1982.tb01276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Darwin KH, Lin G, Chen Z, Li H, et al. Characterization of a Mycobacterium tuberculosis proteasomal ATPase homologue. Mol Microbiol. 2005;55(2):561–571. doi: 10.1111/j.1365-2958.2004.04403.x. [DOI] [PubMed] [Google Scholar]
- 52.Wang T, Li H, Lin G, Tang C, et al. Structural insights on the Mycobacterium tuberculosis proteasomal ATPase Mpa. Structure. 2009;17(10):1377–1385. doi: 10.1016/j.str.2009.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Djuranovic S, Hartmann MD, Habeck M, Ursinus A, et al. Structure and activity of the N-terminal substrate recognition domains in proteasomal ATPases. Mol Cell. 2009;34(5):580–590. doi: 10.1016/j.molcel.2009.04.030. [DOI] [PubMed] [Google Scholar]
- 54.Wang T, Darwin KH, Li H. Binding-induced folding of prokaryotic ubiquitin-like protein on the Mycobacterium proteasomal ATPase targets substrates for degradation. Nat Struct Mol Biol. 2010;17(11):1352–1357. doi: 10.1038/nsmb.1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang F, Hu M, Tian G, Zhang P, et al. Structural insights into the regulatory particle of the proteasome from Methanocaldococcus jannaschii. Mol Cell. 2009;34(4):473–484. doi: 10.1016/j.molcel.2009.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Smith DM, Kafri G, Cheng Y, Ng D, et al. ATP binding to PAN or the 26S ATPases causes association with the 20S proteasome, gate opening, and translocation of unfolded proteins. Mol Cell. 2005;20(5):687–698. doi: 10.1016/j.molcel.2005.10.019. [DOI] [PubMed] [Google Scholar]
- 57.Striebel F, Hunkeler M, Summer H, Weber-Ban E. The mycobacterial Mpa-proteasome unfolds and degrades pupylated substrates by engaging Pup’s N-terminus. EMBO J. 2010;29(7):1262–1271. doi: 10.1038/emboj.2010.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pearce MJ, Arora P, Festa RA, Butler-Wu SM, et al. Identification of substrates of the Mycobacterium tuberculosis proteasome. EMBO J. 2006;25(22):5423–5432. doi: 10.1038/sj.emboj.7601405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kusmierczyk AR, Kunjappu MJ, Kim RY, Hochstrasser M. A conserved 20S proteasome assembly factor requires a C-terminal HbYX motif for proteasomal precursor binding. Nat Struct Mol Biol. 2011;18(5):622–629. doi: 10.1038/nsmb.2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Smith DM, Chang SC, Park S, Finley D, et al. Docking of the proteasomal ATPases’ carboxyl termini in the 20S proteasome’s alpha ring opens the gate for substrate entry. Mol Cell. 2007;27(5):731–744. doi: 10.1016/j.molcel.2007.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hochstrasser M. Ubiquitin-dependent protein degradation. Annu Rev Genet. 1996;30:405–439. doi: 10.1146/annurev.genet.30.1.405. [DOI] [PubMed] [Google Scholar]
- 62.Vijay-Kumar S, Bugg CE, Wilkinson KD, Cook WJ. Three-dimensional structure of ubiquitin at 2.8 A resolution. Proc Natl Acad Sci U S A. 1985;82(11):3582–3585. doi: 10.1073/pnas.82.11.3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wilkinson KD. Regulation of ubiquitin-dependent processes by deubiquitinating enzymes. FASEB J. 1997;11(14):1245–1256. doi: 10.1096/fasebj.11.14.9409543. [DOI] [PubMed] [Google Scholar]
- 64.Ozkaynak E, Finley D, Solomon MJ, Varshavsky A. The yeast ubiquitin genes: a family of natural gene fusions. EMBO J. 1987;6(5):1429–1439. doi: 10.1002/j.1460-2075.1987.tb02384.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kerscher O, Felberbaum R, Hochstrasser M. Modification of proteins by ubiquitin and ubiquitin-like proteins. Annu Rev Cell Dev Biol. 2006;22:159–180. doi: 10.1146/annurev.cellbio.22.010605.093503. [DOI] [PubMed] [Google Scholar]
- 66.Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. 1998;67:425–479. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- 67.Johnson ES. Protein modification by SUMO. Annu Rev Biochem. 2004;73:355–382. doi: 10.1146/annurev.biochem.73.011303.074118. [DOI] [PubMed] [Google Scholar]
- 68.Rotin D, Kumar S. Physiological functions of the HECT family of ubiquitin ligases. Nat Rev Mol Cell Biol. 2009;10(6):398–409. doi: 10.1038/nrm2690. [DOI] [PubMed] [Google Scholar]
- 69.Deshaies RJ, Joazeiro CA. RING domain E3 ubiquitin ligases. Annu Rev Biochem. 2009;78:399–434. doi: 10.1146/annurev.biochem.78.101807.093809. [DOI] [PubMed] [Google Scholar]
- 70.Chau V, Tobias JW, Bachmair A, Marriott D, et al. A multiubiquitin chain is confined to specific lysine in a targeted short-lived protein. Science. 1989;243(4898):1576–1583. doi: 10.1126/science.2538923. [DOI] [PubMed] [Google Scholar]
- 71.Hough R, Rechsteiner M. Ubiquitin-lysozyme conjugates. Purification and susceptibility to proteolysis. J Biol Chem. 1986;261(5):2391–2399. [PubMed] [Google Scholar]
- 72.Scholz O, Thiel A, Hillen W, Niederweis M. Quantitative analysis of gene expression with an improved green fluorescent protein. p6. Eur J Biochem. 2000;267(6):1565–1570. doi: 10.1046/j.1432-1327.2000.01170.x. [DOI] [PubMed] [Google Scholar]
- 73.Karimova G, Pidoux J, Ullmann A, Ladant D. A bacterial two-hybrid system based on a reconstituted signal transduction pathway. Proc Natl Acad Sci U S A. 1998;95(10):5752–5756. doi: 10.1073/pnas.95.10.5752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pearce MJ, Mintseris J, Ferreyra J, Gygi SP, et al. Ubiquitin-like protein involved in the proteasome pathway of Mycobacterium tuberculosis. Science. 2008;322(5904):1104–1107. doi: 10.1126/science.1163885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Singh A, Mai D, Kumar A, Steyn AJ. Dissecting virulence pathways of Mycobacterium tuberculosis through protein-protein association. Proc Natl Acad Sci U S A. 2006;103(30):11346–11351. doi: 10.1073/pnas.0602817103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Burns KE, Liu WT, Boshoff HI, Dorrestein PC, et al. Proteasomal protein degradation in Mycobacteria is dependent upon a prokaryotic ubiquitin-like protein. J Biol Chem. 2009;284(5):3069–3075. doi: 10.1074/jbc.M808032200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Striebel F, Imkamp F, Sutter M, Steiner M, et al. Bacterial ubiquitin-like modifier Pup is deamidated and conjugated to substrates by distinct but homologous enzymes. Nat Struct Mol Biol. 2009;16(6):647–651. doi: 10.1038/nsmb.1597. [DOI] [PubMed] [Google Scholar]
- 78.Chen X, Solomon WC, Kang Y, Cerda-Maira F, et al. Prokaryotic ubiquitin-like protein pup is intrinsically disordered. J Mol Biol. 2009;392(1):208–217. doi: 10.1016/j.jmb.2009.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sutter M, Striebel F, Damberger FF, Allain FH, et al. A distinct structural region of the prokaryotic ubiquitin-like protein (Pup) is recognized by the N-terminal domain of the proteasomal ATPase Mpa. FEBS Lett. 2009;583(19):3151–3157. doi: 10.1016/j.febslet.2009.09.020. [DOI] [PubMed] [Google Scholar]
- 80.Liao S, Shang Q, Zhang X, Zhang J, et al. Pup, a prokaryotic ubiquitin-like protein, is an intrinsically disordered protein. Biochem J. 2009;422(2):207–215. doi: 10.1042/BJ20090738. [DOI] [PubMed] [Google Scholar]
- 81.Jackson PK, Eldridge AG, Freed E, Furstenthal L, et al. The lore of the RINGs: substrate recognition and catalysis by ubiquitin ligases. Trends Cell Biol. 2000;10(10):429–439. doi: 10.1016/s0962-8924(00)01834-1. [DOI] [PubMed] [Google Scholar]
- 82.Festa RA, McAllister F, Pearce MJ, Mintseris J, et al. Prokaryotic ubiquitin-like protein (Pup) proteome of Mycobacterium tuberculosis [corrected] PLoS One. 2010;5(1):e8589. doi: 10.1371/journal.pone.0008589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Watrous J, Burns K, Liu WT, Patel A, et al. Expansion of the mycobacterial “PUPylome”. Mol Biosyst. 2010;6(2):376–385. doi: 10.1039/b916104j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Poulsen C, Akhter Y, Jeon AH, Schmitt-Ulms G, et al. Proteome-wide identification of mycobacterial pupylation targets. Mol Syst Biol. 2010;6:386. doi: 10.1038/msb.2010.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Guth E, Thommen M, Weber-Ban E. Mycobacterial ubiquitin-like protein ligase PafA follows a two-step reaction pathway with a phosphorylated pup intermediate. J Biol Chem. 2011;286(6):4412–4419. doi: 10.1074/jbc.M110.189282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cerda-Maira FA, McAllister F, Bode NJ, Burns KE, et al. Reconstitution of the Mycobacterium tuberculosis pupylation pathway in Escherichia coli. EMBO Rep. 2011;12(8):863–870. doi: 10.1038/embor.2011.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ghadbane H, Brown AK, Kremer L, Besra GS, et al. Structure of Mycobacterium tuberculosis mtFabD, a malonyl-CoA:acyl carrier protein transacylase (MCAT) Acta Crystallogr Sect F Struct Biol Cryst Commun. 2007;63(Pt 10):831–835. doi: 10.1107/S1744309107042455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Iyer LM, Burroughs AM, Aravind L. Unraveling the biochemistry and provenance of pupylation: a prokaryotic analog of ubiquitination. Biol Direct. 2008;3:45. doi: 10.1186/1745-6150-3-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cerda-Maira FA, Pearce MJ, Fuortes M, Bishai WR, et al. Molecular analysis of the prokaryotic ubiquitin-like protein (Pup) conjugation pathway in Mycobacterium tuberculosis. Mol Microbiol. 2010;77(5):1123–1135. doi: 10.1111/j.1365-2958.2010.07276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Imkamp F, Rosenberger T, Striebel F, Keller PM, et al. Deletion of dop in Mycobacterium smegmatis abolishes pupylation of protein substrates in vivo. Mol Microbiol. 2010;75(3):744–754. doi: 10.1111/j.1365-2958.2009.07013.x. [DOI] [PubMed] [Google Scholar]
- 91.Burns KE, Cerda-Maira FA, Wang T, Li H, et al. “Depupylation” of prokaryotic ubiquitin-like protein from mycobacterial proteasome substrates. Mol Cell. 2010;39(5):821–827. doi: 10.1016/j.molcel.2010.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Komander D, Clague MJ, Urbe S. Breaking the chains: structure and function of the deubiquitinases. Nat Rev Mol Cell Biol. 2009;10(8):550–563. doi: 10.1038/nrm2731. [DOI] [PubMed] [Google Scholar]
- 93.Imkamp F, Striebel F, Sutter M, Ozcelik D, et al. Dop functions as a depupylase in the prokaryotic ubiquitin-like modification pathway. EMBO Rep. 2010;11(10):791–797. doi: 10.1038/embor.2010.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Burns KE, Pearce MJ, Darwin KH. Prokaryotic ubiquitin-like protein provides a two-part degron to Mycobacterium proteasome substrates. J Bacteriol. 2010;192(11):2933–2935. doi: 10.1128/JB.01639-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lamichhane G, Raghunand TR, Morrison NE, Woolwine SC, et al. Deletion of a Mycobacterium tuberculosis proteasomal ATPase homologue gene produces a slow-growing strain that persists in host tissues. J Infect Dis. 2006;194(9):1233–1240. doi: 10.1086/508288. [DOI] [PubMed] [Google Scholar]
- 96.Gold B, Deng H, Bryk R, Vargas D, et al. Identification of a copper-binding metallothionein in pathogenic mycobacteria. Nat Chem Biol. 2008;4(10):609–616. doi: 10.1038/nchembio.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Gottesman S. Proteolysis in bacterial regulatory circuits. Annu Rev Cell Dev Biol. 2003;19:565–587. doi: 10.1146/annurev.cellbio.19.110701.153228. [DOI] [PubMed] [Google Scholar]
- 98.Festa RA, Jones MB, Butler-Wu S, Sinsimer D, et al. A novel copper-responsive regulon in Mycobacterium tuberculosis. Mol Microbiol. 2011;79(1):133–148. doi: 10.1111/j.1365-2958.2010.07431.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Maciag A, Dainese E, Rodriguez GM, Milano A, et al. Global analysis of the Mycobacterium tuberculosis Zur (FurB) regulon. J Bacteriol. 2007;189(3):730–740. doi: 10.1128/JB.01190-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Siegrist MS, Unnikrishnan M, McConnell MJ, Borowsky M, et al. Mycobacterial Esx-3 is required for mycobactin-mediated iron acquisition. Proc Natl Acad Sci U S A. 2009;106(44):18792–18797. doi: 10.1073/pnas.0900589106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Serafini A, Boldrin F, Palu G, Manganelli R. Characterization of a Mycobacterium tuberculosis ESX-3 conditional mutant: essentiality and rescue by iron and zinc. J Bacteriol. 2009;191(20):6340–6344. doi: 10.1128/JB.00756-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Nanamiya H, Akanuma G, Natori Y, Murayama R, et al. Zinc is a key factor in controlling alternation of two types of L31 protein in the Bacillus subtilis ribosome. Mol Microbiol. 2004;52(1):273–283. doi: 10.1111/j.1365-2958.2003.03972.x. [DOI] [PubMed] [Google Scholar]
- 103.Natori Y, Nanamiya H, Akanuma G, Kosono S, et al. A fail-safe system for the ribosome under zinc-limiting conditions in Bacillus subtilis. Mol Microbiol. 2007;63(1):294–307. doi: 10.1111/j.1365-2958.2006.05513.x. [DOI] [PubMed] [Google Scholar]
- 104.Liu T, Ramesh A, Ma Z, Ward SK, et al. CsoR is a novel Mycobacterium tuberculosis copper-sensing transcriptional regulator. Nat Chem Biol. 2007;3(1):60–68. doi: 10.1038/nchembio844. [DOI] [PubMed] [Google Scholar]
- 105.Ward SK, Abomoelak B, Hoye EA, Steinberg H, et al. CtpV: a putative copper exporter required for full virulence of Mycobacterium tuberculosis. Mol Microbiol. 2010;77(5):1096–1110. doi: 10.1111/j.1365-2958.2010.07273.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ward SK, Hoye EA, Talaat AM. The global responses of Mycobacterium tuberculosis to physiological levels of copper. J Bacteriol. 2008;190(8):2939–2946. doi: 10.1128/JB.01847-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wolschendorf F, Ackart D, Shrestha TB, Hascall-Dove L, et al. Copper resistance is essential for virulence of Mycobacterium tuberculosis. Proc Natl Acad Sci U S A. 2011;108(4):1621–1626. doi: 10.1073/pnas.1009261108. [DOI] [PMC free article] [PubMed] [Google Scholar]